Back to Journals » International Journal of Nanomedicine » Volume 14
Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate
Authors Kim DS, Cho JH, Park JH ![]() , Kim JS, Song ES, Kwon J, Giri BR
, Kim JS, Song ES, Kwon J, Giri BR ![]() , Jin SG, Kim KS
, Jin SG, Kim KS ![]() , Choi HG, Kim DW
, Choi HG, Kim DW ![]()
Received 4 April 2019
Accepted for publication 18 June 2019
Published 5 July 2019 Volume 2019:14 Pages 4949—4960
DOI https://doi.org/10.2147/IJN.S211014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Dong Shik Kim1,*, Jung Hyun Cho1,*, Jong Hyuck Park,1 Jung Suk Kim,1 Eon Soo Song,2 Jaewook Kwon,2 Bhupendra Raj Giri,2 Sung Giu Jin,3 Kyeong Soo Kim,4 Han-Gon Choi,1 Dong Wuk Kim2
1College of Pharmacy & Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, South Korea; 2College of Pharmacy & Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu, South Korea; 3Department of Pharmaceutical Engineering, Dankook University, Cheonan, South Korea; 4Department of Pharmaceutical Engineering, Gyeongnam National University of Science and Technology, Jinju, South Korea
*These authors contributed equally to this work
Purpose: The objective of this study was to exploit a novel methotrexate (MTX)-loaded solid self-microemulsifying drug delivery system (SMEDDS) with enhanced bioavailability and photostability.
Materials and methods: The optimized liquid SMEDDS was composed of castor oil, Tween® 80, and Plurol® diisostearique at a voluminous ratio of 27:63:10. The solid SMEDDS was formulated by spray drying liquid SMEDDS with the solid carrier (calcium silicate). Particle size analyzer, scanning electron microscopy (SEM), differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), and Fourier transform infrared (FTIR) spectroscopy experiments characterized the physiochemical properties of the MTX-loaded solid SMEDDS. These properties include a z-average diameter of emulsion around 127 nm and the amorphous form of the solid SMEDDS. Furthermore, their solubility, dissolution, and pharmacokinetics in Sprague-Dawley rats were analyzed in comparison with the MTX powder.
Results: The final dissolution rate and required time for complete release of solid SMEDDS were 1.9-fold higher and 10 min shorter, respectively, than those of MTX powder. Pharmacokinetic analysis demonstrated 2.04- and 3.41-fold increments in AUC and Cmax, respectively in comparison to MTX powder. The AUC and Cmax were significantly increased in solid SMEDDS. Finally, the photostability studies revealed the substantially enhanced photostability of the MTX-loaded SMEDDS under the forced degradation and confirmatory conditions.
Conclusion: This solid SMEDDS formulation could be an outstanding candidate for improving the oral bioavailability and photostability of MTX.
Keywords: methotrexate, solid SMEDDS, solubility, bioavailability, photostability
Introduction
Methotrexate (MTX), previously known as amethopterin, is an antimetabolite and antifolate drug that is clinically used for the treatment of various cancers and autoimmune disorders, including breast cancer, leukemia, lung cancer, lymphoma, osteosarcoma, psoriasis, rheumatoid arthritis, and Crohn’s disease.1,2 MTX inhibits the metabolism of folic acid3 and competitively inhibits the enzyme activity of dihydrofolate reductase (DHFR), which is related to the synthesis of DNA, RNA, thymidylates, and proteins. MTX is a hydrophobic drug with low permeability, and thus, has been categorized as a class IV compound in the biopharmaceutical classification (BCS) system.4 MTX is chemically unstable and easily degradable after exposure to light.5 Upon exposure to acid/alkaline pH conditions, extreme temperature, or light, MTX produces a degradation product, which makes the formulation development a challenging task.6 Accordingly, a valid protection strategy is needed to produce a robust oral dosage.
Sufficient aqueous solubility is one of the essential requirements for oral administration of a drug. Limited solubility can lead to insufficient dissolution and further reduce the bioavailability of a drug.7,8 As a result, a variety of effort has been made to enhance the oral bioavailability of poorly water soluble drugs through lipid based emulsion systems, specifically the self-emulsifying drug delivery system (SEDDS).9–12 SEDDSs are defined as homogeneous mixtures of natural or synthetic oils, surfactants, and co-surfactants that easily form emulsion upon mild agitation and generate a high surface area of interactions between the SEDDS formulation and the gastrointestinal (GI) fluid.13 Moreover, SEDDS has been identified as a prominent technology for drug delivery, because the formulations have great solubilization capacity and a tiny droplet size, which could improve permeation across the GI membrane. In present, SEDDS is usually prepared as a liquid solution or encapsulated in soft gelatin capsules; however, these preparations have some imperfections, including sensitivity to temperature and humidity, and high production cost.14 Moreover, liquid and capsules are less convenient to the patient than the solid dosage form, and incompatibility problems can occur between the soft gelatin shell and the drug formulation.15 Transformation of the liquid SEDDS into the solid dosage form using specific techniques, such as spray drying, may maintain the advantages of SEDDS but avoid the disadvantages of liquid SEDDS mentioned above. Spray drying is the most commonly used method to prepare a solid formulation. First, the sample is prepared by mixing an optimized ratio of ingredients (oil, surfactants, and co-surfactants), the drug, and a suitable solid carrier. Afterwards, the liquid solution (suspension) is sprayed into a heated air chamber to evaporate the volatile fraction (organic solvent or water) contained in the sample,16 hence the term, spray drying. This process is executed under the optimized operation conditions, such as inlet/outlet temperature, aspiration, and feed rate. In addition, spray drying may solve the stability problems associated with traditional emulsions during storage. The outer film coating and packaging in spray drying has shown improved photostability in various photosensitive drugs.17
The purposes of the present study were to: (1) develop a novel solid self-microemulsifying drug delivery system (SMEDDS) formulation of MTX via spray drying with calcium silicate as the solid carrier and (2) evaluate whether the solid SMEDDS improved the absorption and photosensitivity problems. Reconstitution properties of the spray dried formulation were investigated and compared to the solid state characterizations of the powder using a scanning electron microscope (SEM), differential scanning calorimeter (DSC), powder X-ray diffraction (PXRD), and Fourier transform infrared (FTIR) spectrophotometer. Comparative dissolution, oral bioavailability, and photostability studies were performed.
Materials and methods
Materials
MTX was purchased from Huzhou Zhanwang Pharmaceutical Co. (Huzhou, China). Capryol 90, Peceol, Plurol® diisostearique, Transcutol P, and Labrasol were obtained from Gattefosse (Saint-Priest Cedex, France). Peanut oil, corn oil, soybean oil, sesame oil, castor oil, cotton seed oil, Span 80, and Tween® 80 were purchased from Daejung Chemical Co. (Siheung, South Korea). Coconut oil, linseed oil, mineral oil, and olive oil were purchased from Samchun Chemical Co. (Pyeongtaek, South Korea). Cremophor EL and Cremophor RH40 were obtained from BASF (Ludwigshafen, Germany). Calcium silicate was supplied by Hanmi Pharm. Co. (Suwon, South Korea). All other chemicals and solvents were of reagent grade and were used without further purification.
Solubility study
To select appropriate components for the development of the SMEDDS formulation, solubility studies were conducted for various oils and surfactants.18 Excess amounts of MTX (approximately 50 mg) was transferred to 15 mL conical tube (SPL #50,015) containing 3 mL of pure oils or 10% (w/v) aqueous surfactant solutions. Then, the mixture was vortexed and kept for 7 days at 25 °C in a shaking water-bath to facilitate the solubilization. Subsequently, 1 mL of samples were centrifuged at 10,000 g for 10 min (Hanil Science Industrial Co., South Korea) to separate the undissolved MTX. The supernatant was taken and diluted with mobile phase for high performance liquid chromatography (HPLC) analysis for the quantification of MTX. The concentration of MTX in the sample was quantified by the HPLC system (Agilent 1260 Infinity, Agilent Technologies, USA) consisting of the Chem Station software, G1311C 1260 Quat Pump, and G1314B 1260 VWD VL detector. The Inertsil ODS-4 column (GL Sciences, Japan, 4.6 mmI.D. x 250 mm, 5 μm) was used, and the column temperature was maintained at 25 °C. The mobile phase consisted of 0.1 M dibasic phosphate (pH was adjusted to 3.0 with hydrochloride solution) and methanol at the volume ratio of 74/26, respectively. The mobile phase was eluted at a flow rate of 1 mL/min. The eluent was monitored by the detector at 303 nm.
Construction of pseudo-ternary phase diagram
A pseudo-ternary phase diagram, containing oil, surfactants, and co-surfactants was constructed to identify the regions that could self-emulsify under dilution and gentle agitation. Based on the results of a solubility study, castor oil, Tween® 80, and Plurol® diisostearique were selected as the oil, surfactant, and co-surfactant, respectively. This formulation (0.2 mL) was introduced to 300 mL of water in a glass beaker at 37 °C, and the contents were mixed gently with a magnetic stirring bar (300 rpm). The tendency of this formulation to emulsify spontaneously and the progress of emulsion droplets were visually observed. An emulsion was judged as “good” when introduced droplets dispersed easily in water and formed a fine emulsion. Whereas, it was judged as “bad” when poor or no emulsion formation was observed with immediate coalescence or creaming of the oil droplets.19 Emulsions that were judged as “fine” or “poor” were further examined based on the particle size, and then given the characteristic of “good” or “bad”. All tests were performed in triplicate.
Preparation of liquid and solid SMEDDS
The liquid SMEDDS was formulated by dissolving MTX (100 mg) into 1 mL of the castor oil, Tween® 80, and Plurol® diisostearique mixture at a volume ratio of 27/63/10, respectively. The final mixture was vortexed to obtain a clear solution. The self-emulsification and particle size analysis were completed after examining the formulations for phase separation or signs of turbidity. Afterwards, 0.5 g of the solid carrier (calcium silicate) was well dissolved in 100 mL of ethanol by magnetic stirring; then, 1 mL of liquid SMEDDS (equivalent to 100 mg of MTX) was magnetically stirred continuously. While the solution was continuously stirring, it was spray dried with a laboratory-scale spray dryer (Buchi Mini Spray Dryer, B-290, Switzerland) under the following conditions: inlet temperature of 60 °C, outlet temperature of 40 °C, aspiration of 100% (−45 mbar), spraying air of 4 kg/cm2, and flow rate of 5 mL/min.
Characterization of the solid SMEDDS
Droplet size of emulsion
The droplet size of emulsions were determined by Zetasizer Nano ZS (Malvern Instruments, UK) with the dynamic light scattering particle size analyzer at a wavelength of 635 nm and a scattering angle of 90° at 25 °C. Liquid SMEDDS (100 μL) and solid SMEDDS (160 mg) prepared as described above were introduced to 100 mL of distilled water with gentle stirring on a magnetic stirrer. Beakers were incubated to settle down the undissolved portion for 30 min at 25 °C. All studies were repeated three times, and the values of z-average diameters were used.
Morphological analysis of solid SMEDDS
The outer morphological features of MTX powder, calcium silicate, and MTX-loaded solid SMEDDS were examined by an SEM (S-4800, Hitachi, Tokyo, Japan). The samples were fixed on a brass sampling disc using double-sided adhesive tape. Then, they were rendered electrically conductive with the sputter coating of platinum (6 nm/min) using an EMI Teck Ion Sputter (K575K) under vacuum (8x10−3 mbar) for 4 min at 15 mA.
Solid state characterization of solid SMEDDS
The thermal aspects of MTX in solid SMEDDS were examined using a DSC Q20 (TA Instruments, New Castle, Delaware, USA). About 5 mg of each sample were sealed in the Tzero pan and lid, and were subjected to heating at the rate of 10 °C/min from 60 to 175 °C under a nitrogen gas purge of 50 mL/min. Furthermore, the crystallinity of the samples was evaluated by PXRD (D/MAX-2500, Rigaku, Japan), which was performed at room temperature using monochromatic Cu Kα radiation (λ=1.54178 Å) at 100 mA and 40 kV in the region of 5° ≤2θ ≤45° with an angular increment of 0.02º/sec.
FTIR spectroscopic analysis
The state of MTX molecule in the formulation was determined by the FTIR spectrophotometer (FTIR-4100, JASCO, Pittsburgh, PA, USA). The resulting bands were analyzed using Spectra Manager II software. All of the samples were properly loaded on the sample disc and scanned in the range of 400–4,000 cm−1.
Estimation of methotrexate content
Before carrying out the dissolution test, drug content in solid SMEDDS was determined. Accurately weighed MTX-loaded solid SMEDDS, equivalent to 10 mg MTX, was thoroughly dissolved in 100 mL of mobile phase using a volumetric flask. The solution was filtered (0.45 μm) and assayed for MTX content at 303 nm using the HPLC method described above. This drug content analysis was performed in triplicate. The drug content (%) was estimated by the following formula:
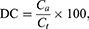
where DC is the percentage of drug content, Ca is the concentration determined by HPLC, and Ct is the theoretical concentration.
Drug release study
The dissolution test was performed using a USP dissolution apparatus II (paddle apparatus) (Vision Classic 6, Hanson Research Co., Los Angeles, CA, USA). Empty hard gelatin capsules were filled with either MTX-loaded solid SMEDDS or MTX powder, equivalent to 50 mg of MTX. The filled capsule was inserted into the sinker and immersed into the dissolution medium, which consisted of 900 mL of water (pH 7.2). The dissolution test was accomplished at 37±0.5 °C using 100 rpm paddle rotations. The dissolution medium-containing vessel was surrounded by an outer water-bath in order to maintain the specific temperature. At predetermined time intervals, 1 mL of dissolution medium was collected and filtered through a nylon syringe filter (0.45 μm). The concentration of MTX in the filtrate (10 μl) was measured by the HPLC method described above.
In vivo study
Care of experimental organisms
Twelve male Sprague-Dawley rats (280±20 g, 7–9 weeks old) were purchased from the Nara Biotech (Seoul, South Korea). Prior to the experiments, the animals were freely provided with standard laboratory food and water. During all of the procedures, the animals were housed in cages under maintained temperature (23–26 °C) and relative humidity (50–55%) environmental conditions. Food was removed approximately 12 hr before the commencement of the pharmacokinetic procedures, but the animals were allowed free access to water. All animal care and procedures were conducted according to the Guiding Principles in the Use of Animals in Toxicology, as adopted in 1989, revised in 1999, and amended in 2008 by the Society of Toxicology (SOT). The protocols for the animal studies were also approved by the Institutional Animal Care and Use Committee (IACUC) at Hanyang University.
Oral administration and blood sampling
The rats were divided into two groups containing six rats per group. Each rat was anesthetized and secured on a surgical board with a thread in the supine position. A polyethylene tube, filled with 50 IU/mL of heparin in saline, was inserted into the right femoral artery of the rat. The rats in each group were orally administered MTX-loaded solid SMEDDS or MTX powder (at the dose of 20 mg/kg) with 1 mL of water. Then, at predetermined time intervals, 0.3 mL of blood was collected via the cannulated tube with heparinised syringes and centrifuged at 3,000 g for 10 min. Plasma samples were stored at −20 °C until further analysis.
Sample preparation and HPLC analysis
The collected plasma samples (90 μL) were deproteinized with methanol (100 μL) in 2 mL microtubes. Afterwards, 10 μL of internal standard solution (theophylline, 1 mg/mL in methanol) were added to each tube and vortexed (1 min). Furthermore, the mixture was centrifuged at 10,000 g for 10 min. The supernatant organic layer was transferred to another clean microtube. Then, the resulting solution (10 μL) was analyzed by HPLC as mentioned above. All pharmacokinetic parameters, including Tmax (time to reach the maximum plasma concentration), Cmax (maximum plasma concentration), AUC (area under the whole blood concentration-time curve), Kel (elimination rate constant), and t1/2 (half-life), were analyzed by the WinNonlin software (Pharsight Corp., Mountain View, CA, USA). Values are reported as mean ± S.D. and the data was statistically significant at P<0.05 between the two formulations checked by the Student’s t-test.
Photostability study
Photostability studies consisted of a forced degradation test and a confirmatory or formal test to evaluate the photostability of MTX powder and MTX-loaded solid SMEDDS. These tests were performed according to the ICH guidelines.20 A forced photodegradation study was carried out first. Therefore, the samples were transferred to chemically inert and transparent containers, and exposed to an artificial light source: 1.2 million lux·hr in 25 °C/60% RH for 20 days using a photostability chamber (Caron Model 6545–2, Caron, Marietta, OH, USA). A confirmatory photostability test was also performed; samples were introduced to transparent containers and placed horizontally towards the light source (indoor light) for even irradiation of the samples for 12 weeks. The drug content in the samples was quantified at predetermined time intervals by the HPLC method described above. The test was performed in triplicate for each sample. Because qualitative evaluation is just as important as the quantitative evaluation in the photostability study, the samples were visually investigated at the end of the test period for any changes in physical properties, such as appearance or color, which can imply the existence of photolytic degradation.
Results and discussion
Solubility study
Self-emulsifying formulations, that consist of oils, surfactants, co-surfactants, and drugs, should be a clear and monophasic liquid when introduced to aqueous phase at ambient temperature and should have good solvent capacity to present the drug into solution.21 The solubility of MTX in various vehicles is shown in Figure 1. The interactions between drugs and excipients could be known from solubility screening results.
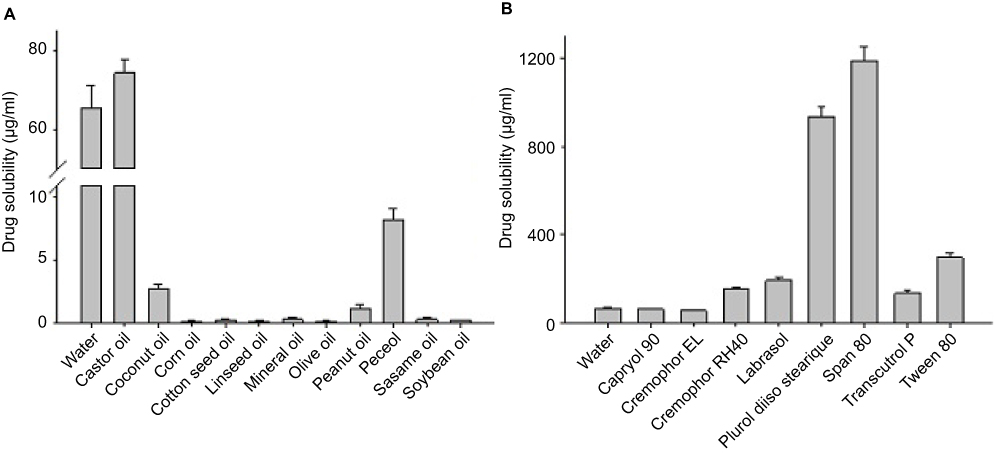 |
Figure 1 Solubility of MTX in crude oil (A) and 10% surfactant solution (B). Each value represents the mean ± S.D. (n=3). |
Among the different tested oils (Figure 1A), castor oil gave the maximum drug solubility (74.40±3.38 μg/mL), and was therefore chosen as the oil for further studies. Among the tested surfactants (Figure 1B), Plurol® diisostearique (triglycerol diisostearate), Span 80 (sorbitan monooleate), and Tween® 80 (polysorbate 80) showed higher drug solubility than the other tested surfactants. In several trial combinations of different surfactants and co-surfactants, the mixture of Tween® 80 (hydrophile-lipophile balance [HLB] 15) and Plurol diisostearique (HLB 4.5) demonstrated excellent viscosity and HLB values.22 Thus, Tween® 80 and Plurol® diisostearique were chosen as the surfactant and co-surfactant, respectively. Although Span 80 (HLB 4.3) showed better solubility than Plurol® diisostearique, it demonstrated high viscosity when mixed with Tween® 80.
All the selected excipients have been widely used in cosmetics and foods and orally, parenterally, and topically in pharmaceutical formulations. It is generally regarded as relatively nontoxic and nonirritant materials, namely biologically safe excipients.
Pseudo-ternary phase diagrams
A series of SMEDDS combinations were prepared, and their self-emulsification properties were observed visually. First, a pseudo-ternary phase diagram was constructed in the absence of MTX to identify optimized concentrations of the oil, surfactant, and co-surfactant. The phase diagram of the system containing castor oil, Tween® 80, and Plurol® diisostearique is shown in Figure 2A. As seen from the pseudo-ternary phase diagram, these combinations produced a reasonably wide self-emulsification region. Fine spontaneous emulsions did not efficiently form when the concentration of the surfactant was less than oil. The efficiency of emulsification was good when the composition of surfactant/co-surfactant was more than 55% v/v of the SMEDDS formulation. The formulations surrounding the self-emulsification region demonstrated poor emulsion forming ability. It has been reported that the drug incorporated into the SMEDDS may influence the self-emulsifying performance.23 However, in our study, no significant differences appeared when comparing the SMEDDS with the MTX-loaded SMEDDS.
 |
Figure 2 Formulation of liquid SMEDDS. Pseudo-ternary phase diagram (A), effect of the ratio of surfactant to oil on the droplet size of emulsion (B), and effect of co-surfactant on the mean emulsion droplet diameter of emulsions containing 7:3 of constant surfactant/oil ratio (C). |
Preparation of liquid and solid SMEDDS
In the SMEDDS systems, visual observation is the primary method for judging self-emulsification. The efficiency of self-emulsification can be evaluated by measuring the rate of emulsification and the droplet size. The emulsion droplet size is an important factor in self-emulsification performance, because it is highly correlated with the amount and rate of drug release, as well as drug absorption.24 The z-average emulsion diameters of various surfactant/oil ratios are shown in Figure 2B. Of the tested formulations, a surfactant/oil ratio of 70/30 (v/v), respectively, showed the most effective emulsion size. When the oil ratio was less than 15%, a smaller diameter micelle was observed instead of an emulsion.25 The effect of the co-surfactant concentration on the emulsion size in SMEDDS was observed (Figure 2C). A co-surfactant ratio of 10% showed the smallest emulsion size, and a rapid increase in emulsion size was observed at co-surfactant ratios of more than 15%. Finally, the liquid SMEDDS formulation, which was composed of 27% castor oil, 63% Tween® 80, and 10% Plurol® diisostearique, gave the smallest z-average diameter of all tested formulations. Furthermore, 10% (w/v) drug was entirely dissolved in this formulation, suggesting that it should be used for further studies.
Subsequently, the ethanolic solution (200 mL) containing 0.5 g of calcium silicate (CaSiO3) and 1 mL of liquid SMEDDS was spray dried to transform liquid SMEDDS into solid SMEDDS. Many kinds of solid carriers, such as silicon dioxide, calcium silicate, and dextran, can be used for the same purpose. However, in a preliminary study testing these solid carriers, calcium silicate was the most effective material. Calcium silicate is capable of absorbing a very large amount of oil (4–6 mL/g) through its numerous pores, and thus, has been commonly used for the solidification of oily formulations.26,27 In addition, the pores show a particular size distribution, which are attributed to interparticle and intraparticle pores.28
Reconstitution properties of solid SMEDDS
The emulsion droplet size is a very important property in SMEDDS, because it is related to the amount and rate of drug release, absorption, and stability.29 The emulsion droplet size was measured to determine the self-emulsification performance of the optimized formulations. The z-average diameter of the drug-loaded liquid SMEDDS (189.33±6.61 nm, PDI: 0.218±0.038) and drug-unloaded liquid SMEDDS (189.50±1.70 nm, PDI: 0.191±0.013) exhibited similar results, suggesting that drug incorporation in the SMEDDS does not have a significant influence on the self-emulsifying performance. Reconstituted microemulsion from the solid SMEDDS had the smallest z-average diameter (126.73±4.32 nm, polydispersity index (PDI): 0.226±0.006) among the tested formulations. Incorporation of liquid SMEDDS into the narrow pore of the solid carrier may have affected the decreased particle size after reconstitution. However, all of the z-average diameters of the liquid and solid SMEDDS showed an acceptable size of less than 200 nm and a low PDI of less than 0.230.
Morphological analysis of solid SMEDDS
Scanning electron micrographs of the MTX powder, solid carrier (calcium silicate), and solid SMEDDS are shown in Figure 3. This analysis revealed that MTX powder (Figure 3A) had an even surfaced and angulated crystals in shape. On the other hand, calcium silicate was uneven and highly porous surface (Figure 3B), which probably allowed the influx of the aqueous phase into the substance. Furthermore, solid SMEDDS (Figure 3C) had a relatively smooth surface of calcium silicate particles, suggesting that liquid SMEDDS is absorbed or coated inside the pores of calcium silicate.
 |
Figure 3 SEM images of (A) MTX powder (x2,000), (B) calcium silicate (×5,000), and (C) solid SMEDDS (×5,000). |
Solid state characterization of solid SMEDDS
DSC is the most widely utilized thermal analysis method to monitor endothermic processes (ie, melting, phase transition, and chemical degradation) as well as exothermic processes. Also, DSC can indicate the existence of drug/carrier interactions. The thermal behavior of the MTX powder, calcium silicate, physical mixture, and solid SMEDDS are shown in Figure 4. The DSC curves showed that MTX had a distinct endothermic peak around 155 °C, indicating its exact melting point and confirming its crystalline nature (Figure 4A). Moreover, a small endothermic peak corresponding to the drug was observed in the physical mixture (Figure 4C), which was mixed with 100 mg of MTX and 500 mg of calcium silicate using pestle and mortar. On the contrary, calcium silicate (Figure 4B) and solid SMEDDS (Figure 4D) had no intrinsic peak of the drug, indicating that the drug must exist in a molecularly dissolved state in the solid SMEDDS.
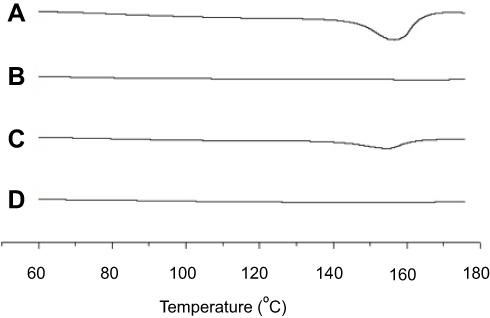 |
Figure 4 DSC curves for MTX powder (A), calcium silicate (B), physical mixture (C), and solid SMEDDS (D). |
From the powder X-ray diffractometric profiles shown in Figure 5, the molecularly dissolved state of MTX in the solid SMEDDS was further verified. MTX showed a typical crystalline pattern (Figure 5A) similar with a reported literature,1 but these representative peaks were not seen in the PXRD pattern of solid SMEDDS, because the drug existed as an amorphous form in solid SMEDDS (Figure 5D).
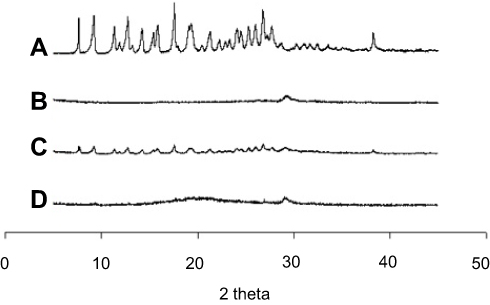 |
Figure 5 PXRD curves for MTX powder (A), calcium silicate (B), physical mixture (C), and solid SMEDDS (D). |
Ftir
FTIR spectroscopic results are shown in Figure 6. MTX produced major distinguishing bands at 3,450 cm−1 (O-H stretching from carboxyl groups), 1,670–1,600 cm−1 (C=O stretching from carboxylic group and amidic group, so the band split into a doublet), 1495 cm−1 (N-H bending from amide group), 1,400–1,200 cm−1 (C-O stretching from carboxylic group), and 832 cm−1 (C-H - two adjacent hydrogens on aromatic ring, para substitution) (Figure 6A). These specific bands, such as 3,450, 1,670–1,600, 1,495, 1,400–1,200, and 832 cm−1 also appeared in the FTIR patterns of the physical mixture (Figure 6C) and of the solid SMEDDS (Figure 6D). There was no apparent shift of bands or an emergence of new bands, confirming the absence of changes in the chemical bond formation between the drug and excipients.
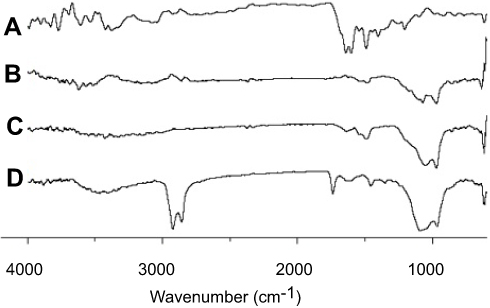 |
Figure 6 FTIR bands for MTX powder (A), calcium silicate (B), physical mixture (C), and solid SMEDDS (D). |
In vitro dissolution test
The prepared SMEDDS formulation showed significantly enhanced aqueous solubility (Figure 7A) and dissolution profiles (Figure 7B) than those of the MTX powder. In the self-emulsifying systems, the required energy to form an emulsion is very low, thereby enabling spontaneous formations at an interface between the oil droplets and water. In the dissolution process, the oil/surfactant/co-surfactant mixture expanded effectively in water; then, the oil droplet size decreased, and eventually MTX was released at an increased rate.30 Dissolution studies were performed for solid SMEDDS and MTX powder, and the results of the release profiles in water are shown in Figure 7B. Within 20 min, the solid SMEDDS formulation reached the maximum percentage of released drug. The final dissolution percentage was almost 100%. However, only 53% of drug was released from the MTX powder at 60 min. These results verify that the solid SMEDDS enhanced the dissolution of MTX.
 |
Figure 7 Drug solubility (A) and dissolution profiles (B) of MTX powder and MTX-loaded solid SMEDDS. Each value represents the mean ± S.D. (n=3). |
Pharmacokinetics study
The mean plasma concentration-time profiles of MTX powder and MTX-loaded solid SMEDDS, equivalent to a 20 mg/kg MTX dose, are represented in Figure 8. Solid SMEDDS showed a significantly higher plasma concentration of MTX than MTX powder for first 4 hr after oral administration. The pharmacokinetic parameters are shown in Table 1. The AUC and Cmax of solid SMEDDS were significantly higher as compared to those of the MTX powder. In particular, the AUC values of MTX powder and solid SMEDDS were 1738.71±294.65 and 3542.69±670.73 hr·ng/mL, respectively, whereas the Cmax values were 265.63±57.05 and 904.99±256.47 ng/mL, respectively. On the other hand, the time to reach the maximum plasma concentration (Tmax), t half-life (t1/2), and elimination rate constant (Kel) were not significantly different. These results suggest that MTX-loaded solid SMEDDS improved the bioavailability of MTX.
 |
Figure 8 Mean plasma concentration–time profiles of MTX after oral administration of MTX powder and MTX-loaded solid SMEDDS in rats. *P<0.05 when compared with MTX powder. Each value represents the mean ± S.D. (n=6). |
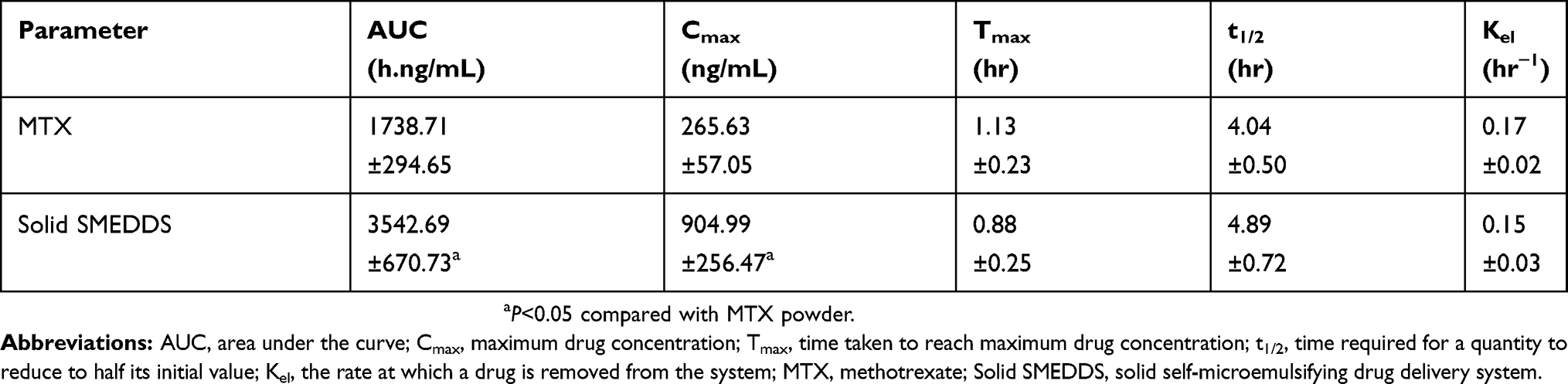 |
Table 1 Pharmacokinetic parameters of MTX powder and MTX-loaded solid SMEDDS |
Stability study
The preservation and robustness of a product against degradation by light is an important property for developing new formulations.31 Time-dependent changes in the MTX content under light irradiation was monitored to evaluate whether solid SMEDDS formulation protected MTX from photodegradation. The photostability of MTX was tested by a forced degradation method and confirmatory analysis. In order to evaluate the overall photosensitivity of the formulation, a forced photodegradation study was performed for 20 days. It was conducted by exposing an excess amount of artificial light using a photostability chamber until significant degradation had occurred. At the same time, the photostability of MTX powder and MTX-loaded solid SMEDDS was tested by exposing the respective formulations to daylight for 12 weeks in the confirmatory analysis. Data obtained after the forced photodegradation and confirmatory test are represented in Figure 9.
 |
Figure 9 Residual concentration of MTX after exposure to photolytic conditions. Forced photodegradation condition for 20 days (A) and daylight for 12 weeks (B). Each value represents the mean ± S.D. (n=3). |
The concentration of the MTX in the solid SMEDDS slightly decreased in the forced photodegradation test after 20 days (87.44±1.93%) and the confirmatory test after 12 weeks (90.57±1.46%) compared to the initial concentration of pure MTX. Whereas, a sharp decrease by nearly 40% in the concentration of MTX (63.21±1.75 and 61.52±2.37%, respectively) was observed.
Moreover, tiny changes in the color of MTX from yellow to orange implied that the formation of photolytic degradants was present in pure MTX samples. It can be concluded that prepared solid SMEDDS is a very effective formulation for maintaining photostability of MTX.32 There are no specific data available that provides a specific limitations for this type of study. Depending on the extent of the change, special labeling or packaging may be needed to mitigate the exposure to light.
Conclusion
In this study, liquid SMEDDS was spray dried using calcium silicate as the solid carrier to produce the MTX-loaded solid SMEDDS formulation. The morphology of the solid SMEDDS formulation revealed well dispersed particles with a smooth surface. The self-emulsification ability of the solid SMEDDS was similar to that of the liquid SMEDDS, and both DSC and PXRD measurements indicated that MTX-loaded solid SMEDDS maintained its molecular dispersion state. Moreover, FTIR analysis confirmed that there were no interactions between the drug and excipients. An in vitro dissolution test revealed that the MTX release was faster and more enhanced in the solid SMEDDS than in the MTX powder. An in vivo study in rats showed that the solid SMEDDS formulation gave a significantly increased bioavailability compared to the MTX powder. Furthermore, the forced degradation and confirmatory photostability studies demonstrated that the solid SMEDDS formulation protected MTX from light-induced degradation efficiently. Thus, this solid SMEDDS formulation may provide an oral solid dosage of the water insoluble and photosensitive drug, MTX.
Acknowledgments
This work was supported by the National Research Foundation (NRF) of South Korea grants funded by the South Korean government (MEST) (No. 2018R1D1A1B07050598). This paper was presented at the 2018 PharmSci 360 as a poster presentation with interim findings.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Thapa RK, Kim JH, Jeong JH, et al. Silver nanoparticle-embedded graphene oxide-methotrexate for targeted cancer treatment. Colloids Surf B Biointerfaces. 2017;153:95–103. doi:10.1016/j.colsurfb.2017.02.012
2. Vadia N, Rajput S. Study on formulation variables of methotrexate loaded mesoporous MCM-41 nanoparticles for dissolution enhancement. Eur J Pharm Sci. 2012;45:8–18. doi:10.1016/j.ejps.2011.10.016
3. Chibber S, Hassan I, Farhan M, Naseem I. In vitro pro-oxidant action of methotrexate in presence of white light. J Photochem Photobiol B. 2011;104:387–393. doi:10.1016/j.jphotobiol.2011.04.005
4. Musmade KP, Deshpande PB, Musmade PB, et al. Methotrexate-loaded biodegradable nanoparticles: preparation, characterization and evaluation of its cytotoxic potential against U-343 MGa human neuronal glioblastoma cells. B Mater Sci. 2014;37:945–951. doi:10.1007/s12034-014-0030-5
5. Bourkaib N, Zhou J, Yao J, Fang Z, Mezghrani O. Combination of β-cyclodextrin inclusion complex and self-microemulsifying drug delivery system for photostability and enhanced oral bioavailability of methotrexate: novel technique. Drug Dev Ind Pharm. 2013;39:918–927. doi:10.3109/03639045.2012.718785
6. Ray S, Joy M, Sa B, Ghosh S, Chakraborty J. pH dependent chemical stability and release of methotrexate from a novel nanoceramic carrier. RSC Adv. 2015;5(49):39482–39494. doi:10.1039/C4RA14244F
7. Na YG, Byeon JJ, Wang M, et al. Strategic approach to developing a self-microemulsifying drug delivery system to enhance antiplatelet activity and bioavailability of ticagrelor. Int J Nanomedicine. 2019;14:1193–1212. doi:10.2147/IJN.S198376
8. Hwang I, Kang CY, Park JB. Advances in hot-melt extrusion technology toward pharmaceutical objectives. J Pharm Investig. 2017;47(2):123–132. doi:10.1007/s40005-017-0309-9
9. Madhav KV, Kishan V. Self microemulsifying particles of loratadine for improved oral bioavailability: preparation, characterization and in vivo evaluation. J Pharm Investig. 2018;48(4):497–508. doi:10.1007/s40005-017-0344-6
10. Zhao J, Xie P, Ye C, et al. Outside-in synthesis of mesoporous silica/molybdenum disulfide nanoparticles for antitumor application. Chem Eng J. 2018;351:157–168. doi:10.1016/j.cej.2018.06.101
11. Rahman MA, Mujahid M, Hussain A, Iqbal Z. Development and pharmacokinetic evaluation of spray-dried self-nanoemulsifying drug delivery system of sertraline. J Pharm Investig. 2017;47(4):325–333. doi:10.1007/s40005-016-0263-y
12. Rashid R, Kim DW, Yousaf AM, et al. Comparative study on solid self-nanoemulsifying drug delivery and solid dispersion system for enhanced solubility and bioavailability of ezetimibe. Int J Nanomedicine. 2015;10:6147–6159.
13. Seo YG, Kim DW, Yousaf AM, et al. Solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced oral bioavailability of poorly water-soluble tacrolimus: physicochemical characterisation and pharmacokinetics. J Microencapsul. 2015;32:503–510. doi:10.3109/02652048.2015.1073392
14. Onoue S, Uchida A, Kuriyama K, et al. Novel solid self-emulsifying drug delivery system of coenzyme Q 10 with improved photochemical and pharmacokinetic behaviors. Eur J Pharm Sci. 2012;46:492–499. doi:10.1016/j.ejps.2012.03.015
15. Nazzal S, Smalyukh I, Lavrentovich O, Khan MA. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: mechanism and progress of emulsion formation. Int J Pharm. 2002;235:247–265.
16. Oh DH, Kang JH, Kim DW, et al. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int J Pharm. 2011;420:412–418. doi:10.1016/j.ijpharm.2011.08.005
17. Bayomi MA, Abanumay KA, Al-Angary AA. Effect of inclusion complexation with cyclodextrins on photostability of nifedipine in solid state. Int J Pharm. 2002;243:107–117. doi:10.1016/S0378-5173(02)00263-6
18. Kim DW, Kwon MS, Yousaf AM, et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohyd Polym. 2014;114:365–374. doi:10.1016/j.carbpol.2014.08.034
19. Balakrishnan P, Lee B-J, Oh DH, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72:539–545. doi:10.1016/j.ejpb.2009.03.001
20. Tonnesen HH. Photostability of Drugs and Drug Formulations. 2nd Ed. Boca Raton (FL): CRC Press; 2004.
21. Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15:958–965. doi:10.1016/j.drudis.2010.08.007
22. Kadu PJ, Kushare SS, Thacker DD, Gattani SG. Enhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS). Pharm Dev Technol. 2011;16:65–74. doi:10.3109/10837450903499333
23. Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’drug delivery systems. Eur J Pharm Sci. 2000;11:S93–S98.
24. Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–182. doi:10.1016/j.biopha.2004.02.001
25. Narang AS, Delmarre D, Gao D. Stable drug encapsulation in micelles and microemulsions. Int J Pharm. 2007;345:9–25. doi:10.1016/j.ijpharm.2007.08.057
26. Ito Y, Kusawake T, Prasad YR, Sugioka N, Shibata N, Takada K. Preparation and evaluation of oral solid heparin using emulsifier and adsorbent for in vitro and in vivo studies. Int J Pharm. 2006;317:114–119. doi:10.1016/j.ijpharm.2006.02.056
27. Weerapol Y, Limmatvapirat S, Takeuchi H, Sriamornsak P. Fabrication of spontaneous emulsifying powders for improved dissolution of poorly water-soluble drugs. Powder Technol. 2015;271:100–108. doi:10.1016/j.powtec.2014.10.037
28. Kinoshita M, Baba K, Nagayasu A, et al. Improvement of solubility and oral bioavailability of a poorly water-soluble drug, TAS-301, by its melt-adsorption on a porous calcium silicate. J Pharm Sci. 2002;91:362–370.
29. Qureshi MJ, Mallikarjun C, Kian WG. Enhancement of solubility and therapeutic potential of poorly soluble lovastatin by SMEDDS formulation adsorbed on directly compressed spray dried magnesium aluminometasilicate liquid loadable tablets: a study in diet induced hyperlipidemic rabbits. Asian J Pharm Sci. 2015;10(1):40–56. doi:10.1016/j.ajps.2014.08.003
30. Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. Oral self emulsifying powder of lercanidipine hydrochloride: formulation and evaluation. Powder Technol. 2012;221:375–382. doi:10.1016/j.powtec.2012.01.032
31. Bani-Yaseen AD, Al-Rawashdeh NF, Al-Momani I. Influence of inclusion complexation with β-cyclodextrin on the photostability of selected imidazoline-derived drugs. J Incl Phenom Macro. 2009;63:109–115. doi:10.1007/s10847-008-9493-6
32. Lv X, Liu T, Ma H, et al. Preparation of essential oil-based microemulsions for improving the solubility, pH stability, photostability, and skin permeation of quercetin. AAPS PharmSciTech. 2017;18(8):3097–3104. doi:10.1208/s12249-017-0798-x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.