")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Sequential delivery of an anticancer drug and combined immunomodulatory nanoparticles for efficient chemoimmunotherapy
Authors Heo MB, Kim S, Yun W, Lim YT
Received 8 June 2015
Accepted for publication 18 July 2015
Published 24 September 2015 Volume 2015:10(1) Pages 5981—5993
DOI https://doi.org/10.2147/IJN.S90104
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Min Beom Heo,1,2 Sun-Young Kim,1 Wan Soo Yun,3 Yong Taik Lim1
1SKKU Advanced Institute of Nanotechnology (SAINT), School of Chemical Engineering, Sungkyunkwan University, Suwon, 2Center for Nanosafety Metrology, Division of Convergence Technology, Korea Research Institute of Standards and Science, Daejeon, 3Department of Chemistry, Sungkyunkwan University, Suwon, Republic of Korea
Abstract: Chemoimmunotherapy combines chemotherapy based on anticancer drugs with immunotherapy based on immune activators to eliminate or inhibit the growth of cancer cells. In this study, water-insoluble paclitaxel (PTX) was dispersed in water using hyaluronic acid (HA) to generate a tumor-associated antigen in the tumor microenvironment. Cytosine–phosphate–guanosine oligodeoxynucleotides (CpG ODNs) were used to enhance the T helper (Th) 1 immune response. However, CpG ODNs also induced the secretion of interleukin-10 (IL-10) that reduces the Th1 response and enhances the T helper 2 (Th2) response. Therefore, RNA interference was used to downregulate IL-10 secretion from bone marrow-derived dendritic cells (BMDCs). For the combined immunomodulation of BMDCs, we fabricated two types of poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) containing CpG ODNs to activate BMDCs via Toll-like receptor 9 (CpG ODN-encapsulated PLGA NPs, PCNs) or a small interfering RNA to silence IL-10 (IL-10 small interfering RNA-encapsulated PLGA NPs, PINs). Treatment of BMDCs with both types of PLGA NPs increased the Th1/Th2 cytokine (IL-12/IL-10) expression ratio, which is important for the effective induction of an antitumor immune response. After primary injection with the HA/PTX complex, the tumor-associated antigen was generated and taken up by tumor-recruited BMDCs. After a secondary injection with immunomodulating PCNs and PINs, the BMDCs became activated and migrated to the tumor-draining lymph nodes. As a result, the combination of chemotherapy using the HA/PTX complex and immunotherapy using PCNs and PINs not only efficiently inhibited tumor growth but also increased the animal survival rate. Taken together, our results suggest that the sequential treatment of cancer cells with a chemotherapeutic agent and immunomodulatory nanomaterials represents a promising strategy for efficient cancer therapy.
Keywords: nanoparticles, chemoimmunotherapy, dendritic cells, cancer, siRNA
Introduction
Cancer is usually treated using surgery, chemotherapy, radiotherapy, or immunotherapy.1,2 Surgery, which involves removal of the tumor and the surrounding tissue, is the oldest form of cancer treatment and remains the most common treatment for various types of cancer. However, it is not typically used to treat blood cancers such as leukemia and lymphoma. Moreover, the combination of surgery with other therapies is necessary because most neoplasms can metastasize into other organs.3–5 Chemotherapy is generally used to treat cancer with anticancer drugs such as paclitaxel (PTX) or doxorubicin (DOX). However, chemical substances can suppress the immune system by diminishing immune cells.6 PTX, one of the most effective antineoplastic drugs, displays excellent therapeutic efficacy against a broad range of cancers, including acute leukemia, ovarian cancer, and breast cancer.7–10 One limitation of PTX is its poor solubility in water.11–14 In recent years, the solubility of PTX (commercial brand name TAXOL® [Bristol-Myers Squibb, New York, NY, USA]) has been improved by mixing the drug with surfactants such as Cremophor EL and ethanol.15 However, the use of Cremophor EL in TAXOL® causes acute hypersensitivity reactions and occasionally leads to neurotoxicity, nephrotoxicity, vasodilatation, labored breathing, lethargy, and hypotension.16,17 Therefore, many researchers have attempted to formulate PTX-based compounds lacking these side effects using nanoparticles (NPs), liposomes, and emulsions.18–21 In fact, nab-PTX (an albumin-bound 130 nm NP form of PTX) was approved by the US Food and Drug Administration (FDA) in 2005 for the treatment of refractory, metastatic, or relapsed breast cancer.22
Cancer immunotherapy uses the immune system to fight the tumor. Activating the immune system for the treatment of cancer is an important goal in immunotherapy. The activity of immune cells, such as dendritic cells (DCs), macrophages, natural killer cells, and T-cells, is enhanced by various immune activators, including cytokines, immune adjuvants, and Toll-like receptor (TLR) ligands.23–26 The TLR ligand unmethylated cytosine–phosphate–guanosine oligodeoxynucleotide (CpG ODN) binds to TLR9 receptors on the endosomal membranes of DCs or macrophages and consequently activates the immune response by inducing the secretion of various cytokines.27,28 These immune responses can increase anticancer activity by generating T helper 1 (Th1) cells and cytotoxic T-lymphocytes (CTLs); however, these responses simultaneously result in the expression of anti-inflammatory cytokines that downregulate the secretion of Th1 cytokines, major histocompatibility complex class II antigens, and costimulatory molecules from DCs or macrophages. The immunosuppressive cytokine interleukin-10 (IL-10) suppresses Th1 cell differentiation and the activity of antigen-presenting cells (APCs). Moreover, IL-10 polarizes the immune system toward a Th2 response and promotes the induction of immunosuppressive regulatory T-cells in the tumor microenvironment.29–31 Recently, many researchers have attempted to silence the expression of IL-10 in DCs using small interfering RNA (siRNA). The decrease in IL-10 expression in DCs induces the enhancement of Th1 and CTL responses and consequently enhances the antitumor immune response.32,33
A combined chemoimmunotherapeutic approach may be beneficial for the efficient elimination of cancer. Chemotherapy using anticancer drugs can kill the tumor cells, causing the cancer to shrink. As a result, tumor-derived antigens, such as whole tumor cells, peptides, or proteins isolated from the tumor cells, can be efficiently internalized by APCs, thereby increasing the antitumor immunity of CTLs.34,35 In this in vivo study, we sequentially subjected tumor-bearing mice to chemotherapy consisting of PTX dissolved in hyaluronic acid (HA) and to immunotherapy using CpG ODNs and IL-10 siRNA incorporated into poly(lactic-co-glycolic acid) (PLGA) NPs (Figure 1). The sequential treatment with chemotherapeutic drugs followed by a combined immunostimulation strategy resulted in a synergistic effect against solid tumors. The Institutional Review Board of the Sungkyunkwan University does not require approvals for these case studies.
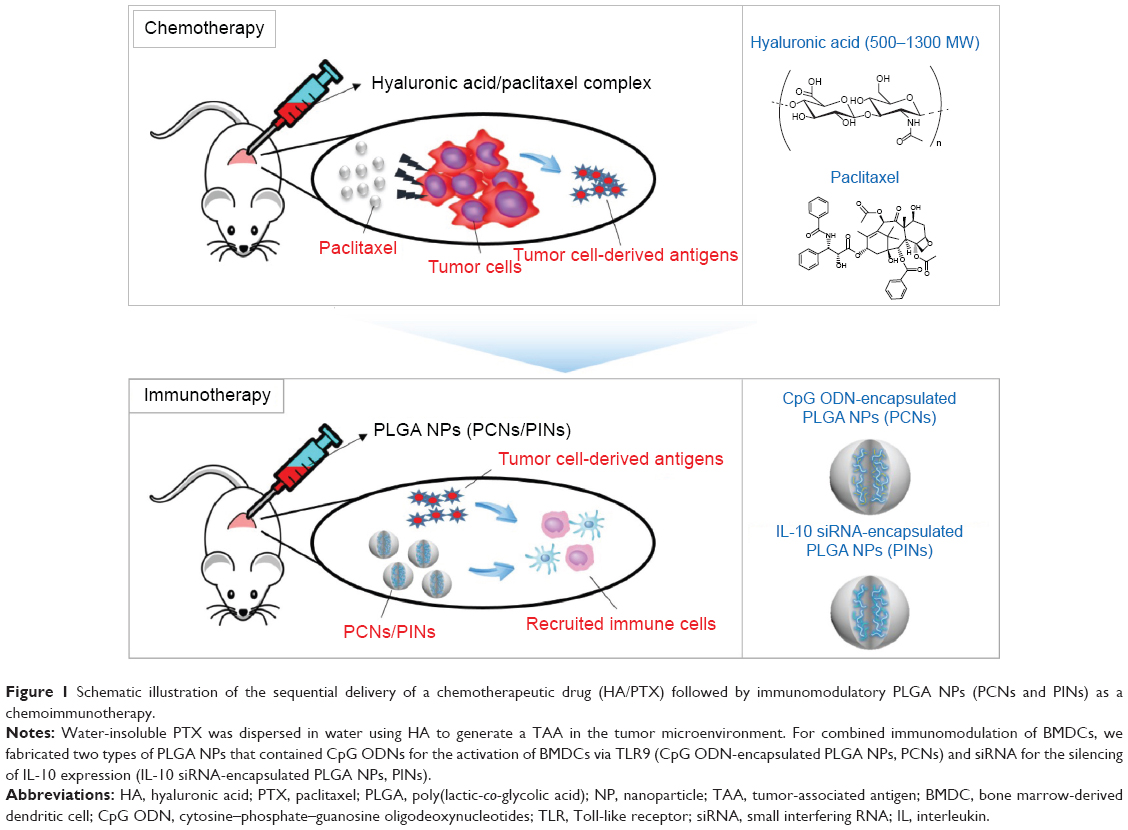 | Figure 1 Schematic illustration of the sequential delivery of a chemotherapeutic drug (HA/PTX) followed by immunomodulatory PLGA NPs (PCNs and PINs) as a chemoimmunotherapy. |
Materials and methods
Preparation of the polymer–drug complex
For the preparation of the HA/PTX complex, 4 mg of PTX (ChemieTek, Indianapolis, IN, USA) in 0.2 mL of methanol was mixed with 4 mg of HA (LMW; Bioland, Cheonan, Korea) in 4 mL of distilled water (DW) at room temperature for 1 hour. The above solution was lyophilized to remove organic solvents. The lyophilized powder was redispersed in DW to obtain a stable dispersion.
Preparation of immunomodulatory PLGA NPs
PLGA NPs containing CpG ODN or IL-10 siRNA (PCNs or PINs) were synthesized by the double-emulsion solvent evaporation method. Briefly, 0.5 mg of indocyanine green (ICG, MW =774 Da; Dongindang Pharm Co, Siheung, Korea) was dissolved in 0.3 mL of DW. A 176 μg of poly-l-lysine (PLL; Sigma-Aldrich, St Louis, MO, USA) was mixed with 140 μg of CpG ODNs or IL-10 siRNA (Bioneer, Daejeon, Korea) (amine group [N]:phosphate group [P] ratio =2:1) in ICG-dissolved DW (0.3 mL) for 30 minutes. Then, the PLL/CpG ODN or PLL/IL-10 siRNA complex was mixed with 2 mL of chloroform containing 60 mg of PLGA (Resomer® RG502H, monomer ratio 50:50, MW =7–17 kDa and inherent viscosity 0.16–0.24 dL/g; Boehringer Ingelheim, Ingelheim, Germany) using a microtip probe sonicator at 4°C for 1 minute. The primary emulsion was further emulsified with a secondary aqueous phase (10 mL of 2.5% w/v polyvinyl alcohol, 80% hydrolyzed, Sigma-Aldrich) in endotoxin-free water at 4°C for 2 minutes to form a secondary (w/o/w) emulsion. The resultant emulsion was agitated using a magnetic stirrer at room temperature overnight until the chloroform was completely evaporated. After 16 hours, the suspension of PLGA NPs was washed three times with DW at 4°C (13,000 rpm, 20 minutes) and freeze-dried. To quantify the encapsulated CpG ODNs or IL-10 siRNA, the PLGA NPs were dissolved in 0.1 M NaOH and 0.1% sodium dodecyl sulfate, incubated at room temperature overnight and analyzed under UV light (260 nm). The binding efficiency between PLL and CpG ODNs or IL-10 siRNA was evaluated using a gel retardation assay. Electrophoresis was performed on a 1% agarose gel at a current of 100 V for 20 minutes in TAE buffer (40 mM Tris–HCl, 1% (v/v) acetic acid, and 1 mM EDTA [ethylenediaminetetraacetic acid]). The migration of the PLL/CpG ODN or PLL/IL-10 siRNA complex was visualized by staining with ethidium bromide.
Characterization of PLGA NPs
The size and surface morphology of PLGA NPs was characterized via field emission scanning electron microscopy (JSM-7000F; JEOL, Tokyo, Japan). The size distribution and zeta potential of the PLGA NPs were analyzed via dynamic light scattering (DLS) using an electrophoretic light scattering photometer (ELS-Z; Otsuka Electronics, Osaka, Japan).
Mice and cell lines
Female C57BL/6 (H-2b) mice 5–6 weeks of age were purchased from KOATECH (Pyeongtaek, Korea). The mice were maintained under pathogen-free conditions. All experiments employing mice were performed in accordance with the Korean NIH guidelines for the care and use of laboratory animals. The murine melanoma cell line B16-F10 was purchased from American Type Culture Collection and was cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific) and 1% antibiotic-antimycotic solution (Thermo Fisher Scientific) at 37°C in 5% CO2/95% humidified air.
Generation of DCs from mouse bone marrow cells
Femurs and tibiae were collected, and the muscle attachments were carefully removed. Intact bones were incubated in 70% (v/v) ethanol for 1 minute for disinfection and were then washed with phosphate-buffered saline (PBS; HyClone). Next, both ends of the bones were cut with scissors, and the marrow was flushed out with RPMI 1640 medium (Thermo Fisher Scientific) using a 1 mL syringe connected to a 26-gauge needle. Clusters within the marrow suspension were disintegrated via vigorous pipetting. After one wash (15,000 rpm, 5 minutes) in RPMI 1640 medium, the red blood cells were depleted using 0.83 M NH4Cl buffer (Sigma-Aldrich). Bone marrow cells (2×106 cells) were collected and cultured in 100 mm Petri dishes containing 10 mL of RPMI medium supplemented with 10% heat-inactivated fetal bovine serum, 50 IU/mL penicillin, 50 μg/mL streptomycin, and 20 ng/mL mouse recombinant granulocyte-macrophage colony-stimulating factor (R&D Systems, Inc., Minneapolis, MN, USA). After 7 days, the nonadherent and loosely adherent cells (imDCs) were harvested, washed, and used for in vitro and in vivo experiments.
Silencing of IL-10 protein in bone marrow-derived dendritic cells
Bone marrow-derived dendritic cells (BMDCs) (2×106 cells) were transfected with IL-10 siRNA (100 nM, 300 nM, or 500 nM) in PLGA NPs for 24 hours at 37°C. Then, the siRNA-transfected DCs were stimulated with 5 μg/mL of PCNs for 24 hours at 37°C. The level of IL-10 messenger RNA in the DCs was estimated via reverse transcriptase–polymerase chain reaction (RT-PCR) and real-time PCR analyses. Total RNA from the cells was isolated using TRIzol reagent (TaKaRa, Japan); a total of 2 μg of total RNA was reverse-transcribed using an oligo(dT) primers and monkey murine leukemia virus RT (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s protocols. The complementary DNA product was amplified via PCR. Primers were designed as follows: mouse IL-10: 5′-GTACAGCCGGGAAGACAATAA-3′ (forward) and 5′-GCATTAAGGAGTCGGTTAGCA-3′ (reverse); mouse GAPDH: 5′-AGGCCGGTGCTGAGTATGTC-3′ (forward) and 5′-TGCCTGCTTCACCACCTTCT-3′ (reverse) (Bioneer). The expression of the housekeeping gene GAPDH was used to normalize the complementary DNA input.
Cytokine and maturation assays
BMDCs (2×106 cells) were transfected with IL-10 siRNA (100 nM, 300 nM, or 500 nM) in PINs for 24 hours at 37°C. Then, the siRNA-transfected DCs were stimulated with 5 μg/mL CpG ODNs in PCNs for 24 hours at 37°C. The expression of tumor necrosis factor (TNF)-α, IL-6, IL-12p70, and IL-10 in the DCs was analyzed using cytokine-specific enzyme-linked immunosorbent assays (ELISAs) (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions. Cytokine concentrations were quantified using a VICTOR3™ microplate reader (PerkinElmer, Inc., Waltham, MA, USA) by measuring the optical density (OD) at 450 nm and at the reference wavelength 570 nm. The expression of DC maturation markers was measured via flow cytometry after staining with FITC-anti-CD40, FITC-anti-CD80, and FITC-anti-CD86 antibodies (BD Biosciences).
Evaluation of in vivo antitumor activity
B16-F10 melanoma cells (1×105) were inoculated into the right flank of 5- to 6-week-old C57BL/6 mice. Ten days after tumor implantation, animals bearing tumors with an average diameter of 100 mm3 were selected. These mice were divided into five treatment groups (n=12 each) and were numbered accordingly. Each drug was administered via intratumoral injection. The HA/PTX complex was injected at day 10 after tumor implantation, and the PLGA NP treatment was continued from day 14 to 22 at 3-day intervals (on days 14, 18, and 22). The tumor diameters were measured until the 26th day after tumor implantation using a sliding caliper. The tumor volume was calculated using the following formula: tumor volume (mm3) = length × (width)2/2. For survival analysis, the treated animals were observed for 36 days.
Evaluation of the in vivo immune status of the tumor-draining lymph nodes
Mice were inoculated with cancer cells as described earlier and were treated via intratumoral injection. On day 19, the tumor-draining lymph node was isolated and filtered through a 70-μm cell strainer in complete RPMI medium for the preparation of a single cell suspension. The cells were fixed and then labeled with the following fluorescent marker-conjugated antibodies for specific cell types: FITC-anti-mouse CD14 for macrophages (BD Biosciences) and FITC-anti-mouse CD11c for DCs (BD Biosciences). The cells were incubated at 4°C for 30 minutes, followed by two washes with PBS. Then, a minimum of 104 cells were analyzed via fluorescence using the BD Accuri™ C6 flow cytometer (BD Biosciences).
In vivo near-infrared imaging of the tumor-draining lymph nodes
Mice were inoculated with cancer cells as described earlier and were treated via intratumoral injection. On day 19, the mice were anesthetized and placed in a sealed dark chamber that was connected to a charge-coupled device camera. Near-infrared (NIR) images of the tumor-draining lymph node were acquired using a 760-nm light-emitting diode light as the excitation light source and an 845/55 nm emission filter.
Results and discussion
Preparation and characterization of the HA/PTX complex
In this study, a low dose of the anticancer drug PTX was dispersed in water with the aid of HA. We examined whether treatment with this complex combined with immunostimulatory NPs exerted a synergistic effect against solid tumors. PTX was dispersed in aqueous solutions for an extended period using the HA matrix. However, PTX that was dispersed in DW alone aggregated and sedimented due to its hydrophobicity (Figure 2). Based on our previous results,36 we prepared the HA/PTX complex for the injection of 200 μg of PTX per mouse, resulting in weak anticancer activity.
 | Figure 2 Digital images of PTX dispersed in distilled water (DW) and in aqueous HA solution. |
Preparation and characterization of PLGA NPs
Two types of PLGA NPs (PCNs and PINs) encapsulating immunomodulatory molecules (CpG ODNs and IL-10 siRNA, respectively) and an NIR imaging tracer (ICG) were fabricated using the emulsion evaporation method. The average size and surface charge of the NPs were measured via DLS (Table 1). The average diameters of the PCNs and PINs were 142.9 nm and 151.0 nm, respectively. Based on zeta potential analysis, the surface charge of the synthesized PCNs and PINs was strongly negative, ranging from −15 to −25 mV. The amount of CpG ODNs encapsulated by PCNs was 4.7±0.32 μg/mg of PCN. IL-10 siRNA was efficiently encapsulated by PINs at a load of 3.69±0.69 μg/mg of PINs. A cationic polymer (PLL) was combined with the CpG ODNs or the IL-10 siRNA to enhance the loading efficiency of the small-sized siRNA onto the hydrophobic PLGA matrix.37 The capacity of the binding of CpG ODNs or IL-10 siRNA to PLL prepared at various N:P ratios was evaluated using a gel retardation assay (Figure 3A). PLL/DNA or RNA complexes were well combined at N:P ratios ranging from 2:1 to 5:1. The positively charged amine groups of PLL neutralized the negatively charged phosphate groups in the DNA or RNA backbone, thus retarding the mobility of the CpG ODNs and the IL-10 siRNA based on ethidium bromide staining. Naked CpG ODN or IL-10 siRNA was used as a control, and the band intensity decreased at increasing N:P ratios. Based on the scanning electron microscopy images, the PCNs and the PINs appeared to be spherical in shape with a smooth surface (Figure 3B). The size of the PCNs and the PINs was approximately 100–200 nm, which was consistent with the result obtained via DLS.
 | Table 1 Characterization of immunomodulatory PLGA NPs |
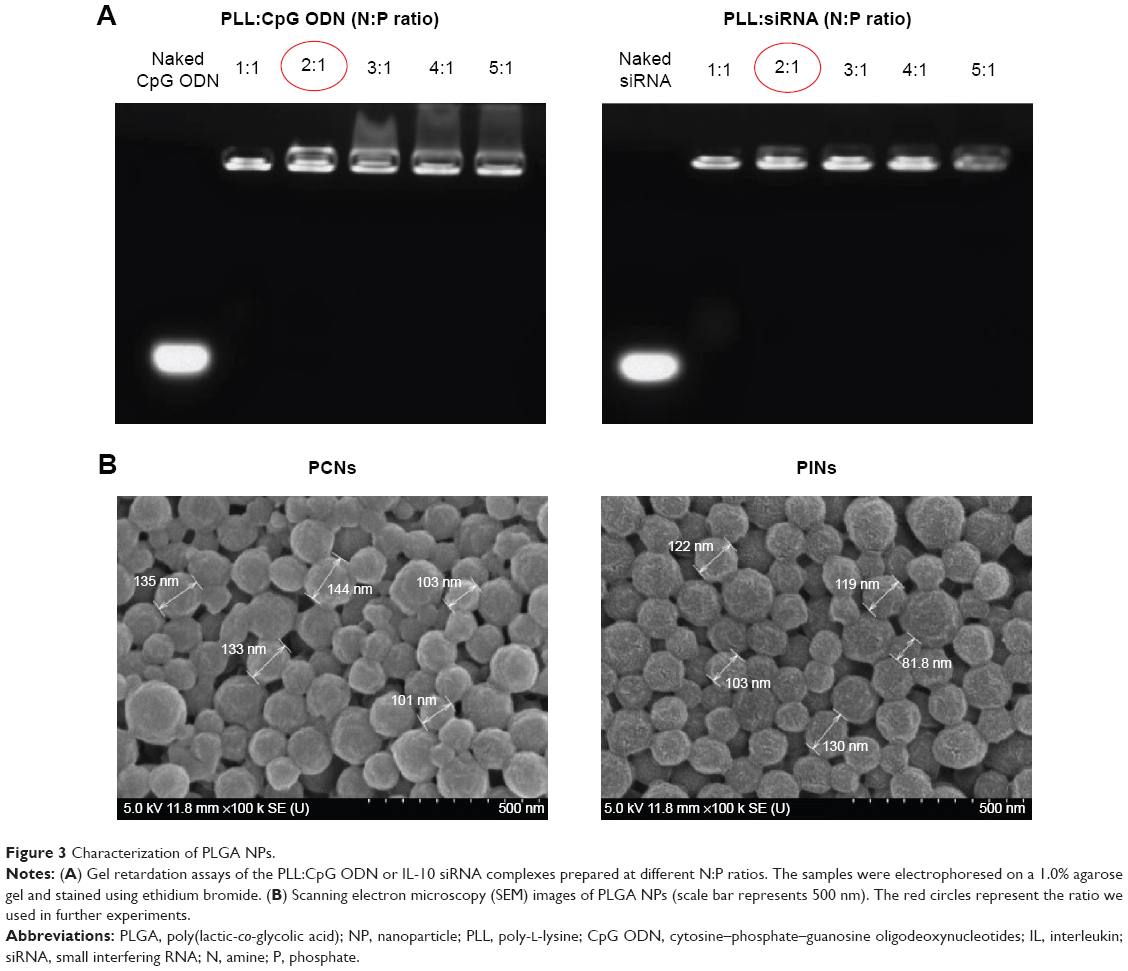 | Figure 3 Characterization of PLGA NPs. |
In vitro gene silencing of BMDCs treated with PINs
The specificity of siRNA for gene inhibition was investigated after transfecting BMDCs with PINs containing various concentrations of IL-10 siRNA. Immature BMDCs were transfected with empty PLGA NPs (mock) or with PINs encapsulating 100 nM, 300 nM, or 500 nM siRNA. After 24 hours, the BMDCs were matured via the addition of 5 μg/mL PCNs encapsulating CpG ODNs. After maturation in CpG ODNs for 24 hours, the in vitro gene-silencing effects of PINs were determined via RT-PCR (Figure 4A) and real-time PCR (Figure 4B) analyses. The level of knock down was dependent on the treated concentration of siRNA (100–500 nM). The PINs containing 500 nM IL-10 siRNA reduced the IL-10 gene expression level by ~70% compared with the mock treatment.
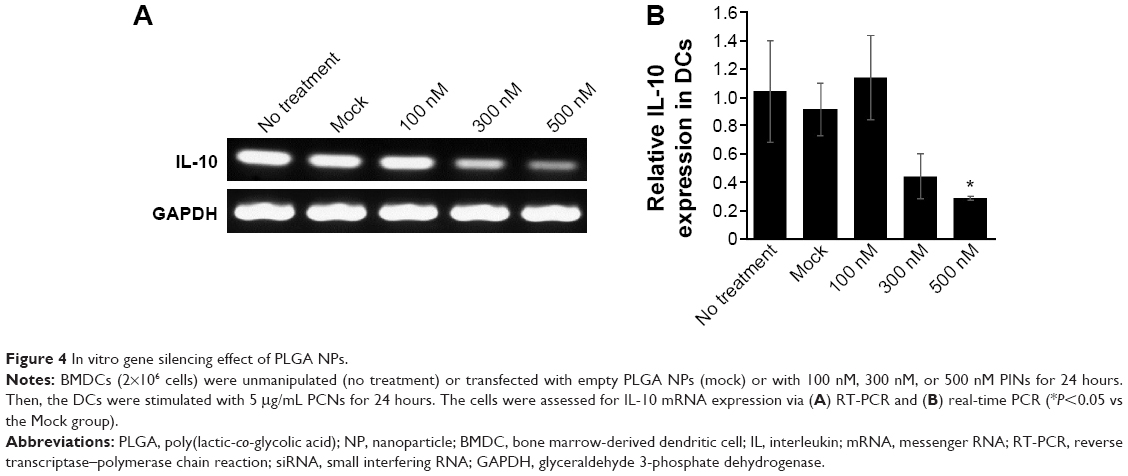 | Figure 4 In vitro gene silencing effect of PLGA NPs. |
In vitro activation and maturation of BMDCs treated with PCNs and PINs
To assess the IL-10-silencing effect of the PINs, we measured the level of pro-inflammatory cytokines (TNF-α, IL-6, and IL-12p70) secreted by BMDCs transfected with PINs containing various concentrations of IL-10 siRNA. Then, the TNF-α, IL-6, and IL-12p70 cytokine levels in the culture supernatants were determined via ELISA following incubation in 5 μg/mL PCNs encapsulating CpG ODNs for 24 hours. As shown in Figure 5A–C, cytokine expression in BMDCs was increased at increasing concentrations of IL-10 siRNA. These results suggested that the IL-10 siRNA in the PINs enhanced the activation of BMDCs, resulting in the expression of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-12p70, in vitro.
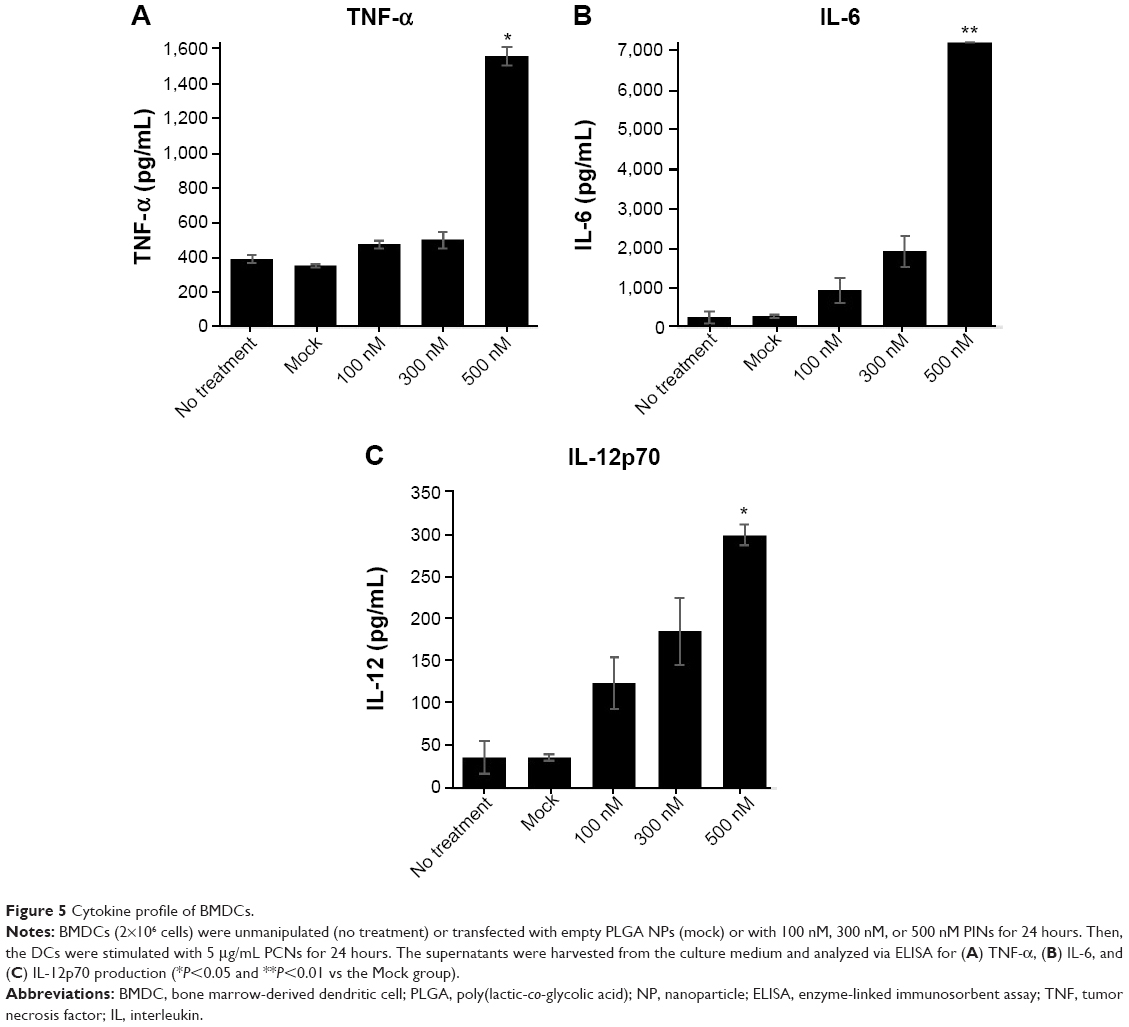 | Figure 5 Cytokine profile of BMDCs. |
Figure 6 shows the balance of Th1/Th2 cytokine expression in BMDCs treated with PCNs or both PCNs and PINs. In the tumor microenvironment, Th1-dominant immunity is critically important for the induction of antitumor cellular immunity in vivo. Because Th2 cytokines have been shown to inhibit Th1 responses, IL-10 (a representative Th2 cytokine) may be considered as a CTL-inhibitory cytokine. Treatment of BMDCs with PCNs enhanced the Th1 response (IL-12p70) and induced an elevated Th2 response (IL-10) (Figure 6A and B). The increased production of IL-10 promoted a shift in the Th1/Th2 balance toward a Th2 response, thereby inhibiting the Th1 response. However, treatment of the BMDCs with both PCNs and PINs resulted in the inhibition of CpG ODN-induced IL-10 production due to gene silencing. Thus, these results indicated that the cotransfection of PCNs and PINs enhances the shift in the Th1/Th2 balance toward a Th1 response compared with transfection with PCNs alone (Figure 6C).
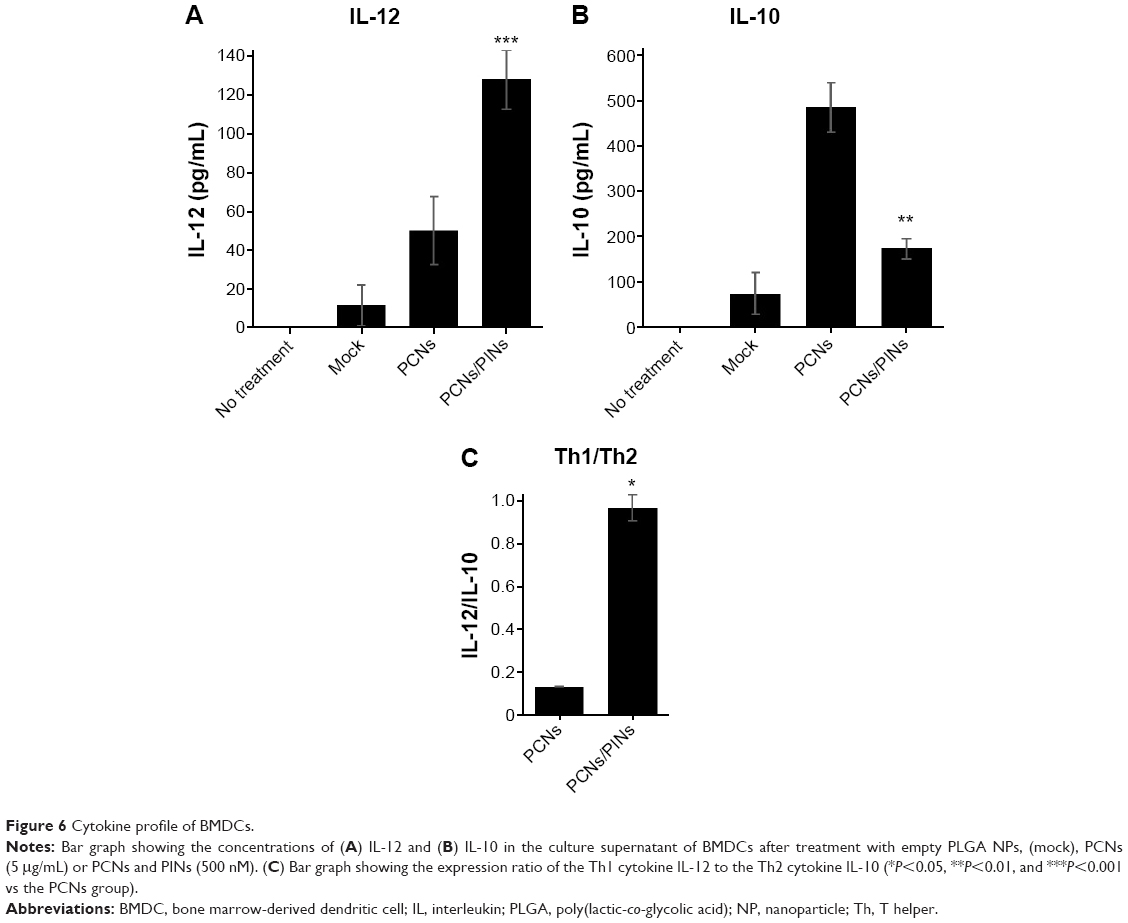 | Figure 6 Cytokine profile of BMDCs. |
To confirm the effects of IL-10 silencing using PINs on the BMDC phenotype after maturation, we analyzed the expression of cell surface markers via flow cytometry (Figure 7). In general, CpG ODN treatment induced the upregulation of maturation markers, including adhesion-related (CD40) and costimulatory (CD80 and CD86) molecules, in immature BMDCs. Therefore, BMDCs treated with PCNs were confirmed to display upregulated CD40, CD80, and CD86 expression levels on their surfaces. The siRNA-mediated knock down of IL-10 did not affect BMDC maturation.
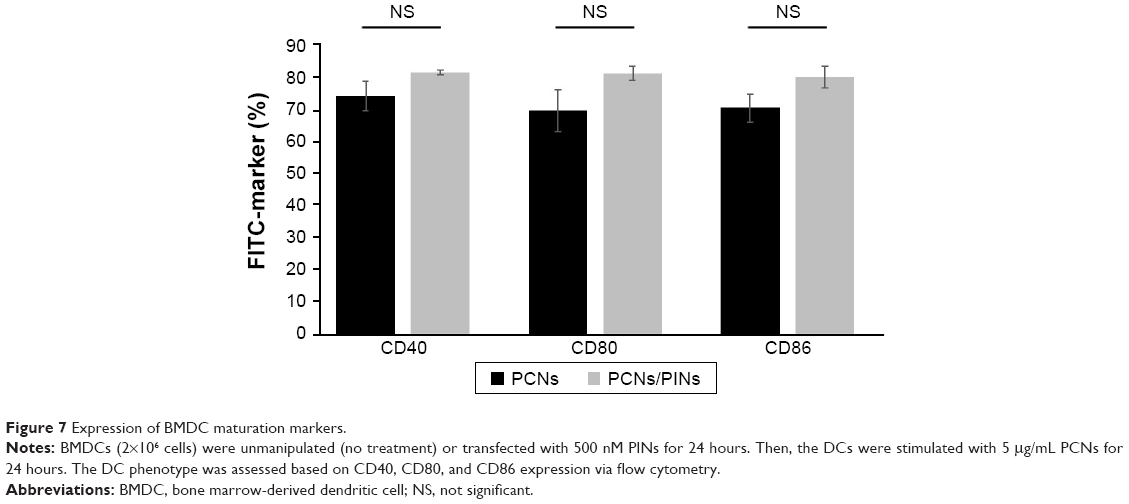 | Figure 7 Expression of BMDC maturation markers. |
In vivo antitumor efficacy of chemoimmunotherapy
To evaluate the in vivo antitumor effects of chemoimmunotherapy using the HA/PTX complex and two immunomodulatory NPs (PCNs and PINs), the tumor volume, which was initially ~120 mm3, was monitored for 26 days after intratumoral injection (at 3-day intervals) with the HA/PTX complex or with PCNs and PINs (Figure 8A). The HA/PTX complex inhibited tumor growth to some extent, whereas HA alone exerted no effect. Additionally, no difference in tumor growth inhibition was observed between cotreatment of PCNs and the HA/PTX complex and treatment with the HA/PTX complex alone. These results indicated that treatment with PCNs alone might be insufficient to induce an antitumor response due to IL-10 expression by tumor-infiltrating DCs or macrophages. Interestingly, the combined treatment with the HA/PTX complex and both PCNs and PINs drastically inhibited the growth of the solid tumors. Figure 8B shows Kaplan–Meier survival curves of the mice treated with various therapeutic materials. The group of mice treated with the HA/PTX complex and both PCNs and PINs exhibited >40% survival at 36 days postinjection, whereas in the other groups, all mice died within 36 days.
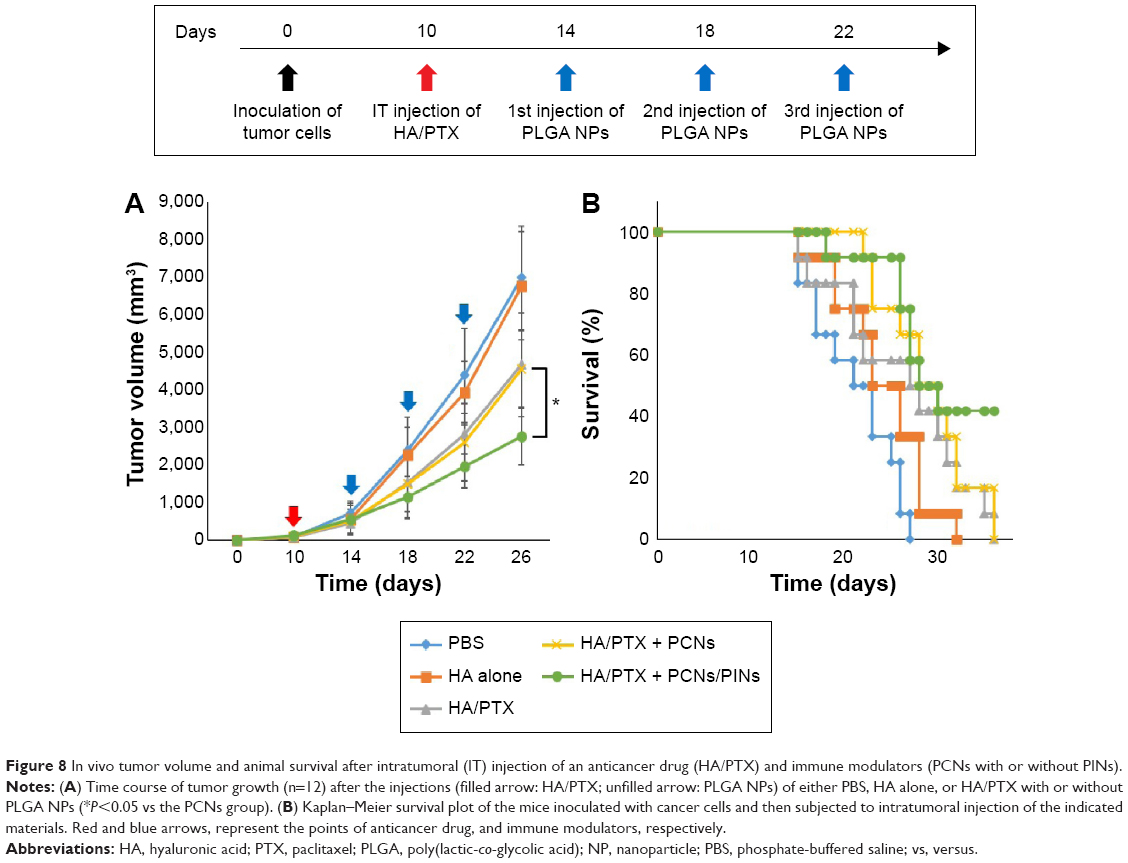 | Figure 8 In vivo tumor volume and animal survival after intratumoral (IT) injection of an anticancer drug (HA/PTX) and immune modulators (PCNs with or without PINs). |
In vivo tracking of APCs using NIR fluorescence imaging and the status of immune cells in the tumor-draining lymph nodes
APCs such as macrophages and DCs capture and process tumor-derived antigens in the tumor microenvironment following maturation. Then, the mature APCs migrate to secondary lymphoid tissues, such as the draining lymph nodes. We investigated whether the injected NPs migrated to the tumor-draining lymph nodes following their internalization by APCs. ICG-loaded NPs were injected at the tumor site, and their migration was analyzed using an NIR optical imaging system. The ICG-loaded NPs efficiently migrated to tumor-draining lymph nodes and generated strong NIR signals (Figure 9A). These results suggested that the NPs were taken up by recruited APCs after intratumoral injection. The NP-loaded DCs migrated to a lymph node for the priming of naive T-cells that, in turn, induce activation marker expression, cytokine secretion, and proliferation. We also confirmed the presence of an ex vivo NIR signal in the inguinal lymph nodes dissected from the NP-injected mice (Figure 9B). To confirm the in vivo immune status of APCs after treatment with PCNs and PINs, the tumor-draining lymph nodes were analyzed for the presence of populations of CD14+ macrophages and CD11c+ DCs (Figure 10). After combined injection of the HA/PTX complex and both PCNs and PINs in the tumor microenvironment, the percentage of CD14+ macrophages detected in the tumor-draining lymph nodes was increased to 2.9% compared to 1.3% in those from the PBS-injected controls (Figure 10A). Moreover, the percentage of CD11c+ DCs was increased to 9.9% following this combined treatment compared to 8.7% for the control PBS injection (Figure 10B). The percentage of APCs was increased to some extent in the PCNs-treated group; however, as observed in the tumor challenge experiment, PCNs treatment alone may be insufficient for the inhibition of tumor growth. In fact, we have analyzed immunostimulatory Th1 cytokines (TNF-α, IL-6, and IL-12) in DCs isolated from the tumor-draining lymph nodes in our previous study. Cytokine expression was increased when the DCs were treated with CpG ODN-encapsulated PLGA NPs.38 The current in vivo experimental results are also consistent with the previous analysis. Therefore, the sequential treatment of cancer cells with a chemotherapeutic agent and immunomodulatory nanomaterials represents a promising strategy for efficient cancer therapy. The combination of the HA/PTX complex and both PCNs and PINs displayed excellent potential to activate APCs at the injection site and naive T-cells in the lymph nodes, thereby generating a potent antitumor immune response.
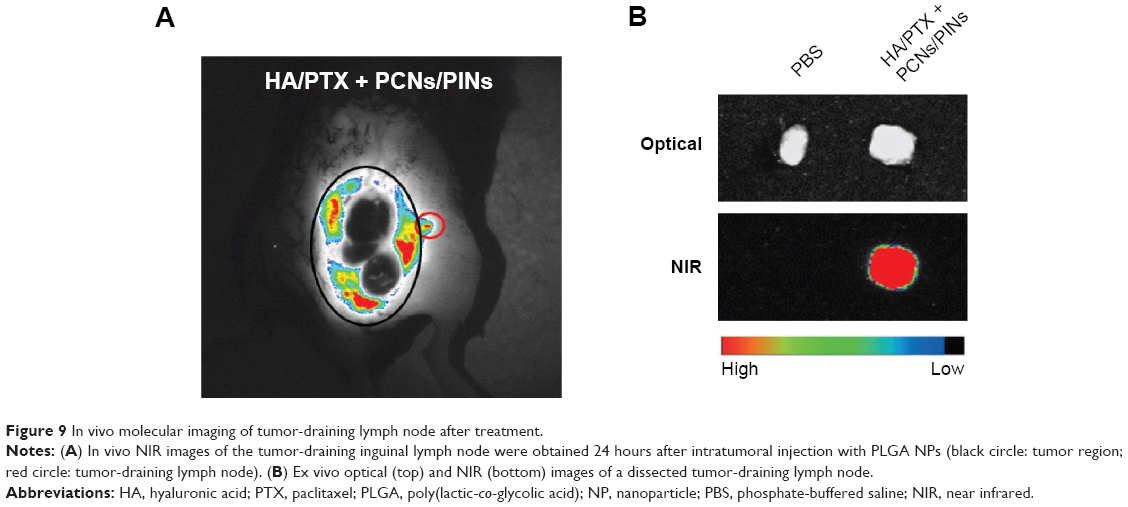 | Figure 9 In vivo molecular imaging of tumor-draining lymph node after treatment. |
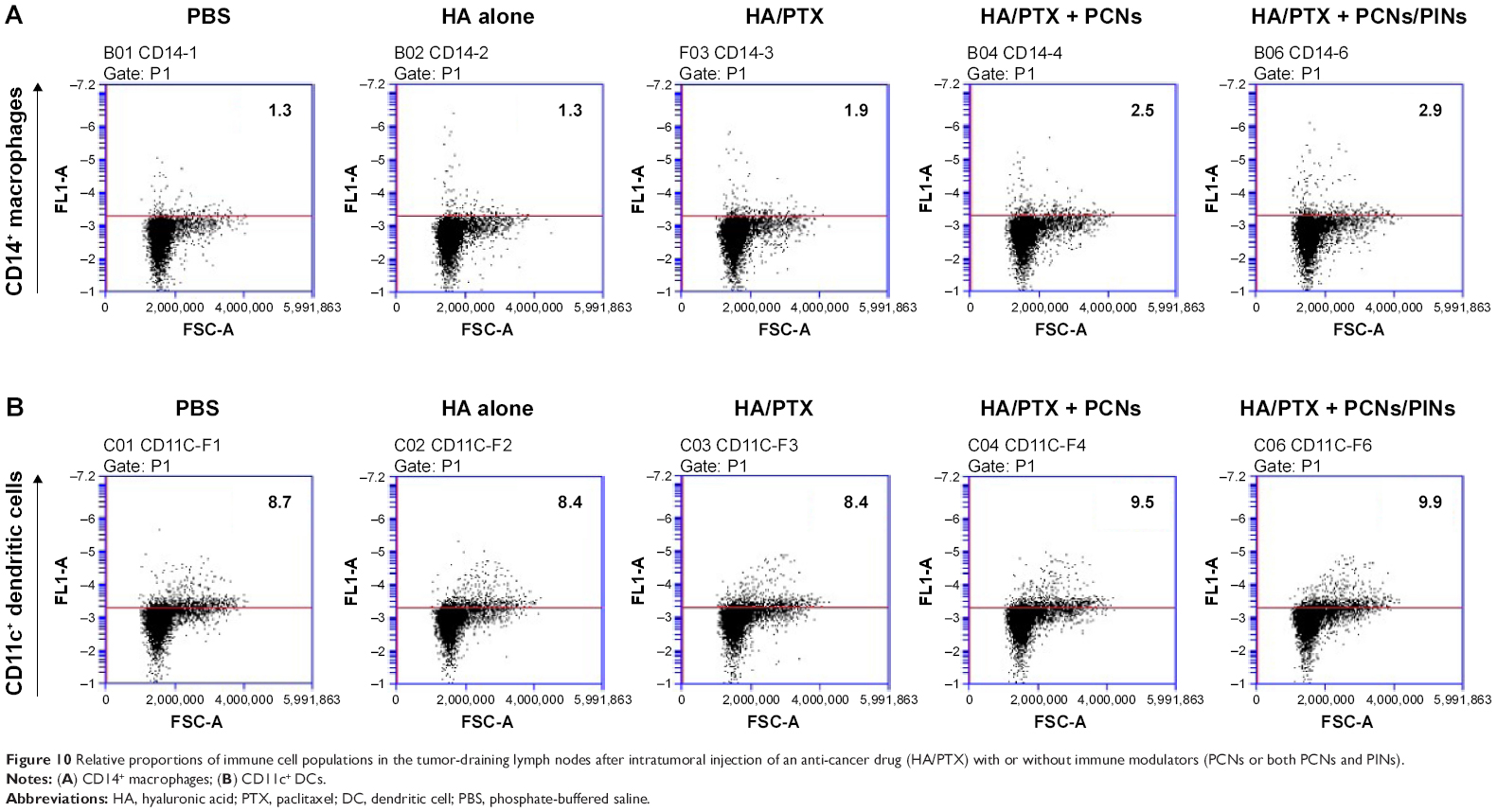 | Figure 10 Relative proportions of immune cell populations in the tumor-draining lymph nodes after intratumoral injection of an anti-cancer drug (HA/PTX) with or without immune modulators (PCNs or both PCNs and PINs). |
Conclusion
In this study, we developed a system utilizing an HA/anticancer drug complex and polymer NPs loaded with TLR-based immunostimulating material and a silencer of an immunosuppressive gene. The water-insoluble anticancer drug PTX was stably dispersed using HA. CpG ODNs and IL-10 siRNA were encapsulated in PLGA NPs, and these materials were used as immune cell activators. Treatment of mice with CpG ODNs alone in vivo exerted a negligible anticancer effect. However, the suppression of the Th2 response by IL-10 siRNA increased immune cell activity. The sequential delivery of the HA/PTX complex (chemotherapy) followed by PCNs and PINs (immunotherapy) powerfully induced immunogenic tumor cell death.
Acknowledgments
This work was supported by financial support from the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (grant no 2014R1A2A1A10049960, 2012M3A9C6050070).
Disclosure
The authors report no conflicts of interest in this work.
References
Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230–252. | ||
Peng XH, Qian X, Mao H, et al. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. Int J Nanomedicine. 2008;3(3):311–321. | ||
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56(11):1649–1659. | ||
Ozols RF, Herbst RS, Colson YL, et al. Clinical cancer advances 2006: major research advances in cancer treatment, prevention, and screening – a report from the American society of clinical oncology. Jpn J Clin Oncol. 2006;25(1):146–162. | ||
Arruebo M, Fernández-Pacheco R, Ibarra MR, Santamaría J. Magnetic nanoparticles for drug delivery. Nano Today. 2007;2(3):22–32. | ||
Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV. ChemoImmunoModulation: immune regulation by the antineoplastic chemotherapeutic agents. Curr Med Chem. 2012;19(12):1792–1803. | ||
Spencer CM, Faulds D. Paclitaxel. Drugs. 1994;48(5):794–847. | ||
de Bree E, Theodoropoulos PA, Rosing H, et al. Treatment of ovarian cancer using intraperitoneal chemotherapy with taxanes: from laboratory bench to bedside. Cancer Treat Rev. 2006;32(6):471–482. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1):179–192. | ||
Fjällskog M-L, Frii L, Bergh J. Paclitaxel-induced cytotoxicity – the effects of cremophor EL (castor oil) on two human breast cancer cell lines with acquired multidrug resistant phenotype and induced expression of the permeability glycoprotein. Eur J Cancer. 1994;30(5):687–690. | ||
Meerum Terwogt J, Nuijen B, Huinink WW, Beijnen JH. Alternative formulations of paclitaxel. Cancer Treat Rev. 1997;23(2):87–95. | ||
Chun C, Lee SM, Kim SY, Yang HK, Song S-C. Thermosensitive poly (organophosphazene)–paclitaxel conjugate gels for antitumor applications. Biomaterials. 2009;30(12):2349–2360. | ||
Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release. 2004;97(2):249–257. | ||
Xiong W, Peng L, Chen H, Li Q. Surface modification of MPEG-b-PCL-based nanoparticles via oxidative self-polymerization of dopamine for malignant melanoma therapy. Int J Nanomedicine. 2015;10:2985–2996. | ||
Wood AJ, Rowinsky EK, Donehower RC. Paclitaxel (taxol). N Engl J Med. 1995;332(15):1004–1014. | ||
Singh S, Dash AK. Paclitaxel in cancer treatment: perspectives and prospects of its delivery challenges. Crit Rev Ther Drug Carrier Syst. 2009;26(4):333–372. | ||
Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. | ||
Hennenfent K, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17(5):735–749. | ||
Yu D-H, Lu Q, Xie J, Fang C, Chen H-Z. Peptide-conjugated biodegradable nanoparticles as a carrier to target paclitaxel to tumor neovasculature. Biomaterials. 2010;31(8):2278–2292. | ||
Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Van S, Das SK, Wang X, et al. Synthesis, characterization, and biological evaluation of poly(L-γ-glutamyl-glutamine)-paclitaxel nanoconjugate. Int J Nanomedicine. 2010;5:825–837. | ||
Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. | ||
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. | ||
Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411(6835):380–384. | ||
Schuster M, Nechansky A, Kircheis R. Cancer immunotherapy. Biotechnol J. 2006;1(2):138–147. | ||
Serda RE. Particle platforms for cancer immunotherapy. Int J Nanomedicine. 2013;8:1683–1696. | ||
Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10(4):499–511. | ||
Alving CR, Peachman KK, Rao M, Reed SG. Adjuvants for human vaccines. Curr Opin Immunol. 2012;24(3):310–315. | ||
Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180(9):5771–5777. | ||
O’Garra A, Vieira P. TH1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7(6):425–428. | ||
Moore KW, O’garra A, Malefyt RDW, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11(1):165–190. | ||
Pradhan P, Qin H, Leleux JA, et al. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35(21):5491–5504. | ||
Liu G, Ng H, Akasaki Y, et al. Small interference RNA modulation of IL-10 in human monocyte-derived dendritic cells enhances the Th1 response. Eur J Immunol. 2004;34(6):1680–1687. | ||
Roy A, Singh MS, Upadhyay P, Bhaskar S. Combined chemo- immunotherapy as a prospective strategy to combat cancer: a nanoparticle based approach. Mol Pharm. 2010;7(5):1778–1788. | ||
Ma W, Chen M, Kaushal S, et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine. 2012;7:1475–1487. | ||
Seth A, Heo MB, Lim YT. Poly (γ-glutamic acid) based combination of water-insoluble paclitaxel and TLR7 agonist for chemo-immunotherapy. Biomaterials. 2014;35(27):7992–8001. | ||
Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35(1):590–600. | ||
Kim JH, Noh YW, Heo MB, Cho MY, Lim YT. Multifunctional hybrid nanoconjugates for efficient in vivo delivery of immunomodulating oligonucleotides and enhanced antitumor immunity. Angew Chem Int Ed Engl. 2012;51(38):9670–9673. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.