Back to Journals » OncoTargets and Therapy » Volume 13
Self-Renewal Signalling Pathway Inhibitors: Perspectives on Therapeutic Approaches for Cancer Stem Cells
Authors Zhu Q, Shen Y, Chen X, He J, Liu J, Zu X
Received 25 July 2019
Accepted for publication 7 November 2019
Published 16 January 2020 Volume 2020:13 Pages 525—540
DOI https://doi.org/10.2147/OTT.S224465
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Qingyun Zhu, 1,* Yingying Shen, 1,* Xiguang Chen, 1 Jun He, 2 Jianghua Liu, 1 Xuyu Zu 1
1Institute of Clinical Medicine, The First Affiliated Hospital of University of South China, Hengyang, Hunan 421001, People’s Republic of China; 2Department of Spine Surgery, The Affiliated Nanhua Hospital of University of South China, Hengyang, Hunan 421001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuyu Zu
Institute of Clinical Medicine, The First Affiliated Hospital of University of South China, 69 Chuanshan Road, Hengyang, Hunan 421001, People’s Republic of China
Tel +86-734-8279018
Fax +86-734-8279009
Email [email protected]
Jianghua Liu
Institute of Clinical Medicine, The First Affiliated Hospital of University of South China, 69 Chuanshan Road, Hengyang, Hunan 421001, People’s Republic of China
Email [email protected]
Abstract: The poor survival and prognosis of individuals with cancer are often attributed to tumour relapse and metastasis, which may be due to the presence of cancer stem cells (CSCs). CSCs have the characteristics of self-renewal, differentiation potential, high carcinogenicity, and drug resistance. In addition, CSCs exhibit many characteristics similar to those of embryonic or tissue stem cells while displaying persistent abnormal activation of self-renewal pathways associated with development and tissue homeostasis, including the Wnt, Notch, Hedgehog (Hh), TGF-β, JAK/STAT3, and NF-κB pathways. Therefore, we can eliminate CSCs by targeting these self-renewal pathways to constrain stem cell replication, survival and differentiation. At the same time, we cannot neglect the ping-pong effect of the tumour microenvironment, which releases cytokines and promotes self-renewal pathways in CSCs. Recently, meaningful progress has been made in the study of inhibitors of self-renewal pathways in tumours. This review primarily summarizes several representative and novel agents targeting these self-renewal signalling pathways and the tumour microenvironment and that represent a promising strategy for treating refractory and recurrent cancer.
Keywords: cancer stem cells, signalling pathway inhibitors, targeted therapy, small-molecule chemicals, tumour microenvironment
Introduction
A growing collection of evidence has demonstrated that cancer is the primary threat to human health. To date, there are 18.1 million newly diagnosed cases and 9.6 million cancer-related deaths.1 Due to its high fatality rate, cancer remains one of the toughest health challenges human beings face.
Cancer therapy is primarily hindered by recurrence and chemoresistance. Accumulating evidence has suggested that cancer stem cells (CSCs), which initiate and maintain tumour growth, are a small subset of tumour cells. CSCs are thought to cause tumour relapse, metastasis, and chemo-resistance.2,3 In 1994, Lapidot et al first isolated human acute myeloid leukaemia stem cells (LSCs) using specific cell surface markers. Their research revealed that only LSCs possessed the high self-renewal capacity necessary to maintain the malignant phenotype, strongly supporting the objective existence of CSCs.4 Subsequently, CSCs were identified in many types of solid tumours, including pancreatic,5 breast,6 lung,7 and liver tumors.8 Traditional chemotherapy eliminates the bulk of tumour cells but cannot eradicate CSCs, which have enhanced repair and renewal abilities.9,10 Due to their self-renewal ability and therapy resistance, CSCs are considered the root cause of tumorigenesis, progression, drug resistance and recurrence.11 Previous studies have found that CSCs are enriched after chemotherapy.12 Multiple signalling pathways are abnormally activated in CSCs.13 Furthermore, changes in the tumour microenvironment (TME) after treatment, such as anti-angiogenic tumour tube neonatal drug treatment, can cause tumour tissue to become hypoxic, which induces tumour stem cell proliferation.14 Therefore, targeting CSCs is a more effective approach for treating cancer. Numerous studies have shown that abnormalities in different signalling pathways exist in CSCs, including the Notch, Hedgehog (Hh), and Wnt pathways, which play vital roles in embryonic development and differentiation of normal stem cells.15 In addition, the TME releases cytokines that increase activation of these signalling pathways to enhance the cancer stem cell population.16 Therefore, targeting these pathways and the TME represents a promising therapy to suppress CSC self-renewal and proliferation and thus the tumour development promoted by CSCs.
Herein, we focus on six key self-renewal CSC signalling pathway inhibitors for the Wnt, Hh, Notch, TGF-β, JAK/STAT3 and NF-κB pathways as well as the TME, with the hope that this discussion may provide new insight for advances in clinical oncology.
Signalling Pathway Inhibitors
Tumours are prone to recurrence and metastasis due to the existence of CSCs, which convey a poor prognosis. CSCs demonstrate persistent abnormal activation of self-renewal pathways. Hence, targeting these dysregulated signalling pathways is expected to be useful for cancer treatment.17 It has been hypothesized that cancer can be eliminated or perpetually inhibited by inhibiting CSC signalling pathways while avoiding serious impacts on normal tissue renewal.18 Therefore, signalling pathway inhibitors are a promising strategy for cancer therapy.19
Targeting the Wnt Signalling Pathway
The evolutionarily conserved Wnt pathway regulates the pluripotency of stem cells20 and plays a crucial role in self-renewal and differentiation of cells.21,22 In this signalling pathway, the Axin/GSK-3β/APC complex promotes degradation of the intracellular signalling molecule β-catenin. However, when the Wnt ligand is activated by binding to Frizzled and the low-density lipoprotein-related receptor (LRP), the Axin/GSK-3β/APC complex decomposes. Then, intracytoplasmic β-catenin becomes stable and can enter the nucleus to facilitate transcription of target genes21,23 (Figure 1). Abnormal activation of Wnt signalling is thought to promote CSC development, leading to malignant transformation.24 Therefore, many small-molecule inhibitors that specifically target these key factors in the pathway, such as Frizzled, Dishevelled, Porcupine, or Tankyrase, can be designed via drug development.25
 |
Figure 1 Schematic representation of the key CSC signalling pathways Notch, Wnt, and Hedgehog. Some of the current drugs that target these pathways in CSCs are shown. Abbreviations: NICD, Notch intracellular domain; CBF-1, C-promoter binding factor-1; TCF/LEF, T cell factor/lymphoid enhancer factor; LRP, Low-density lipoprotein-related receptor; GSK-3β, glycogen synthase kinase 3β; APC, adenomatous polyposis coli; CPB, cAMP-response element-binding binding protein; Gli, glioma-associated oncogene. |
LGK974 (Wnt974)
LGK974 is a compound that specifically targets Porcupine,21 which performs posttranslational Wnt acylation that is necessary for Wnt secretion.26 LGK974 significantly blocks Wnt signalling27 and is well tolerated in rodent tumour models. It has been reported that treatment with LGK974 significantly inhibits the proliferation of breast cancer stem cells (BCSCs).28 LGK974 is a new strategy for effectively targeting glioblastoma stem-like cells.27 The latest research suggests that cutaneous squamous cell carcinoma (cSCC) mice given LGK974 exhibit differentiation of cancer cells and an obvious reduction in CSCs. Therefore, the Wnt signalling pathway is crucial for initiation, progression and maintenance of the CSC niche in cSCC. Thus, interfering with Wnt secretion is a promising therapeutic approach.29 LGK-974 is currently undergoing phase I clinical trials.30
ICG-001(PRI 724)
ICG-001 is a small-molecule inhibitor that selectively targets cAMP-response element-binding (CREB) binding protein (CBP). The CBP/ß-catenin/FOXM1 transcriptional complex leads to increased CSC numbers, drug resistance and a poor prognosis. Therefore, CBP may represent a novel therapeutic target for eliminating CSCs.31 In light of its ability to suppress the Wnt signalling pathway, ICG-001 has anti-tumour effects in certain tumour types. It was shown that ICG-001 represents a potential therapeutic strategy for gastric cancer because of its ability to restrain cell growth, metastasis, gastric cancer stem cell-like properties and chemoresistance.32 Moreover, ICG-001 effectively suppressed tumour metastasis of aggressive subpopulations of oral squamous cell carcinoma stem-like cells in zebrafish embryos.33 By disrupting Wnt signalling, ICG-001 also exerts potent effects against leukaemia stem cells.34 Using clinical databases and patient tumour samples, dual inhibition of Wnt and YAP using ICG-001 and simvastatin, respectively, powerfully suppressed CSC populations in triple negative breast cancer (TNBC).35 Collectively, these data suggest that ICG-001 is a potential agent for eliminating or inhibiting CSCs in various cancers.
Ipafricept (OMP-54F28)
Ipafricept is a recombinant fusion protein comprising the ligand-binding domain of frizzled receptor 8 and an immunoglobulin G1 (IgG1) Fc fragment. Ipafricept performs the role of a decoy receptor that prevents the Wnt receptor from binding to Wnt ligands, thereby inhibiting Wnt signalling.36 Recent studies have shown that the Wnt inhibitor Ipafricept can suppress the invasiveness, sphere formation and growth of CSCs in head and neck squamous cell carcinoma (HNSCC).37 Ipafricept is being used in phase I clinical trials for treatment of cancer patients.38
Targeting the Notch Signalling Pathway
The Notch signalling pathway is an evolutionarily conserved pathway that influences the survival and differentiation of CSCs.39,40 It also plays an important role in tumour initiation and progression.41 When the ligand protein binds to the Notch receptor, the Notch intracellular domain (NICD) is released into the nucleus through three cleavages mediated by γ-secretase, activating transcription of Notch target genes42 (Figure 1). The Notch pathway is composed of a series of signalling molecules, including NICD, Hes1 and Hey1, which play a vital role in cell development and maintenance of stemness.43 Activation of Notch signalling promotes tumour proliferation and metastasis, while inhibition of this pathway can eliminate CSCs and increase sensitivity to drugs. Thus, genes in the Notch signalling pathway may represent potential cancer therapeutic targets.44,45 Notch inhibitors can be used singly or in combination with chemotherapy agents to treat cancer and prevent recurrence.46 For example, chemotherapeutic drugs combined with NOTCH1 inhibitors synergistically impair chemotherapy-enriched CSC populations both in vitro and in vivo. Therefore, Notch signalling pathway inhibitors exhibit anti-proliferative tumour effects and represent a novel treatment for cancer.47
DAPT (GSI-IX)
DAPT (GSI-IX) is a novel γ-secretase inhibitor that suppresses Aβ with an IC50 of 20 nM in HEK 293 cells. Currently, it is primarily used to treat Alzheimer’s disease, but a study found that it had potential as an anti-CSC drug. In fact, the Notch signalling pathway is significant in the self-renewal of osteosarcoma stem cells (OSCs). DAPT is a cell-permeable dipeptide that blocks ligand-induced Notch proteolysis and inhibits Notch activity, targeting OSCs and increasing tumour sensitivity to platinum.48 A previous study demonstrated that DAPT suppresses LSC growth by inhibiting proliferation in colony formation assays and regulates LSC self-renewal, potentially providing a novel agent for cancer therapy.49 In addition, DAPT suppresses the self-renewal ability and stemness of ovarian cancer cells.50 Therefore, DAPT is a prospective anti-tumour agent due to its high specificity, minimal side effects and attenuated toxicity. DAPT has currently entered phase II clinical trials.
MK-0752
MK-0752 is a moderately effective γ-secretase inhibitor that targets Aβ40 with an IC50 of 5 nM. CSCs can be targeted by MK-0752 combined with chemotherapy agents to treat recurrent, metastatic and refractory cancer. Studies have shown that MK-0752 combined with tocilizumab significantly reduces BCSC numbers and suppresses tumour growth,51 For patients with advanced solid tumours, treatment with MK-0752 combined with dalotuzumab was shown to inhibit cell proliferation, angiogenesis and stem cell propagation in a phase I trial.52 Likewise, patients with advanced breast cancer were treated with MK-0752 and docetaxel in clinical trials, and tumours were serially biopsied. This study found that the index of CSCs, which identified CD44+/CD24− and ALDH+ cells, and mammosphere formation ability were significantly reduced.53 These studies suggest that the combination of MK-0752 and chemotherapy may represent a novel approach to treating various cancers. At present, MK-0752 has completed a phase I clinical trial.
Demcizumab (OMP-21M18)
Demcizumab is an IgG2 humanized monoclonal antibody agent that targets the Notch ligand DLL4.54 DLL4 is an important ligand in the Notch signalling pathway and plays a critical role in the maintenance and proliferation of CSCs as well as tumour angiogenesis.55 Thus, the anti-CSC and anti-angiogenesis activities of Demcizumab contribute to its anti-tumour efficacy. Furthermore, Demcizumab is the first antibody agent targeting the Notch signalling pathway that has entered the clinical trial phase. The anti-tumour activity of Demcizumab monotherapy was demonstrated in phase I trials in patients previously treated for solid tumours, and doses of ≤ 5 mg per week were generally well tolerated.54 Demcizumab combined with pemetrexed and carboplatin as first-line treatment for metastatic non-squamous non-small cell lung cancer has undergone a phase IB trial.56
Targeting the Hedgehog Signalling Pathway
The classical Hh signalling pathway is responsible for embryonic development, regulating stem cell renewal and tissue homeostasis in cancer.57 Hh has three gene homologs: Sonic Hh (SHh), Desert Hh (DHh), and Indian Hh (IHh).58,59 When extracellular Hh ligands (SHh, IHh, DHh) bind to PTCH, the inhibitory effect of PTCH on Smoothened (Smo) is relieved, allowing Gli to translocate to the nucleus and induce transcription of target genes.15 (Figure 1). Abnormal activation of Hh signalling is a key driver of tumorigenesis in breast, prostate, small cell lung cancer, stomach and haematopoietic malignancies.60,61 Hh signalling pathway inhibitors have demonstrated efficacy in early clinical trials. Furthermore, the development of Hh inhibitors has aroused great interest in the pharmaceutical exploitation field.15,30 Recently, inhibition of Hh signalling was reported to suppress self-renewal and chemoresistance in pancreatic CSCs.62 The Hh pathway also regulates self-renewal of BCSCs via Bmi-1.63 In addition, treatment with Smo inhibitors downregulated the expression of CSC markers and sensitized tumours to docetaxel treatment in a patient-derived xenograft model, with significantly improved survival and reduced metastasis.64 Therefore, Smo receptor and Gli family proteins are useful as molecular targets for suppressing CSC self-renewal by inhibiting the Hh pathway.
Cyclopamine
Cyclopamine, a natural compound, is a Hh signalling pathway inhibitor that specifically targets Smo.65 Hh signalling, which is overactivated in CSCs, is involved in embryonic development and tumorigenesis. A previous study demonstrated that cyclopamine is an effective method to reverse gemcitabine resistance in pancreatic cancer cells, significantly suppressing the expression of CSC markers and certain Hh members.66 Treatment with cyclopamine downregulated the expression of stemness and epithelial-mesenchymal transition (EMT) markers, as well as sHh downstream gene expression, in HCT-116 spheres.67 A previous study showed that the SHh inhibitor cyclopamine has potent antitumour activity against bladder tumorigenesis and inhibits self-renewal of bladder cancer stem cells.68 Increasing evidence indicates that cyclopamine has anti-cancer activity against a variety of human cancer types by suppressing CSC proliferation, emphasizing the druggability of the Hh pathway and providing new opportunities for discovery of novel anticancer drugs.
Vismodegib (GDC-0449)
Vismodegib (GDC-0449) is a first-in-class small molecular antagonist of the Hh pathway that binds to Smo and inhibits abnormal activation of the Hh pathway.69,70 Vismodegib was the first agent to target the Hh pathway and received US Food and Drug Administration (FDA) approval for treatment of advanced basal cell carcinoma (BCC).71 Vismodegib promotes differentiation of tumour cells and inhibits a hair follicle-like fate, thereby mediating regression of BCC.72 Currently, vismodegib has been used as a monotherapy or in combination with other chemotherapeutic drugs in clinical trials for many cancer types, including small-cell lung cancer, glioblastoma, basal cell carcinoma, metastatic pancreatic cancer and prostate cancer.73 The biological activities of pancreatic CSCs that are involved in conveying malignancy, such as cell viability, Gli-DNA binding and transcriptional activities, are suppressed by vismodegib. Furthermore, vismodegib activates caspase 3 and induces PARP cleavage, promoting apoptosis of pancreatic CSCs. Therefore, by targeting pancreatic CSCs, vismodegib can be applied for effective inhibition of pancreatic cancer.74 In addition, vismodegib has several effects on BCSCs, such as anti-proliferative, anti-invasive, pro-apoptotic and inhibitory effects. In vivo studies have also found that treatment with this drug leads to pronounced inhibition of tumour formation and growth.75 Although vismodegib is effective in therapy against BCC, its clinical application has been impeded by severe side effects, low selectivity for CSCs, and drug resistance.73 Drug-resistant Smo mutations and abnormal activation of Hh signalling downstream of Smo have been observed clinically, suggesting that alternative approaches are urgently needed.76
GANT61
GANT61 was the first small molecular inhibitor of glioma-associated oncogene homologue 1 (Gli1).77 Meanwhile, it is the most extensively used antagonist that targets Gli proteins to specifically block Hh signalling.76 GANT61 has a strong inhibitory influence on cell proliferation and migration, which has been confirmed in both in vivo and in vitro experiments in breast cancer, pancreatic cancer and other malignancies.78–80 Because basal cell carcinoma patients were previously reported to develop resistance to Smo inhibitors, the Gli inhibitor GANT61 is one of the most promising drugs for inhibiting Hh signalling and has been studied in various cancers.81 Accumulating experimental evidence supports the use of GANT61 as an anti-cancer agent to suppress cell proliferation, survival, apoptosis, DNA damage repair, CSCs, autophagy, EMT, and immune responses in multiple malignant tumour types.81 GANT61 not only effectively inhibited the growth of paclitaxel-resistant cells but also markedly reduced the proportion of CSCs in TNBC cells.82 In addition, combining GANT61 with mTOR inhibition provides an effective method for inhibiting pancreatic CSCs, thereby improving pancreatic cancer treatment83 (the chemical structures of Notch, Wnt, and Hh inhibitors are shown in Figure 2).
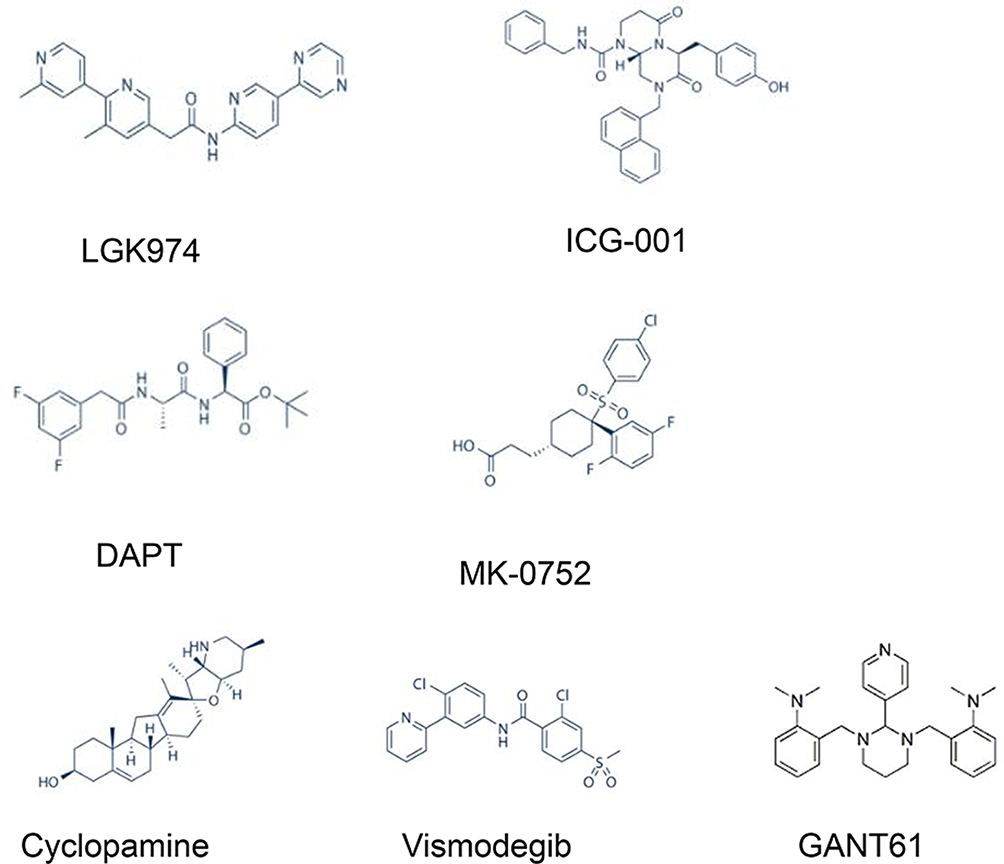 |
Figure 2 Chemical structures of key inhibitors of CSC signalling pathways, including the Notch, Wnt, and Hedgehog pathways. |
Targeting the TGF-β Signalling Pathway
TGF-β represents a family of polypeptides involved in various pathophysiological processes, including cell proliferation, migration, viability, angiogenesis, immunosurveillance, and regulation of stem cell pluripotency and differentiation.84–86 Furthermore, TGF-β plays a significant role in cancer development.87 The TGF-β ligand binds to the type II receptor, which recruits and phosphorylates the type I receptor. The type I receptor then phosphorylates the SMAD protein (R-SMAD), and R-SMAD and coSMAD subsequently combine. The R-SMAD/coSMAD complex then translocates to the nucleus, where it activates transcription of various target genes88,89 (Figure 3). Studies have demonstrated that the TGF-β signalling pathway may be involved in regulation of cancer stem cell-like properties. Inhibition of TGF-β signalling prevents cancer cell proliferation and EMT and reduces the CSC population, impeding cancer progression.90 TGF-β was shown to possess anticancer effects, which are mediated by inhibition of CSC survival, and may represent a novel treatment for hepatocellular carcinoma.91 Furthermore, use of TGF-β signalling pathway inhibitors reinforces tumour penetration by nanoparticles, thereby effectively killing CSCs.92 Therefore, TGF-β pathway inhibitors represent a novel treatment paradigm against CSCs.
 |
Figure 3 Schematic representation of the key CSC signalling pathways TGF-β, JAK/STAT3, and Nf-κB. Some of the current drugs targeting these pathways in CSCs are shown. Abbreviations: TGF-β, transforming growth factor-β; R-Smad, receptor-regulated Smad; Co-Smad, common Smad; JAK, Janus kinase; STAT, signal transducer and activator of transcription; NF-κB, nuclear factor kappa-B; IκB, inhibitor of NF-κB; IKK, inhibitor of NF-κB kinase. |
LY2109761
As a novel TGF-β receptor type I/II (TβRI/II) dual inhibitor, LY2109761 exhibits an inhibitory spectrum that targets SMAD2 and exerts antitumour activity in various tumour models, including pancreatic cancer,93 colorectal cancer,94 and hepatocellular carcinoma.95 Previous experiments indicated that LY2109761 is an effective therapeutic method to reduce clonogenicity and enhance radiation sensitivity in glioblastoma multiforme cancer stem-like cells.96 Recent studies have demonstrated that HSP47 promotes glioblastoma multiforme (GBM) stem-like cell survival, while blocking the TGF-β pathway using LY2109761 inhibits HSP47-induced tumorigenesis and stemness, providing a broad spectrum for GBM treatment.97
Galunisertib (LY2157299)
Galunisertib (LY2157299) is a TGFβRI/ALK5 inhibitor with highly selective activity and is the only TGF-β pathway inhibitor currently undergoing clinical studies in patients with hepatocellular carcinoma (HCC). It was reported that galunisertib decreases clonogenic ability, liver spheroid formation and the invasive ability of HCC cells in vitro. Recent studies have suggested that downregulation of CD44 in response to galunisertib may represent one of the mechanisms by which the agent mediates its effects on stemness and invasion of HCC cells. Galunisertib may have great prospects for clinical application98 and is currently in phase I and phase II clinical trials.
Targeting the JAK/STAT3 Signalling Pathway
The JAK-STAT signalling pathway is involved in many important biological processes, such as cell proliferation, differentiation, apoptosis and immune regulation. Certain cytokines and growth factors bind to the receptor, which induces STAT phosphorylation via JAK activation. Activated STAT translocates into the nucleus as a dimer and binds to target genes to regulate transcription (Figure 2). Abnormal activation of JAK/STAT3 signalling facilitates proliferation and survival of cancer cells.99 Therefore, the JAK/STAT3 signalling pathway is a potential anti-tumour target that has been extensively studied in breast and other cancer types. Previous studies have demonstrated that targeting JAK2 and STAT3 may result in more specific and effective breast cancer treatment.100 The IL-6/JAK2/STAT3 pathway is activated in BCSCs, and inhibition of JAK2 reduced this cell population and suppressed the growth of xenograft tumours. Recently, studies have shown that leptin-JAK/STAT3 facilitates breast cancer cell stemness and chemoresistance, while its inhibition leads to chemosensitization and a reduction in BCSCs in vivo.101 Thus, targeting the JAK/STAT3 signalling pathway may represent an efficient cancer treatment strategy.
Ruxolitinib (INCB018424)
Ruxolitinib, previously referred to as INCB018424 or INC424, is a selective and potent oral antagonist of Janus kinase (JAK) 1 and JAK2. In myelofibrosis (MF) and polycythemia vera (PV), abnormal activation of the JAK/STAT pathway is observed. In 2011, ruxolitinib was approved by the FDA for treatment of MF and was subsequently approved for treatment of PV in 2014.102 In addition, as an inhibitor of JAK, ruxolitinib suppresses the growth and activity of CSCs. It was demonstrated that myxoid liposarcoma (MLS) is a subpopulation of cells with CSC characteristics. Rosobinib combined with doxorubicin to treat proliferating cells and CSCs provides a new method for evading chemoresistance in patients with MLS.103 Ruxolitinib is currently undergoing phase II clinical trials.
Napabucasin
Napabucasin (BBI608) is an oral CSC inhibitor that targets Stat3.104,105 In both preclinical toxicology studies and clinical trials, napabucasin was well tolerated and showed no signs of adverse effects on haematopoiesis or other normal adult stem cells. Napabucasin effectively downregulated stemness gene expression and eliminated cancer cells exhibiting high stemness marker expression, leading to inhibition of cancer cell spherogenesis. In addition, napabucasin had strong anti-metastasis activity in a model of spontaneous liver metastasis of colorectal cancer and inhibited cancer recurrence in a model of pancreatic cancer.106 Recent studies have also shown that napabucasin significantly reduces colon cancer cell growth and tumoursphere formation, decreasing the expression of CSC markers at both the mRNA and protein level.107 In summary, napabucasin may prove useful in targeting CSCs, with the potential to attenuate cancer relapse and metastasis in patients with various cancer types.105 Napabucasin is currently being investigated in a phase III clinical trial.
Targeting the Nf-κB Pathway
The NF-κB family consists of five proteins: p65 (RelA), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52).108 The biological roles of NF-κB involve immune responses, proliferation, development, survival, invasion and metastasis.109 In general, the NF-κB dimer binds to IκB (inhibitory protein of NF-κB), which inactivates NF-κB. IKK acts as a kinase that phosphorylates IκB, leading to its degradation. NF-κB dimers can expose nuclear localization signals (NLS), rapidly enter the nucleus from the cytoplasm, and bind to specific DNA sequences, inducing transcription of target genes.110 (Figure 2). Abnormal activation of NF-κB is found in CSCs in various cancers and may be involved in maintenance, proliferation, expansion and survival of CSCs.111 The NF-κB pathway reportedly activates pathways in pancreatic CSCs, whereas stem cell-like properties of cells are observably decreased in response to treatment with NF-κB pathway inhibitors.112 Therefore, inhibition of NF-κB may provide a promising strategy to eliminate CSCs and improve anti-cancer therapies.
GTCpFE
GTCpFE is an ASA prodrug for which fumarate is the ancillary pharmacophore. This prodrug potently suppresses NF-κB activity with no congenital cytotoxicity. GTCpFE displays apparent anti-CSC activity by inhibiting mammosphere formation and impairing the CSC-associated CD44+/CD24− immunophenotype. These findings established a viable strategy for development of improved anti-inflammatory agents for chemoprevention and cancer treatment.113 To improve drug efficacy and minimize toxicity, one study found that the fumarate-based ASA prodrug GTCpFE represents a prototype for development of novel anti-inflammatory and anti-CSC drugs for treatment of potentially aggressive breast cancers.114
BMS-345541
BMS-345541 is an IKK complex inhibitor that has been used to inactivate NF-κB in a variety of cancer types, including melanoma and leukaemia.115,116 BMS-345541 effectively inhibits NF-κB activity at concentration ranges from 0.4 μM to 10.0 μM. It was reported that BMS-345541 is a potential therapeutic agent for restraining breast tumorigenesis and metastasis through targeting of CSCs.117 Recent studies have also suggested that BMS-345541 potently inhibits stemness and self-renewal of lung CSCs. In addition, the expression of EMT-related genes and apoptosis resistance were significantly decreased in response to treatment, indicating that NF-κB inhibition is sufficient to both induce apoptosis and prevent EMT in lung CSCs.118 These findings suggest that BMS-345541 decreases the CSC population within the tumour, potentially reducing cancer progression, recurrence, and chemoresistance (the chemical structures of TGF-β, JAK/STAT3, and Nf-κB inhibitors are presented in Figure 4).
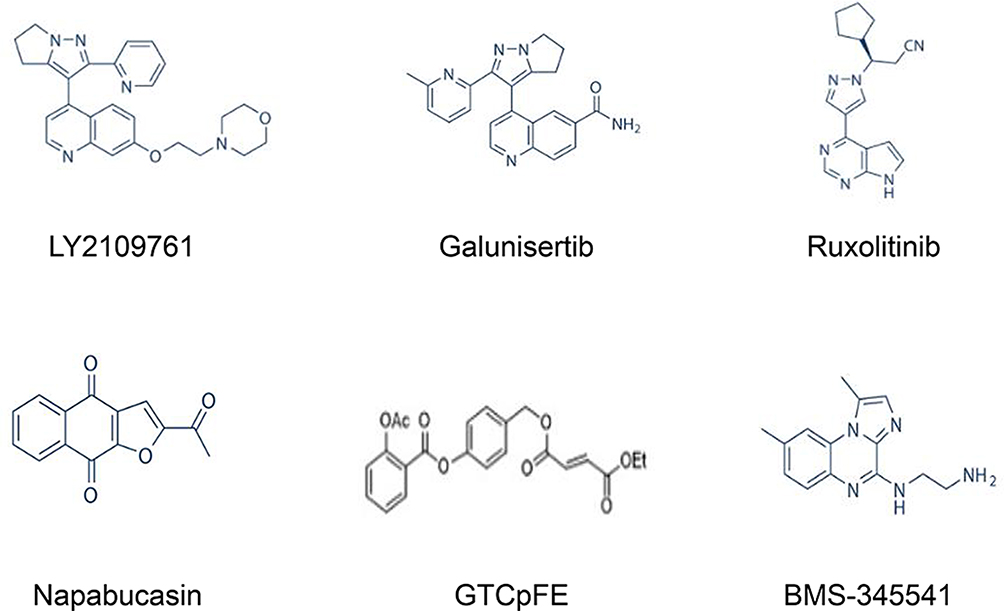 |
Figure 4 Chemical structures of key CSC signalling pathway inhibitors, including TGF-β, JAK/STAT3, and Nf-κB inhibitors. |
Cross-Talk Between Signalling Pathways
Due to the complexity of tumour development, most tumours do not rely on a single signalling pathway to maintain their growth and survival, and there is cross-talk between signalling pathways. Multipathway inhibitors can achieve dual functions for synergistic therapy and overcome drug resistance by inhibiting multiple signalling pathways.
EC-70124
EC-70124, a novel chimeric indolocarbazole compound, acts as a broad-spectrum multikinase inhibitor, displaying strong anti-proliferative effects. Because it targets many elements of the NF-κB, JAK/STAT, PI3K/AKT/mTOR and FLT3 pathways, EC-70124 shows antitumour activity in various solid tumours, including breast, colorectal and prostate tumours, and in acute myeloid leukaemia (AML).119–122 Accordingly, the effect of EC-70124 on CSCs has been detected in other cancer types. Accumulating evidence indicates that EC-70124 exerts an inhibitory effect on the NF-κB and STAT3 signalling pathways, reversing the characteristics of tumorigenesis in prostate CSCs. EC-70124 provides an innovative approach for advanced prostate cancer treatment by interfering with CSCs in prostate tumours.120 EC-70124 has currently completed safety assessments in preclinical models and is beginning clinical development.123
Niclosamide
Niclosamide is an FDA-approved anthelmintic drug124,125 that suppresses multiple CSC signalling pathways, such as the Wnt, Notch, STAT3 and NF-κB pathways.126 In addition to targeting mitochondria to induce cell cycle arrest, growth suppression, and apoptosis in cancer cells, niclosamide inhibits CSCs, providing further evidence that it represents a promising agent for cancer therapy. Previous reports have indicated that niclosamide inhibits breast and ovarian CSCs.127,128 Recently, niclosamide was shown to inhibit the CSC population and their self-renewal activity in colorectal cancer (CRC) cells, leading to irreversible destruction of tumour-initiating potential in vivo.124 In a trial of cisplatin-induced oral CSC enrichment, application of niclosamide suppressed this process. Consequently, ALDH+ OSCC cells are more sensitive to cisplatin therapy.129 Presently, niclosamide is undergoing phase I/II clinical trials (the chemical structures of EC-70124 and niclosamide are presented in Figure 5).
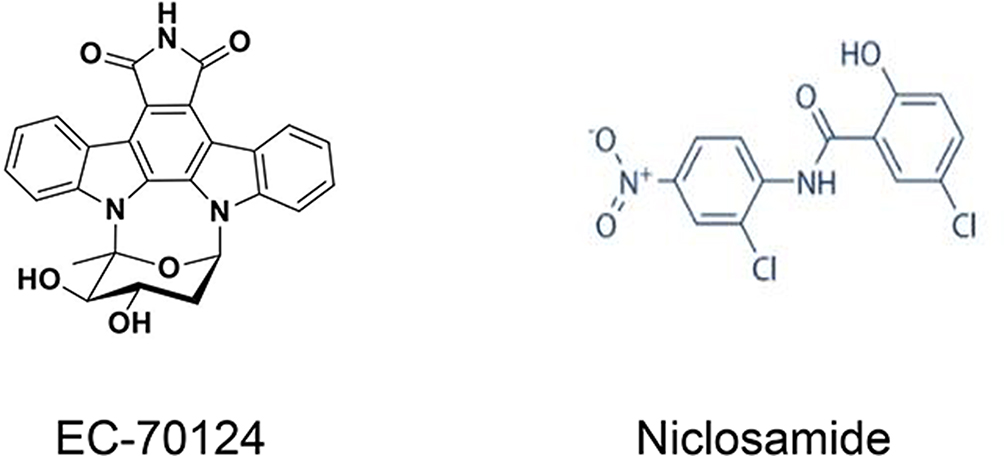 |
Figure 5 Chemical structures of the multipathway inhibitors EC-70124 and niclosamide. |
Targeting the Tumour Microenvironment
CSCs can regulate and control tumour development and metastasis through the interaction between abnormally activated self-renewal signalling pathways and cytokines in the TME.130 The TME releases certain cytokines that increase activation of these signalling pathways to enhance the cancer stem cell population. Studies have found that microenvironmental cytokines, such as IL-6, can promote Notch-Jagged signalling, increasing the proportion of CSCs.16 Studies have found that CAFs can secrete hepatocyte growth factor (HGF) to activate the Wnt signalling pathway and regulate stemness in colon cancer cells.131 Tumour-associated macrophages can secrete IL-6 and other cytokines to activate the STAT3 and Hh signalling pathways, thereby affecting stem cell self-renewal and chemoresistance.132 In turn, the cytokines produced by CSCs recruit and regulate various cells in the TME. For example, CSCs can regulate the expression of cytokines (such as mip-1 alpha/CCL3 and icam-1) through SOX2, activate the NF-κB and Stat signalling pathways, regulate the TME, recruit tumour-related macrophages (TAMs), and create an environment for tumour development.133 Simultaneous treatment is needed when inhibiting CSCs. It is possible to use drugs that inhibit the TME and self-renewal pathways or a combination of these drugs, which plays an important role in inhibiting the development of CSCs and tumour recurrence and metastasis. Studies have shown that ZL170 specifically targets the TGFβ/BMP signalling pathway and may produce an inhibitory TME characterized by decreased ECM deposition and impaired recruitment and proliferation of stromal cells (tumour-associated fibroblasts, endothelial cells, and immune cells), thereby inhibiting stemness, EMT and metastasis of TNBC cells.134 One study showed that CAF secretory factors enhance Wnt signalling and that the Wnt inhibitor Ipafricept (OMP-54f28) suppresses proliferation of patient-derived xenografts (PDXs) and inhibits CSC tumour initiation by inhibiting Wnt signalling involved in the cancer-TME interaction in head and neck squamous cell carcinoma (HNSCC).37 Other research on breast cancer indicates that paclitaxel treatment of breast cancer cells leads to relatively high levels of ROR1 expression and that accessory cells in the TME induce activation of the Wnt5a signalling pathway by expressing ROR1, thereby promoting the stemness of breast cancer cells. Furthermore, the humanized anti-ROR1 drug cirmtuzumab inhibits activation of the ROR1 signalling pathway, which hinders the development and metastasis of breast cancer.135 Therefore, targeting the TME to block CSC proliferation holds great therapeutic potential.
Current and Future Developments
Cancer is a major medical problem throughout the world, and CSCs have been confirmed to be an important cause of tumour resistance and tumorigenesis. Successful targeting of CSCs is expected to cure cancer. CSCs exhibit many characteristics similar to those of embryonic or tissue stem cells while demonstrating persistent abnormal activation of self-renewal pathways associated with development and tissue homeostasis, which can cause cancer. Therefore, signalling pathway inhibitors can be developed for this small subpopulation of cells. Distinct inhibitors targeting key elements of these signalling pathways are currently in different clinical stages of development for the treatment of human malignancies (Table 1). However, the development of signalling pathway inhibitors comes with several challenges. First, these pathways contain too many elements that are difficult to control. Second, some signalling pathways are highly expressed in normal tissues, and their inhibition will affect normal biological functions. Therefore, studies on how to effectively eradicate CSCs without destroying normal stem cell signals are needed. The possible reason for the relative success of some signalling pathway inhibitors, such as Hh CSC inhibitors, is that Hh pathway-related genes are barely expressed in normal cells; thus, blocking this pathway has little effect on the biological function of normal tissues. Therefore, pathway inhibitors with high selectivity for tumour tissues are preferred. At the same time, we can also strengthen the development of multipathway inhibitors, such as niclosamide and EC-70124, which simultaneously act on multiple overactivated signalling pathways in cancer, producing effects superior to those of single pathway inhibitors. Furthermore, the use of single pathway inhibitors can activate compensation mechanisms, resulting in the development of drug resistance. More importantly, multipathway inhibitors have a wider range of effects and serum concentrations than single-pathway inhibitors, leading to reduced toxicity and side effects. In addition, we cannot neglect the ping-pong effect of the TME, which releases cytokines and promotes activation of self-renewal pathways in CSCs. Simultaneous treatment is needed when inhibiting CSCs. It is possible to use drugs that inhibit the TME and self-renewal pathways or a combination of them, which plays an important role in inhibiting the development of CSCs and tumour recurrence and metastasis. Recently, an increasing number of studies have demonstrated that targeting of CSCs has potential for development of novel anticancer agents. As self-renewal signalling pathway inhibitors continue to be refined, cancer patients are expected to have improved treatment options in the future.
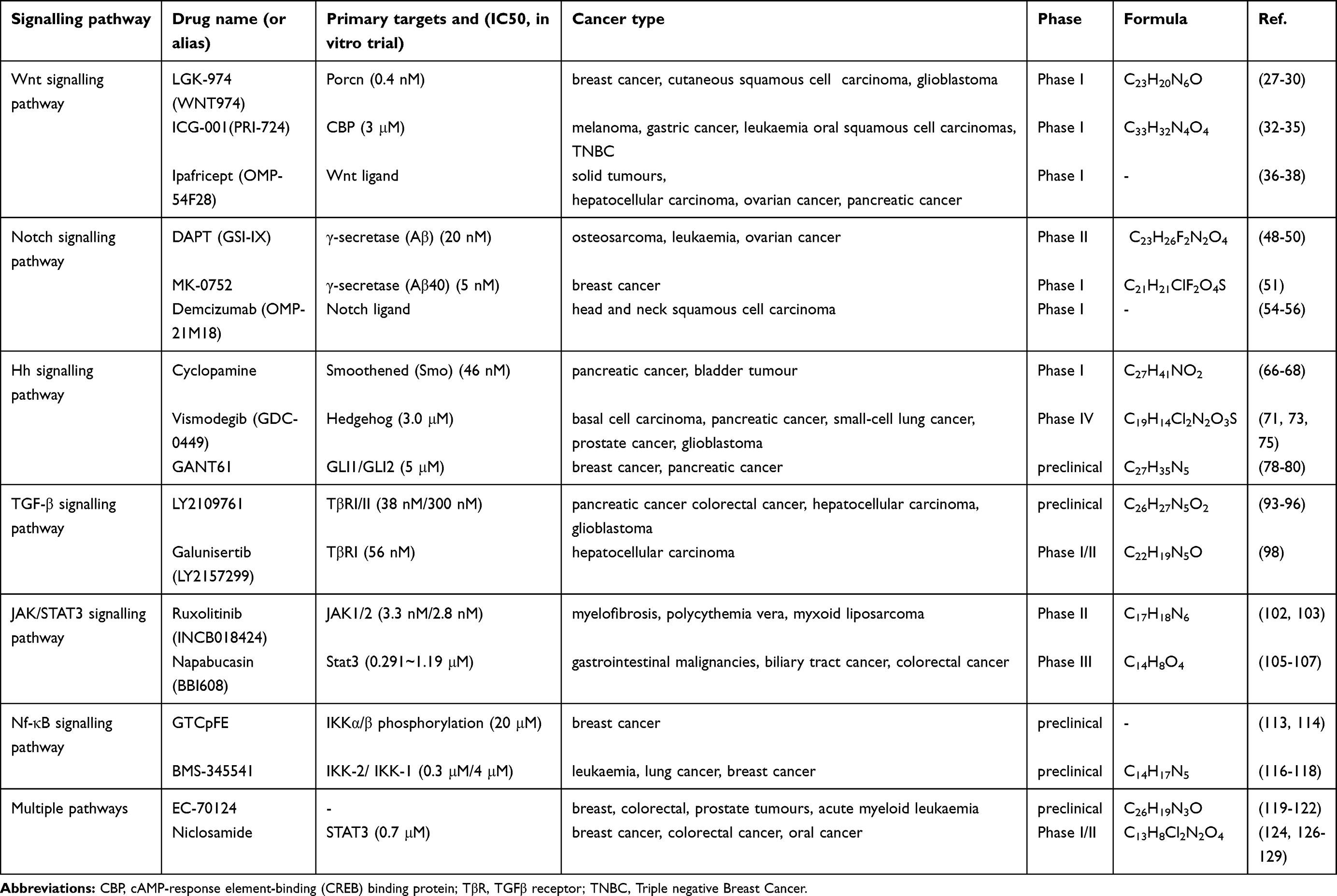 |
Table 1 Summary of the Signalling Pathway Inhibitors and Their Characteristics |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81972487, No. 81873651, No. 81502276), Major Projects of Science and Technology of Health and Family Planning Commission of Hunan Province (No. A2017013), Natural Science Foundation of Hunan Province (No. 2019JJ50552, No. 2019JJ80100), Education Department of Hunan Province (No.18A255, No.18C0473), and Hunan Provincial Innovation Foundation For Postgraduate (No. CX2018B639).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807. doi:10.1182/blood-2008-08-077941
3. Matteucci C, Balestrieri E, Argaw-Denboba A, et al. Human endogenous retroviruses role in cancer cell stemness. Semin Cancer Biol. 2018;53:17–30. doi:10.1016/j.semcancer.2018.10.001
4. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi:10.1038/367645a0
5. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. doi:10.1158/0008-5472.Can-06-2030
6. Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Nat Acad Sci U S A. 2003;100(7):3983–3988. doi:10.1073/pnas.0530291100
7. Alamgeer M, Peacock CD, Matsui W, et al. Cancer stem cells in lung cancer: evidence and controversies. Respirology. 2013;18(5):757–764. doi:10.1111/resp.12094
8. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911–1918. doi:10.1172/jci66024
9. Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66(19):9339–9344. doi:10.1158/0008-5472.Can-06-3126
10. Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–4320. doi:10.1158/0008-5472.Can-08-0364
11. Chang JC. Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016;95(1 Suppl 1):S20–25. doi:10.1097/md.0000000000004766
12. Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi:10.1093/jnci/djn123
13. Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–497. doi:10.1172/jci39397
14. Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Nat Acad Sci U. S. A. 2012;109(8):2784–2789. doi:10.1073/pnas.1018866109
15. Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–3140. doi:10.1158/1078-0432.Ccr-09-2846
16. Bocci F, Gearhart-Serna L, Boareto M, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Nat Acad Sci U. S. A. 2019;116(1):148–157. doi:10.1073/pnas.1815345116
17. Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi:10.1038/35102167
18. Sell S. Cancer and stem cell signaling: a guide to preventive and therapeutic strategies for cancer stem cells. Stem Cell Rev. 2007;3(1):1–6. doi:10.1007/s12015-007-0015-5
19. Oren O, Smith BD. Eliminating cancer stem cells by targeting embryonic signaling pathways. Stem Cell Rev. 2017;13(1):17–23. doi:10.1007/s12015-016-9691-3
20. Sokol SY. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development. 2011;138(20):4341–4350. doi:10.1242/dev.066209
21. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
22. Saito-Diaz K, Chen TW, Wang X, et al. The way Wnt works: components and mechanism. Growth Factors. 2013;31(1):1–31. doi:10.3109/08977194.2012.752737
23. Fleming HE, Janzen V, Lo Celso C, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2(3):274–283. doi:10.1016/j.stem.2008.01.003
24. Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells?. Clin Cancer Res. 2010;16(12):3153–3162. doi:10.1158/1078-0432.Ccr-09-2943
25. Tran FH, Zheng JJ. Modulating the wnt signaling pathway with small molecules. Protein Sci. 2017;26(4):650–661. doi:10.1002/pro.3122
26. Takada R, Satomi Y, Kurata T, et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11(6):791–801. doi:10.1016/j.devcel.2006.10.003
27. Kahlert UD, Suwala AK, Koch K, et al. Pharmacologic Wnt inhibition reduces proliferation, survival, and clonogenicity of glioblastoma cells. J Neuropathol Exp Neurol. 2015;74(9):889–900. doi:10.1097/nen.0000000000000227
28. Zhao Z, Lu P, Zhang H, et al. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014;16(4):408. doi:10.1186/s13058-014-0408-8
29. Zimmerli D, Cecconi V, Valenta T, et al. Wnt ligands control initiation and progression of human papillomavirus-driven squamous cell carcinoma. Oncogene. 2018;37(27):3753–3762. doi:10.1038/s41388-018-0244-x
30. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–464. doi:10.1038/nrclinonc.2015.61
31. Ring A, Nguyen C, Smbatyan G, et al. CBP/beta-Catenin/FOXM1 is a novel therapeutic target in triple negative breast cancer. Cancers (Basel). 2018;10(12). doi:10.3390/cancers10120525
32. Liu Y, Chen H, Zheng P, et al. ICG-001 suppresses growth of gastric cancer cells and reduces chemoresistance of cancer stem cell-like population. J Exp Clin Cancer Res. 2017;36(1):125. doi:10.1186/s13046-017-0595-0
33. Kartha VK, Alamoud KA, Sadykov K, et al. Functional and genomic analyses reveal therapeutic potential of targeting beta-catenin/CBP activity in head and neck cancer. Genome Med. 2018;10(1):54. doi:10.1186/s13073-018-0569-7
34. Jiang X, Mak PY, Mu H, et al. Disruption of Wnt/beta-Catenin exerts antileukemia activity and synergizes with FLT3 inhibition in FLT3-mutant acute myeloid leukemia. Clin Cancer Res. 2018;24(10):2417–2429. doi:10.1158/1078-0432.Ccr-17-1556
35. Sulaiman A, McGarry S, Li L, et al. Dual inhibition of Wnt and yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol Oncol. 2018;12(4):423–440. doi:10.1002/1878-0261.12167
36. Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol Ther. 2015;146:1–11. doi:10.1016/j.pharmthera.2014.08.005
37. Le PN, Keysar SB, Miller B, et al. Wnt signaling dynamics in head and neck squamous cell cancer tumor-stroma interactions. Mol Carcinog. 2019;58(3):398–410. doi:10.1002/mc.22937
38. Jimeno A, Gordon M, Chugh R, et al. A first-in-human Phase I study of the anticancer stem cell agent Ipafricept (OMP-54F28), a Decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(24):7490–7497. doi:10.1158/1078-0432.Ccr-17-2157
39. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi:10.1126/science.284.5415.770
40. Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442(7104):823–826. doi:10.1038/nature04940
41. Li JL, Harris AL. Notch signaling from tumor cells: a new mechanism of angiogenesis. Cancer Cell. 2005;8(1):1–3. doi:10.1016/j.ccr.2005.06.013
42. Schweisguth F. Regulation of notch signaling activity. Curr Biol. 2004;14(3):R129–138. doi:10.1016/j.cub.2004.01.023
43. Wang MM. Notch signaling and Notch signaling modifiers. Int J Biochem Cell Biol. 2011;43(11):1550–1562. doi:10.1016/j.biocel.2011.08.005
44. Nefedova Y, Sullivan DM, Bolick SC, et al. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111(4):2220–2229. doi:10.1182/blood-2007-07-102632
45. Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139(2):95–110. doi:10.1016/j.pharmthera.2013.02.003
46. Venkatesh V, Nataraj R, Thangaraj GS, et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018;5:5. doi:10.21037/sci.2018.02.02
47. Locatelli M, Curigliano G. Notch inhibitors and their role in the treatment of triple negative breast cancer: promises and failures. Curr Opin Oncol. 2017;29(6):411–427. doi:10.1097/cco.0000000000000406
48. Dai G, Deng S, Guo W, et al. Notch pathway inhibition using DAPT, a gamma-secretase inhibitor (GSI), enhances the antitumor effect of cisplatin in resistant osteosarcoma. Mol Carcinog. 2019;58(1):3–18. doi:10.1002/mc.22873
49. Gal H, Amariglio N, Trakhtenbrot L, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia. 2006;20(12):2147–2154. doi:10.1038/sj.leu.2404401
50. Jiang LY, Zhang XL, Du P, et al. gamma-secretase inhibitor, DAPT inhibits self-renewal and stemness maintenance of ovarian cancer stem-like cells in vitro. Chin J Cancer Res. 2011;23(2):140–146. doi:10.1007/s11670-011-0140-1
51. Wang D, Xu J, Liu B, et al. IL6 blockade potentiates the anti-tumor effects of gamma-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2018;25(2):330–339. doi:10.1038/cdd.2017.162
52. Brana I, Berger R, Golan T, et al. A parallel-arm phase I trial of the humanised anti-IGF-1R antibody dalotuzumab in combination with the AKT inhibitor MK-2206, the mTOR inhibitor ridaforolimus, or the NOTCH inhibitor MK-0752, in patients with advanced solid tumours. Br J Cancer. 2014;111(10):1932–1944. doi:10.1038/bjc.2014.497
53. Schott AF, Landis MD, Dontu G, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013;19(6):1512–1524. doi:10.1158/1078-0432.Ccr-11-3326
54. Smith DC, Eisenberg PD, Manikhas G, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res. 2014;20(24):6295–6303. doi:10.1158/1078-0432.Ccr-14-1373
55. Gurney A, Hoey T. Anti-DLL4, a cancer therapeutic with multiple mechanisms of action. Vasc Cell. 2011;3(1):18. doi:10.1186/2045-824x-3-18
56. McKeage MJ, Kotasek D, Markman B, et al. Phase IB trial of the anti-cancer stem cell DLL4-binding agent demcizumab with pemetrexed and carboplatin as first-line treatment of metastatic non-squamous NSCLC. Target Oncol. 2018;13(1):89–98. doi:10.1007/s11523-017-0543-0
57. Habib JG, O’Shaughnessy JA. The hedgehog pathway in triple-negative breast cancer. Cancer Med. 2016;5(10):2989–3006. doi:10.1002/cam4.833
58. Micchelli CA, The I, Selva E, et al. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development. 2002;129(4):843–851.
59. Takebe N, Harris PJ, Warren RQ, et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97–106. doi:10.1038/nrclinonc.2010.196
60. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19(11):1410–1422. doi:10.1038/nm.3389
61. Pietanza MC, Litvak AM, Varghese AM, et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer. 2016;99:23–30. doi:10.1016/j.lungcan.2016.04.014
62. Huang FT, Zhuan-Sun YX, Zhuang YY, et al. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int J Oncol. 2012;41(5):1707–1714. doi:10.3892/ijo.2012.1597
63. Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. doi:10.1158/0008-5472.Can-06-0054
64. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897. doi:10.1038/s41467-018-05220-6
65. Chen JK, Taipale J, Young KE, et al. Small molecule modulation of Smoothened activity. Proc Nat Acad Sci U S A. 2002;99(22):14071–14076. doi:10.1073/pnas.182542899
66. Yao J, An Y, Wie JS, et al. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med Wkly. 2011;141(5):w13208. doi:10.4414/smw.2011.13208
67. Batsaikhan BE, Yoshikawa K, Kurita N, et al. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014;34(11):6339–6344.
68. Li C, Du Y, Yang Z, et al. GALNT1-mediated glycosylation and activation of Sonic Hedgehog signaling maintains the self-renewal and tumor-initiating capacity of bladder cancer stem cells. Cancer Res. 2016;76(5):1273–1283. doi:10.1158/0008-5472.Can-15-2309
69. Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171–2179. doi:10.1056/NEJMoa1113713
70. Meiss F, Andrlova H, Vismodegib ZR. Recent results. Cancer Res. 2018;211:125–139. doi:10.1007/978-3-319-91442-8_9
71. Shimizu T, Nakagawa K. Novel signal transduction pathways: the molecular basis for targeted cancer therapies in Hedgehog/Notch/Wnt pathway. Nihon Rinsho. 2015;73(8):1342–1348.
72. Sanchez-Danes A, Larsimont JC, Liagre M, et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature. 2018;562(7727):434–438. doi:10.1038/s41586-018-0603-3
73. Pietrobono S, Stecca B. Targeting the oncoprotein smoothened by small molecules: focus on novel acylguanidine derivatives as potent smoothened inhibitors. Cells. 2018;7(12). doi:10.3390/cells7120272
74. Singh BN, Fu J, Srivastava RK, et al. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS ONE. 2011;6(11):e27306. doi:10.1371/journal.pone.0027306
75. Li W, Yang H, Li X, et al. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol Rep. 2019;41(1):437–446. doi:10.3892/or.2018.6805
76. Calcaterra A, Iovine V, Botta B, et al. Chemical, computational and functional insights into the chemical stability of the Hedgehog pathway inhibitor GANT61. J Enzyme Inhib Med Chem. 2018;33(1):349–358. doi:10.1080/14756366.2017.1419221
77. Lauth M, Bergstrom A, Shimokawa T, et al. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Nat Acad Sci U S A. 2007;104(20):8455–8460. doi:10.1073/pnas.0609699104
78. Kurebayashi J, Koike Y, Ohta Y, et al. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017;108(5):918–930. doi:10.1111/cas.13205
79. Fu J, Rodova M, Roy SK, et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013;330(1):22–32. doi:10.1016/j.canlet.2012.11.018
80. Wickstrom M, Dyberg C, Shimokawa T, et al. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int J Cancer. 2013;132(7):1516–1524. doi:10.1002/ijc.27820
81. Gonnissen A. Isebaert S, Haustermans K. Targeting the Hedgehog signaling pathway in cancer: beyond Smoothened. Oncotarget. 2015;6(16):13899–13913. doi:10.18632/oncotarget.4224
82. Koike Y, Ohta Y, Saitoh W, et al. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer. 2017;24(5):683–693. doi:10.1007/s12282-017-0757-0
83. Miyazaki Y, Matsubara S, Ding Q, et al. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer. 2016;15(1):49. doi:10.1186/s12943-016-0534-2
84. Mullen AC, Orlando DA, Newman JJ, et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011;147(3):565–576. doi:10.1016/j.cell.2011.08.050
85. Xi Q, Wang Z, Zaromytidou AI, et al. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147(7):1511–1524. doi:10.1016/j.cell.2011.11.032
86. Young RA. Control of the embryonic stem cell state. Cell. 2011;144(6):940–954. doi:10.1016/j.cell.2011.01.032
87. Miyamoto K, Ushijima T. Diagnostic and therapeutic applications of epigenetics. Jpn J Clin Oncol. 2005;35(6):293–301. doi:10.1093/jjco/hyi088
88. Liu Z, Bandyopadhyay A, Nichols RW, et al. Blockade of autocrine TGF-beta signaling inhibits stem cell phenotype, survival, and metastasis of murine breast cancer cells. J Stem Cell Res Ther. 2012;2(1):1–8. doi:10.4172/2157-7633.1000116
89. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi:10.1038/nature02006
90. Liang Y, Zhu F, Zhang H, et al. Conditional ablation of TGF-beta signaling inhibits tumor progression and invasion in an induced mouse bladder cancer model. Sci Rep. 2016;6:29479. doi:10.1038/srep29479
91. Kim JB, Lee S, Kim HR, et al. Transforming growth factor-beta decreases side population cells in hepatocellular carcinoma in vitro. Oncol Lett. 2018;15(6):8723–8728. doi:10.3892/ol.2018.8441
92. Zuo ZQ, Chen KG, Yu XY, et al. Promoting tumor penetration of nanoparticles for cancer stem cell therapy by TGF-beta signaling pathway inhibition. Biomaterials. 2016;82:48–59. doi:10.1016/j.biomaterials.2015.12.014
93. Melisi D, Ishiyama S, Sclabas GM, et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7(4):829–840. doi:10.1158/1535-7163.Mct-07-0337
94. Zhang B, Halder SK, Zhang S, et al. Targeting transforming growth factor-beta signaling in liver metastasis of colon cancer. Cancer Lett. 2009;277(1):114–120. doi:10.1016/j.canlet.2008.11.035
95. Fransvea E, Angelotti U, Antonaci S, et al. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47(5):1557–1566. doi:10.1002/hep.22201
96. Zhang M, Kleber S, Rohrich M, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71(23):7155–7167. doi:10.1158/0008-5472.Can-11-1212
97. Jiang X, Zhou T, Wang Z, et al. HSP47 promotes glioblastoma stemlike cell survival by modulating tumor microenvironment extracellular matrix through TGF-beta pathway. ACS Chem Neurosci. 2017;8(1):128–134. doi:10.1021/acschemneuro.6b00253
98. Rani B, Malfettone A, Dituri F, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018;9(3):373. doi:10.1038/s41419-018-0384-5
99. Liu L, Nam S, Tian Y, et al. 6-Bromoindirubin-3ʹ-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011;71(11):3972–3979. doi:10.1158/0008-5472.Can-10-3852
100. Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi:10.1172/jci44745
101. Wang T, Fahrmann JF, Lee H, et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27(1):136–150.e135. doi:10.1016/j.cmet.2017.11.001
102. Ajayi S, Becker H, Reinhardt H, et al. Ruxolitinib. Recent Results Cancer Res. 2018;212:119–132. doi:10.1007/978-3-319-91439-8_6
103. Dolatabadi S, Jonasson E, Linden M, et al. JAK-STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int J Cancer. 2019;145:435–449. doi:10.1002/ijc.32123
104. Karandish F, Froberg J, Borowicz P, et al. Peptide-targeted, stimuli-responsive polymersomes for delivering a cancer stemness inhibitor to cancer stem cell microtumors. Colloids Surf B Biointerfaces. 2018;163:225–235. doi:10.1016/j.colsurfb.2017.12.036
105. Hubbard JM, Grothey A. Napabucasin: an update on the first-in-class cancer stemness inhibitor. Drugs. 2017;77(10):1091–1103. doi:10.1007/s40265-017-0759-4
106. Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Nat Acad Sci U. S. A. 2015;112(6):1839–1844. doi:10.1073/pnas.1424171112
107. Beyreis M, Gaisberger M, Jakab M, et al. The cancer stem cell inhibitor napabucasin (BBI608) shows general cytotoxicity in biliary tract cancer cells and reduces cancer stem cell characteristics. Cancers (Basel). 2019;11(3). doi:10.3390/cancers11030276
108. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. doi:10.1101/cshperspect.a000034
109. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–2224. doi:10.1101/gad.1228704
110. Hassanzadeh P. Colorectal cancer and NF-kappaB signaling pathway. Gastroenterol Hepatol Bed Bench. 2011;4(3):127–132.
111. Vazquez-Santillan K, Melendez-Zajgla J, Jimenez-Hernandez L, et al. NF-kappaB signaling in cancer stem cells: a promising therapeutic target? Cell Oncol (Dordr.). 2015;38(5):327–339. doi:10.1007/s13402-015-0236-6
112. Sun L, Mathews LA, Cabarcas SM, et al. Epigenetic regulation of SOX9 by the NF-kappaB signaling pathway in pancreatic cancer stem cells. Stem Cells. 2013;31(8):1454–1466. doi:10.1002/stem.1394
113. Kastrati I, Delgado-Rivera L, Georgieva G, et al. Synthesis and characterization of an aspirin-fumarate prodrug that inhibits NFkappaB activity and breast cancer stem cells. J Vis Exp. 2017:119. doi:10.3791/54798.
114. Kastrati I, Litosh VA, Zhao S, et al. A novel aspirin prodrug inhibits NFkappaB activity and breast cancer stem cell properties. BMC Cancer. 2015;15:845. doi:10.1186/s12885-015-1868-7
115. Berger A, Quast SA, Plotz M, et al. Sensitization of melanoma cells for TRAIL-induced apoptosis by BMS-345541 correlates with altered phosphorylation and activation of Bax. Cell Death Dis. 2013;4:e477. doi:10.1038/cddis.2012.198
116. Yang J, Amiri KI, Burke JR, et al. BMS-345541 targets inhibitor of kappaB kinase and induces apoptosis in melanoma: involvement of nuclear factor kappaB and mitochondria pathways. Clin Cancer Res. 2006;12(3 Pt 1):950–960. doi:10.1158/1078-0432.Ccr-05-1220
117. Battula VL, Nguyen K, Sun J, et al. IKK inhibition by BMS-345541 suppresses breast tumorigenesis and metastases by targeting GD2+ cancer stem cells. Oncotarget. 2017;8(23):36936–36949. doi:10.18632/oncotarget.16294
118. Zakaria N, Mohd Yusoff N, Zakaria Z, et al. Inhibition of NF-kappaB signaling reduces the stemness characteristics of lung cancer stem cells. Front Oncol. 2018;8:166. doi:10.3389/fonc.2018.00166
119. Cuenca-Lopez MD, Serrano-Heras G, Montero JC, et al. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget. 2015;6(29):27923–27937. doi:10.18632/oncotarget.4736
120. Civenni G, Longoni N, Costales P, et al. EC-70124, a novel glycosylated indolocarbazole multikinase inhibitor, reverts tumorigenic and stem cell properties in prostate cancer by inhibiting STAT3 and NF-kappaB. Mol Cancer Ther. 2016;15(5):806–818. doi:10.1158/1535-7163.Mct-15-0791
121. Puente-Moncada N, Costales P, Antolin I, et al. Inhibition of FLT3 and PIM kinases by EC-70124 exerts potent activity in preclinical models of acute myeloid leukemia. Mol Cancer Ther. 2018;17(3):614–624. doi:10.1158/1535-7163.Mct-17-0530
122. Serrano-Heras G, Cuenca-Lopez MD, Montero JC, et al. Phospho-kinase profile of colorectal tumors guides in the selection of multi-kinase inhibitors. Oncotarget. 2015;6(31):31272–31283. doi:10.18632/oncotarget.5211
123. Sanchez C, Salas AP, Brana AF, et al. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb.). 2009;27:4118–4120. doi:10.1039/b905068j
124. Park SY, Kim JY, Choi JH, et al. Inhibition of LEF1-mediated DCLK1 by niclosamide attenuates colorectal cancer stemness. Clin Cancer Res. 2019;25(4):1415–1429. doi:10.1158/1078-0432.Ccr-18-1232
125. Li Y, Li PK, Roberts MJ, et al. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. 2014;349(1):8–14. doi:10.1016/j.canlet.2014.04.003
126. Pan JX, Ding K, Wang CY. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin J Cancer. 2012;31(4):178–184. doi:10.5732/cjc.011.10290
127. Wang YC, Chao TK, Chang CC, et al. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS ONE. 2013;8(9):e74538. doi:10.1371/journal.pone.0074538
128. Yo YT, Lin YW, Wang YC, et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol Cancer Ther. 2012;11(8):1703–1712. doi:10.1158/1535-7163.Mct-12-0002
129. Wang LH, Xu M, Fu LQ, et al. The antihelminthic niclosamide inhibits cancer stemness, extracellular matrix remodeling, and metastasis through dysregulation of the nuclear beta-catenin/c-Myc axis in OSCC. Sci Rep. 2018;8(1):12776. doi:10.1038/s41598-018-30692-3
130. Saygin C, Matei D, Majeti R, et al. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. 2019;24(1):25–40. doi:10.1016/j.stem.2018.11.017
131. Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010;12(5):468–476. doi:10.1038/ncb2048
132. Jinushi M, Chiba S, Yoshiyama H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Nat Acad Sci U S A. 2011;108(30):12425–12430. doi:10.1073/pnas.1106645108
133. Mou W, Xu Y, Ye Y, et al. Expression of Sox2 in breast cancer cells promotes the recruitment of M2 macrophages to tumor microenvironment. Cancer Lett. 2015;358(2):115–123. doi:10.1016/j.canlet.2014.11.004
134. Di L, Liu LJ, Yan YM, et al. Discovery of a natural small-molecule compound that suppresses tumor EMT, stemness and metastasis by inhibiting TGFbeta/BMP signaling in triple-negative breast cancer. J Exp Clin Cancer Res. 2019;38(1):134. doi:10.1186/s13046-019-1130-2
135. Zhang S, Zhang H, Ghia EM, et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc Nat Acad Sci U S A. 2019;116(4):1370–1377. doi:10.1073/pnas.1816262116
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.