Back to Journals » Drug Design, Development and Therapy » Volume 15
Selective Inhibition of 11β-Hydroxysteroid Dehydrogenase Type 1 Attenuates High-Fat Diet-Induced Hepatic Steatosis in Mice
Authors Li H, Sheng J, Wang J, Gao H, Yu J, Ding G, Ding N, He W ![]() , Zha J
, Zha J ![]()
Received 12 October 2020
Accepted for publication 8 May 2021
Published 31 May 2021 Volume 2021:15 Pages 2309—2324
DOI https://doi.org/10.2147/DDDT.S285828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Professor Anastasios Lymperopoulos
Huashan Li,1,* Jianying Sheng,1,* Jing Wang,1 Haiting Gao,1 Jing Yu,2 Guoxian Ding,2 Ning Ding,1 Weiqi He,1,3 Juanmin Zha1
1Jiangsu Key Laboratory of Neuropsychiatric Diseases and Cambridge-Suda (CAM-SU) Genomic Resource Center, Medical College of Soochow University, Department of Oncology, The First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of Geriatrics, Division of Geriatric Endocrinology, The First Affiliated Hospital to Nanjing Medical University, Nanjing, People’s Republic of China; 3State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weiqi He; Juanmin Zha
Cambridge-Suda (CAM-SU) Genomic Resource Center, Medical College of Soochow University, Department of Oncology, The First Affiliated Hospital of Soochow University, 199 Ren-Ai Road, Suzhou, 205123, People’s Republic of China
Tel +86-512-6588-3545
Fax +86-6588-3562
Email [email protected]; [email protected]
Introduction: The effect of 11β-hydroxysteroid dehydrogenase type1 (11β-HSD1) inhibition on hepatic steatosis is incompletely understood. Here, we aimed to determine the therapeutic effect of BVT.2733, a selective 11β-HSD1 inhibitor, on hepatic steatosis.
Materials and Methods: C57B/6J mice were randomly divided into a low-fat diet (LFD) fed group and a high-fat diet (HFD) fed group. Mice were fed with HFD for 28 weeks which induced obesity and severe hepatic steatosis. The two groups were further divided into four groups as follows: LFD, LFD with BVT.2733, HFD, and HFD with BVT.2733. Mice in LFD+BVT and HFD+BVT groups were intraperitoneally injected with BVT.2733 daily for 30 days. Effects of BVT.2733 on mice body weight, serum lipid profile, serum free fatty acids (FFAs), glucocorticoid levels, gene expression in adipose and liver tissues were assessed.
Results: Injection of a low dose of BVT.2733 (50 mg/kg/day) reduced body weight and hyperlipidemia, but did not improve glucose tolerance and insulin resistance in diet-induced obese mice. The low dose of BVT.2733 attenuated hepatic steatosis, liver injury, and liver lipolytic gene expression in diet-induced obese mice. Besides, the low dose of BVT.2733 reduced fat mass and lipolysis in visceral adipose tissues, hepatic FFAs, and serum corticosterone levels in diet-induced obese mice.
Conclusion: Our study shows that moderate inhibition of 11β-HSD1 by BVT.2733 reduces FFAs and corticosterone synthesis in fatty tissues, thereby attenuates the delivery of corticosterone and FFAs to the liver. Collectively, this prevents high-fat diet-induced hepatic steatosis.
Keywords: 11β-HSD1, hepatic steatosis, inhibitor, adipose tissue, free fatty acids
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a widely distributed clinical disease with a great economic and clinical burden.1 The prevalence of NAFLD ranges from 27–34% in the general population in North America, 25% in Europe, and 15–20% in Asia.2,3 NAFLD is characterized by excessive lipid accumulation in the liver independent of alcoholic abuse, drug toxicity, or virus injury.4 NAFLD encompasses a spectrum of liver conditions, ranging from hepatic steatosis, steatohepatitis, fibrosis to cirrhosis, all of which lead to hepatic carcinoma. NAFLD patients with simple fatty liver carry a low risk of adverse outcomes and have a similar life expectancy to the general population. In contrast, those with steatohepatitis have a higher risk of developing cardiovascular and liver-related diseases which greatly decreases the survival rate.5 Thus, interventions that ameliorate hepatic steatosis in NAFLD patients will improve the clinical outcomes of such patients.
The inability of the liver to regulate free fatty acids (FFAs) is thought to play a central role in the pathogenies of NAFLD. Increased diet-derived FFAs and FFAs originating from lipolysis in adipose tissue elevate FFAs flux to the liver. Excessive FFAs in hepatocytes produce lipotoxic species that induce endoplasmic reticulum (ER) stress and hepatocellular injury.6 Hepatic de novo lipogenesis (DNL) that converts diet-derived carbohydrates to FFAs in hepatocytes is an important source of FFAs. FFAs in the liver are converted into triglyceride (TG) by mitochondrial β-oxidation and esterification. Given that TG formation or reduced lipid export in the liver does not induce hepatic inflammation, TG accumulation in the liver may prevent the production of lipotoxic species.7,8 Adipose tissues, especially the visceral adipose tissue (VAT), serve as an extra-hepatic pathogenic organ for NAFLD. Excess stored fat due to increased lipolysis in obese patients is an important source of hepatic FFAs flux.9,10 Additionally, adipose tissues secrete several adipokines regulated by peripheral and central metabolic signals that impart pathogenic effects on the liver.11
11β-hydroxysteroid dehydrogenase type1 (11β-HSD1) is an ER membrane enzyme. Its hydrophobic N-terminus is anchored to the ER membrane and the catalytic domain faces the luminal space.12 11β-HSD1 was initially identified, purified, and found to be abundantly expressed in the liver where it mainly functions as a reductase that converts inactive 11-dehydrocorticosterone to corticosterone in rodents (cortisone to cortisol in human).13 11β-HSD1 is also expressed in white adipose tissue, and functions as an amplifier of glucocorticoids (corticosterone in rodents and cortisol in humans). Hepatic transgenic overexpression of 11β-HSD1 in mice induced fatty liver and insulin resistance without obesity.14 Transgenic mice with an ~2.5-fold overexpression of 11β-HSD1 in adipose tissue exhibited a metabolic syndrome with insulin resistance, dyslipidemia, and visceral obesity.15 Elevated 11β-HSD1 expression in VAT and increased portal glucocorticoid levels have been reported in ob/ob mice, a classic animal model of NAFLD. In humans, 11β-HSD1 overexpression in VAT correlated with the development of NAFLD.16 Taken together, these cases suggest a pathogenic role of hepatic and adipic 11β-HSD1 in the pathogenesis of NAFLD.
Previously, we have shown that a selective inhibitor of 11β-HSD1, BVT.2733, prevents body weight gain, adipose tissue inflammation, and cardiomyopathy in diet-induced obese mice.17,18 Some preliminary studies indeed showed that a 11β-HSD1 inhibitor, Compound A, reduced liver and serum TG concentrations.19 AZD6925, a selective 11β-HSD1 inhibitor, reduced liver triglyceride levels and prevented hepatic steatosis in db/db mice.20 The 11β-hydroxysteroid dehydrogenase type 1 inhibitor, H8, protects against insulin resistance and hepatic steatosis in db/db mice.21 However, the specific mechanism of 11β-HSD1 inhibitor on hepatic steatosis has not been studied in detail in all the above studies. The effect of inhibitors of 11β-HSD1, RO5093151, has been studied in patients with type 2 diabetes as well as in patients with fatty liver disease. However, these studies on fatty liver and metabolic syndrome have been modest, and the use of the inhibitors will produce some untoward effects. Compared with the placebo group, participants in the RO5093151 group had adverse events including gastrointestinal disorders, infections, and nervous system disorders.22 Therefore, we need to test the efficacy of other inhibitors, such as BVT.2733, to block liver steatosis in animal and human studies.
Previous studies showed that the treatment of 11β-HSD1 inhibitors protected against hepatic steatosis before mice were obese. Different from these kinds of research, we pay attention to the therapeutic effect of BVT.2733, not the preventive effect. Therefore, we fed mice with a high-fat diet and established an obese mice model. Obese mice then received BVT.2733 injection to determine whether BVT.2733 had a protective effect on hepatic steatosis. A high dose of 11β-HSD1 inhibitor may lead to off-target because 11β-HSD1 inhibitor can still improve metabolic phenotype in male 11β-HSD1 knockout mice fed with the high-fat diet.23 In our study, mice were given low-dose inhibitors to reduce the off-target effect and side effects. We demonstrated that moderate inhibition of 11β-HSD1 by the low dose of BVT.2733 protected against high-fat diet-induced hepatic steatosis by inhibiting lipolysis in fatty tissues and lipogenesis in the liver.
Materials and Methods
Ethics Statement
All animal experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals approved by the Animal Ethics Committee of Cambridge-Suda (CAM-SU) Genomic Resource Center, Soochow University (protocol number: WH-2017-1).
Mice and BVT2733 Administration
Male C57BL/6 mice used in this study were housed in a specific-pathogen-free facility, with lights on from 08:00–20:00. Mice were given ad libitum access to food and drinking water. BVT.2733 (synthesized by Guangzhou Isun Pharmaceutical Co., Ltd, purity: ~98%) was dissolved in 12% β-hydroxy propyl cyclodextrin and 0.3% sodium chloride. To test the toxicity of BVT.2733, male 8-week-old mice were separately administered with doses of 50, 100, 200 mg/kg/day BVT.2733, or vehicle, through intraperitoneal injection once a day for 16 consecutive days. The body weight and behavior of mice were recorded.
To explore the therapeutic effects of BVT.2733 on NAFLD, a diet-induced mice model of NAFLD was established. Male mice (8-week old) were fed with a low-fat diet (LFD, Research Diets #12450J, 3.82 KJ/g, 70% energy from carbohydrate, 10% from fat, and 20% from protein), or a high-fat diet (HFD, Research Diets #D12492, 5.21 KJ/g, 20% energy from carbohydrate, 60% from fat, and 20% from protein) for 28 weeks. Mice fed with HFD became obese and developed severe hepatic steatosis. These two groups were further administered with BVT.2733 (50 mg/kg/d) or vehicle through intraperitoneal injection once a day for 30 consecutive days (Figure 1).
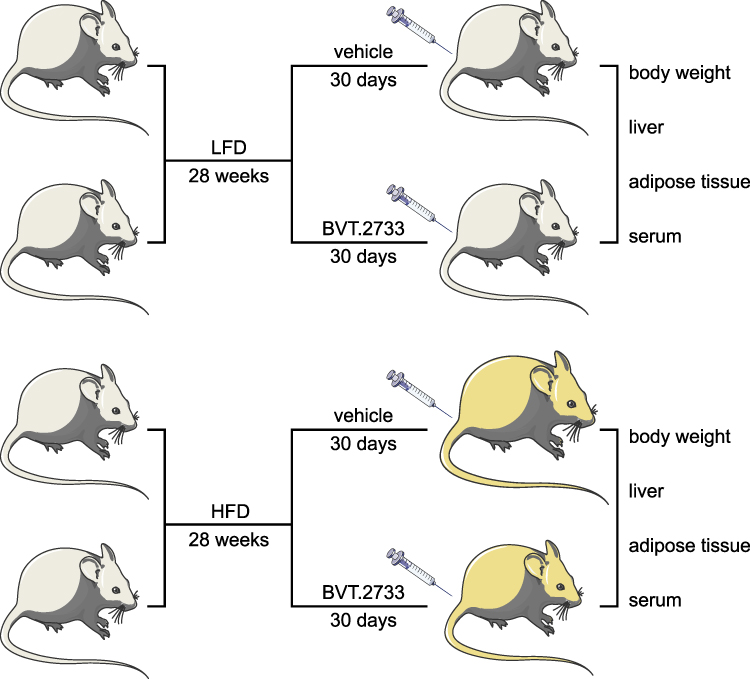 |
Figure 1 Schematic diagram illustrating the major steps of the experiments in the study. The mice were fed with LFD or HFD for 28 weeks. The mice were then injected with vehicle or BVT.2733 for 30 consecutive days. The body weight of each mouse was recorded. The liver and adipose tissue, and serum were collected from each mouse, and were subjected to specific experiments. |
RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
Total RNA was extracted from liver and fat tissues using the GeneJET RNA Purification Kit (Thermo Fisher, K0731). The RNA was reverse-transcribed into cDNA using PrimeScript RT reagent Kit (Takara, RR047B) according to the manufacturer’s instructions. Quantitative Real-time PCR (qRT-PCR) was conducted on the ABI-Stepone instrument using the SYBR Premix Ex Taq reagent (Takara, RR420B). All groups were tested in three replicates and normalized to Gapdh. Primers used for qRT-PCR are listed in Table 1.
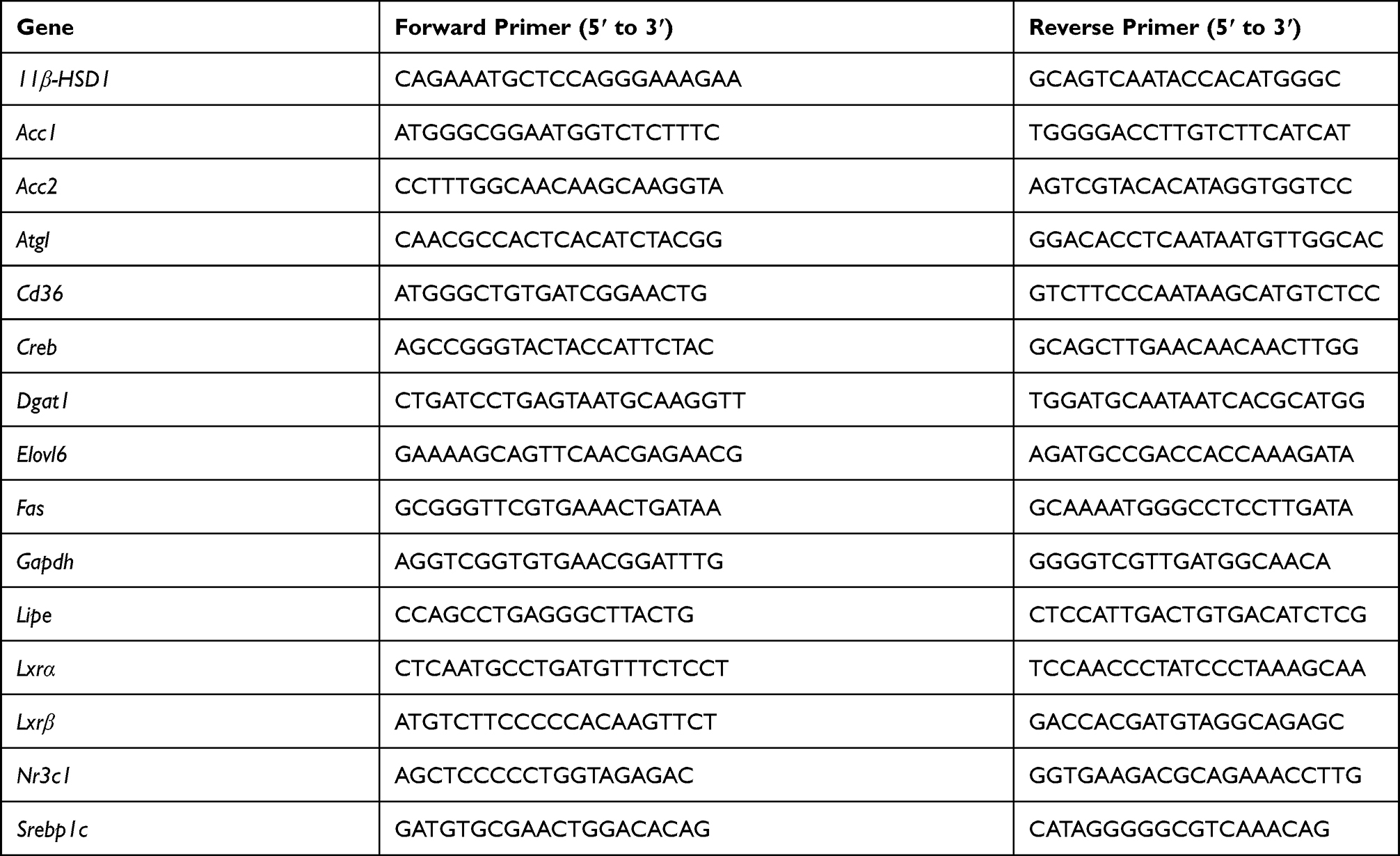 |
Table 1 Primer Pairs Used for qRT-PCR |
Western Blotting Assay
Protein lysates of liver or adipose tissue were isolated as described previously and were separated by SDS–PAGE (Bio-Rad Laboratories) and then transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories). After blocking, the membranes were blotted using the following primary antibody: anti-Fas antibody (3180, 1:1000 dilution), anti-Acc1 antibody (3676, 1:1000 dilution) and anti-β-tubulin antibody (5346, 1:1000 dilution) from Cell Signaling Technology; anti-Dgat1 antibody (11561-1-AP, 1:500 dilution), anti-Srebp1c antibody (14088-1-AP, 1:1000 dilution), anti-Elovl6 antibody (21160-1-AP, 1:1000 dilution), anti-β-actin antibody (66009-1-Ig, 1:10,000 dilution) from Proteintech Group. Afterward, membranes were blotted by peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and detected by the enhanced chemiluminescent method. Signal intensity was assessed using Image J software.
Glucose and Insulin Tolerance Test
The glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed as previously.24 For the glucose tolerance test (GTT), the mice were fasted overnight for about 16 hours. A glucose solution (250 mg/mL D-glucose) was prepared in advance. Blood was collected from the tail vein before (defined as 0) or 15, 30, 60, 90, 120, and 150 min after the intraperitoneal injection of glucose (2.5g/kg body weight). To perform the insulin tolerance test (ITT), the mice were fasted for 4 hours. The insulin solution was pre-prepared at a concentration of 0.1 U/mL. Blood was collected from the tail vein before (defined as 0) or 15, 30, 60, 90, 120, and 150 min after the intraperitoneal injection of insulin (0.75U/kg body weight). Blood glucose was measured with a glucometer (Roche, Accu-Chek Performa).
Hematoxylin and Eosin and Oil-Red O Staining
Liver tissues were washed with cold phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde (PFA) at 4°C for 24 hours, and then embedded in paraffin for sectioning into 5 μm pieces. Hematoxylin & eosin (H&E) staining was performed according to the standard methods. For oil-red O staining, liver tissues were embedded in the Tissue Freezing Medium (Leica, H22136) and sectioned into 8 μm pieces using a Leica Cryostat (Leica, CM1850). The frozen sections were aired dry at room temperature and rinsed briefly with distilled water. Next, the sections were fixed with 1% PFA for 10 min, briefly washed with distilled water for 5 min, and rinsed with 60% isopropanol for 5 min. The tissues were stained with freshly prepared 0.3% Oil Red O working solution (Sigma, O0625, dissolved in 60% isopropanol) for 15 min. After staining, tissue sections were rinsed with 60% isopropanol for 5 min and washed multiple times with distilled water, and mounted in glycerine jelly.
Serum and Liver Biochemistry Profiles
Blood samples were collected from the posterior orbital venous plexus of mice and centrifuged to obtain serum. Serum levels of total cholesterol (TC), triglyceride (TG), low-density cholesterol (LDL), high-density lipoprotein (HDL), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were measured with a biochemistry automatic analyzer (Hitachi, 7100+ISE). Liver specimens were collected, homogenized, and levels of TC and TG were quantified as mentioned above.
Serum Corticosterone Detection
Serum corticosterone was determined using the Corticosterone Parameter Assay kit (R&D, KGE009) following the manufacturer’s instructions.
Statistical Analysis
Quantitative data are shown as mean ± sem. Statistical significance was determined with Software GraphPad Prism (version 8) by unpaired t-test with Welch’s correction or Two-way ANOVA as indicated in each figure legend. P < 0.05, and P < 0.01 were considered significant differences.
Results
The Low Dose of BVT.2733 Does Not Induce Toxicity in Mice
BVT.2733 is a selective inhibitor of 11β-HSD1 that functions in an isoform-selective manner.25 Previously, we showed that administration of BVT.2733 at a dose of 200 mg/kg/day for four weeks significantly reduced body weight and improved glucose tolerance in high-fat diet (HFD)-induced obese mice.17 However, we did not assess the potential toxicity of BVT.2733 in mice fed with a low-fat diet (LFD). Furthermore, it is likely that high doses of 11β-HSD1 inhibitors cause off-target effects in 11β-HSD1 knockout mice fed with the high-fat diet.23 We, therefore, performed a pilot experiment in which mice fed on LFD received a daily intraperitoneal injection of BVT.2733 at 50, 100, or 200 mg/kg doses for 16 days. Although these doses did not affect mice survival or behavior, daily 100 or 200 mg/kg doses significantly induced weight loss (Figure 2A). In addition, there were no significant differences in weight loss between daily doses of 100 and 200 mg/kg (Figure 2A). This result is consistent with a previous study that gave 100 mg/kg BVT.2733 to mice for 16–17 days reduced body weight.26 In contrast, daily injection of BVT.2733 at 50 mg/kg for 16 days did not induce toxicity, in terms of weight loss or behavior in LFD fed mice (Figure 2A). Thus, BVT.2733 at 50 mg/kg/day dose was used in subsequent experiments.
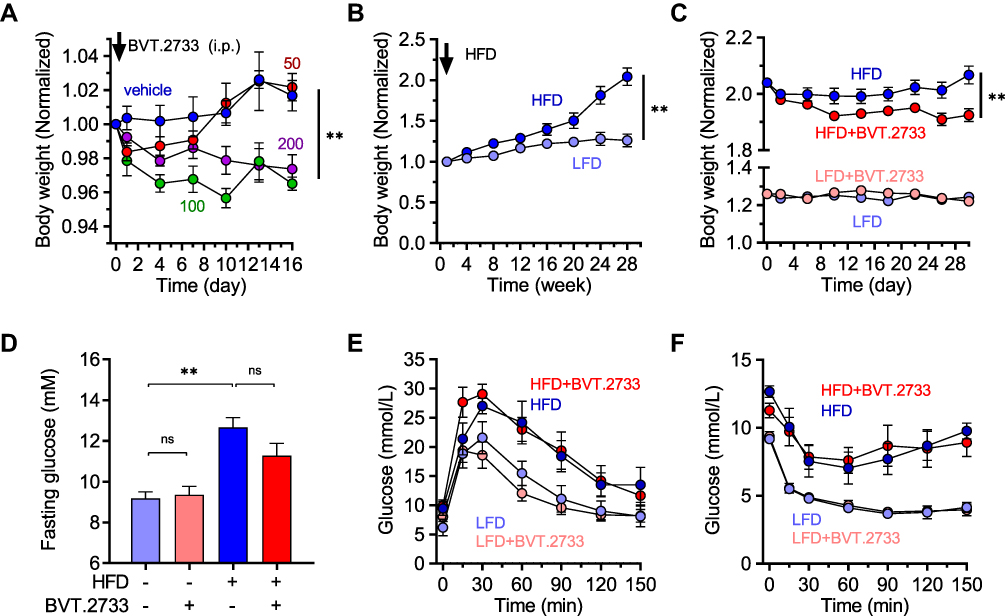 |
Figure 2 Low dose of BVT.2733 reduces body weight in diet-induced obese mice. (A) Mice (12-week old) in four groups were separately administered with vehicle only, 50, 100, or 200 mg/kg BVT.2733 through an intraperitoneal injection given daily for 16 consecutive days. The body weight of each group was recorded to assess BVT.2733 toxicity at different doses. The body weight on day 0 (the day start of BVT.2733 injection) of each mouse was standardized as 1. Data are presented as the means ± sem; vehicle or 50 mg/kg/day groups vs 100 mg/kg/day or 200 mg/kg/day groups, **P < 0.01 by Two-way ANOVA, n=4. (B) Body weight of mice fed either with LFD or HFD for 28 weeks. LFD vs HFD fed mice. The body weight on the day starts of HFD or LFD of each mouse was standardized as 1. Data are presented as the means ± sem; **P < 0.01 by Two-way ANOVA, n=10. (C) Body weight of vehicle or BVT.2733 (50 mg/kg/day) mice fed with either LFD or HFD. BVT.2733 was daily injected for 30 consecutive days at a dose of 50 mg/kg. The body weight on the day starts of HFD or LFD of each mouse was standardized as 1. Data are presented as the means ± sem; vehicle vs 50 mg/kg/day groups. *P < 0.05 by Two-way ANOVA, n=4–5. (D) The concentration of fasting glucose in vehicle or BVT.2733 mice fed with either LFD or HFD. Data are presented as the means ± sem; **P < 0.01 by unpaired t-test with Welch’s correction, n=4–5. (E and F) Glucose tolerance test (E) and insulin tolerance test (F) in vehicle or BVT.2733 injected mice fed with either LFD or HFD. D-F, experiments were performed on day 30 after BVT.2733 or vehicle injection. Data are presented as the means ± sem; mice fed with LFD vs mice fed with HFD, **P < 0.01 by Two-way ANOVA, n=4–5. |
The Low Dose of BVT.2733 Reduces Body Weight, but Does Not Improve Glucose Tolerance and Insulin Resistance in Diet-Induced Obese Mice
Mice were fed with the high-fat diet for 28 weeks to establish a diet-induced animal model of NAFLD. Mice fed with HFD gained body weight by 1.6-fold compared with mice fed with LFD (Figure 2B). To explore whether 11β-HSD1 inhibition could control the effects of LFD or HFD, mice fed with either LFD or HFD were injected daily with BVT.2733 at 50 mg/kg or vehicle for 30 days. Consistent with the pilot toxicity experiment, BVT.2733 administration did not affect the body weight of mice fed with LFD, indicating 30 days of BVT.2733 treatment was also safe for mice. In contrast, BVT.2733 slightly reduced the body weight of mice fed with HFD by 6.9% (Figure 2C), suggesting that the low dose of BVT.2733 reduced body weight in HFD-induced obese mice as efficient as the high dose of BVT.2733 (200 mg/kg/day) as we reported previously.
The fasting blood glucose level was significantly higher in HFD fed mice than in mice fed on LFD (Figure 2D). Inconsistent with this, HFD fed mice developed glucose tolerance and insulin resistance as revealed by intraperitoneal glucose tolerance test (GTT) and insulin tolerance test (ITT) (Figure 2E and F). The fasting blood glucose level was not affected by administration of the low dose of BVT.2733 either in LFD or HFD fed mice (Figure 2D). Besides, the low dose of BVT.2733 failed to improve glucose tolerance and insulin resistance in HFD-fed mice (Figure 2E and F).
The Low Dose of BVT.2733 Attenuates Hyperlipidemia, Hepatic Steatosis, and Liver Injury in a Mouse Model of Diet-Induced Obese Mice
The liver plays an important role in lipid metabolism, and excessive lipid accumulation in the liver affects its histology and function. As expected, serum cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were higher by 1.6-, 2.8-, 1.3-, 1.6-, and 1.8-fold, respectively, in mice fed with HFD compared to mice fed with LFD (Table 2). Interestingly, the low dose of BVT.2733 remarkably reduced serum cholesterol, LDL, ALT, AST, and alkaline phosphatase (ALP), by 20.8%, 35.6%, 40.7%, 41.1%, and 30.0%, respectively, in mice fed with HFD (Table 2). These results suggested that the low dose of BVT.2733 improved high-fat diet-induced hyperlipidemia and liver injury.
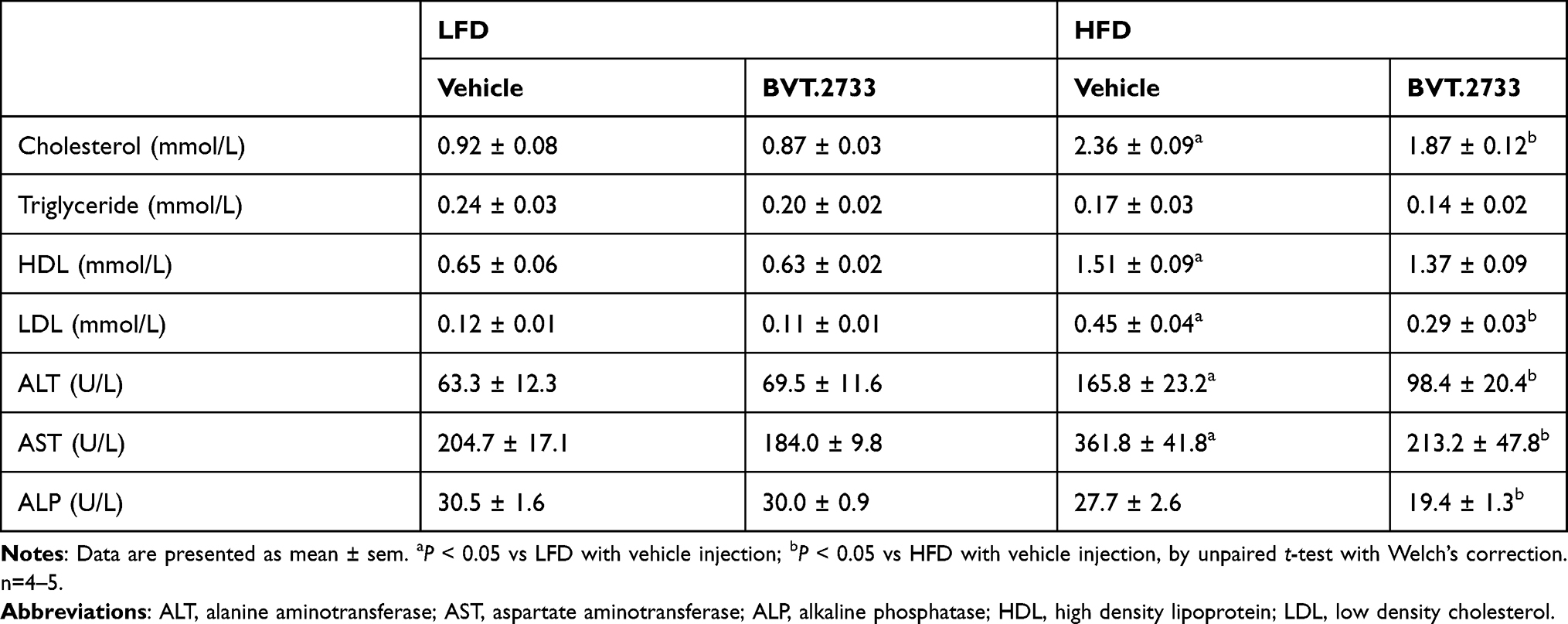 |
Table 2 The Effect of BVT.2733 on Serum Lipid Profiles and Liver Function |
The successful establishment of the NAFLD mice model was confirmed by morphological observation and histopathological analysis of the liver. The livers excised from mice fed with HFD for 28 weeks were larger and appeared pale (Figure 3A and B). H&E staining demonstrated typical macrovesicular steatosis in hepatocytes of HFD fed mice (Figure 3C and D). Consistently, Oil-red O staining revealed excessive lipid accumulation in the liver (Figure 3E and F). Surprisingly, administration of BVT.2733 significantly reduced the weight of the liver by 38.6% (Figure 3A and B). Administration of BVT.2733 significantly reduced typical macrovesicular steatosis area from 42.0 ± 2.9% to 21.7 ± 6.1% in the liver of mice fed with HFD (**P < 0.01, Figure 3C and D). Consistently, administration of BVT.2733 significantly reduced oil-red O stained area from 44.6 ± 4.6% to 22.2 ± 3.2% in the liver of mice fed with HFD (**P < 0.01, Figure 3E and F). BVT.2733 administration, however, did not affect the morphology or weight of the liver in mice fed with LFD (Figure 3A–F). These results suggested that BVT.2733 treatment specifically attenuated hepatic steatosis in an HFD-induced NAFLD model.
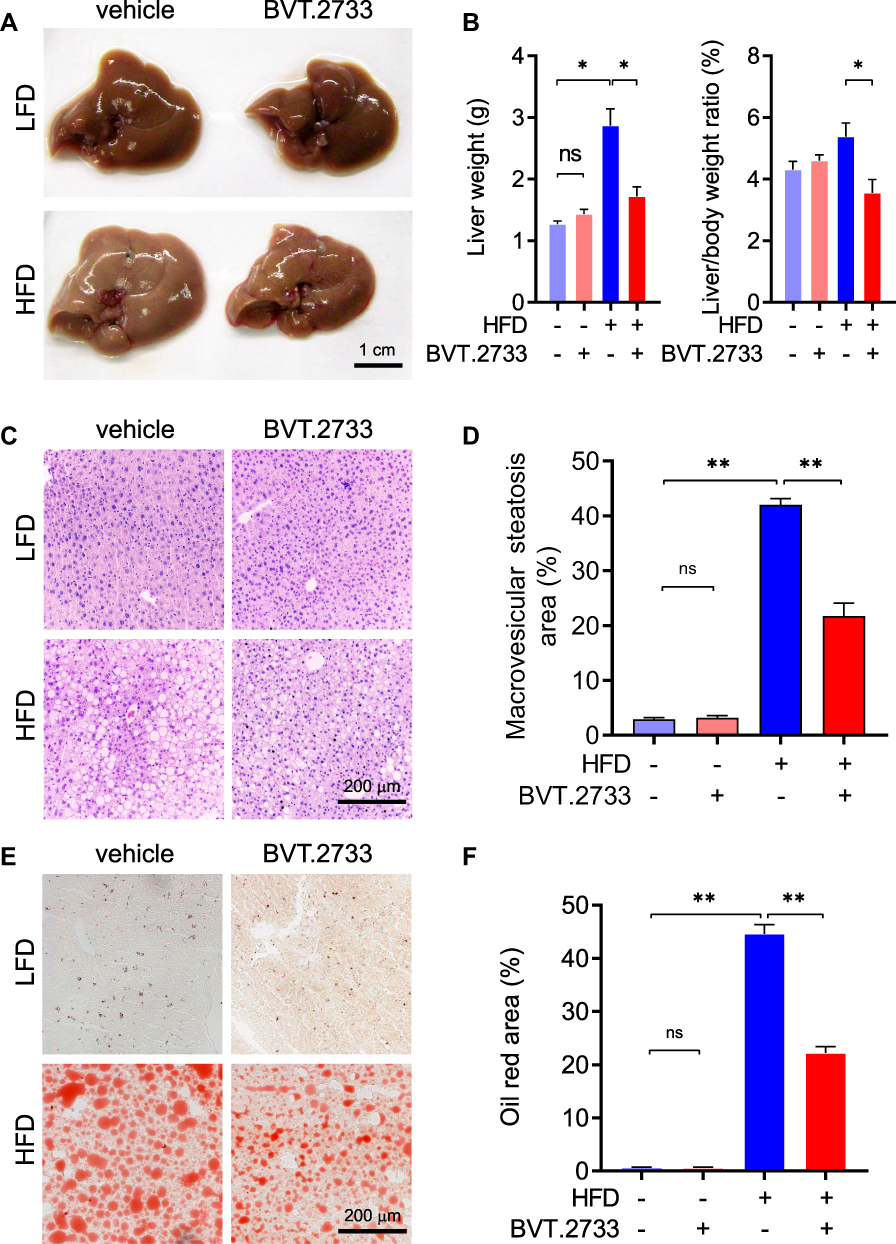 |
Figure 3 BVT.2733 attenuates hepatic steatosis in diet-induced NAFLD mice. (A and B) The morphology of liver (A) and weight of liver (B) in vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. Data are presented as the means ± sem; *P < 0.05 by unpaired t-test with Welch’s correction, n=4–5. (C–F) H&E staining (C) and Oil-red O staining (E) of liver sections in the vehicle or BVT.2733 (50 mg/kg/day) group of mice fed with either LFD or HFD. Bars=200 μm. Quantification of macrovesicular steatosis area (D) and Oil-red O positive area (F) of liver sections. Data are presented as the means ± sem, n=8. *P < 0.05; **P < 0.01; by unpaired t-test with Welch’s correction. Abbreviation: ns, not significant. |
The Low Dose of BVT.2733 Reduces the Expression of the Lipolytic Gene in Liver Tissue
High fat diet-induced hepatic lipid accumulation stems from increased de novo lipogenesis (DNL) in the liver, in which diet-derived carbohydrates are converted to free fatty acids (FFAs) in the liver. We, therefore, examined the expression of de novo lipogenesis-related proteins in liver tissues. Our results showed that a high-fat diet did not alter the expression of de novo lipogenesis-related proteins including Fas, Acc1, Srebp1c, Dgat1, and Elovl6 in the liver tissues. The low dose of BVT.2733 significantly reduced the protein expression of Fas, Acc1, Srebp1c, and Dgat1 in liver tissues of mice fed with LFD. This is consistent with the results from antisense-mediated inhibition of 11β-HSD1 in mice.27 Importantly, BVT.2733 reduced the protein expression of Acc1 in liver tissues of mice fed with HFD (Figure 4A and B).
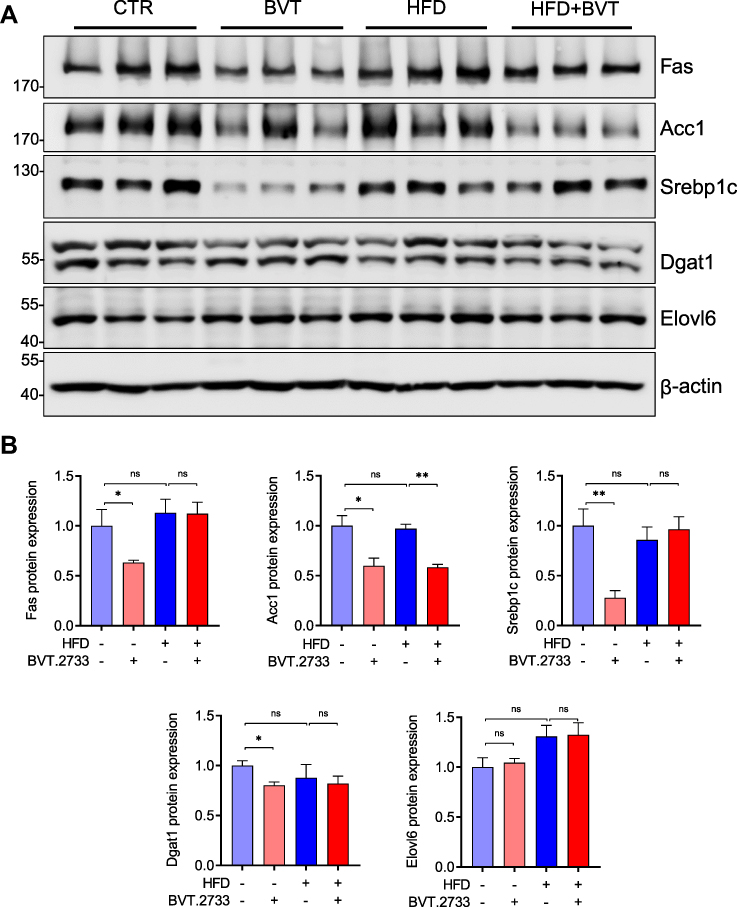 |
Figure 4 BVT.2733 reduces the expression of the lipolytic gene in liver tissue. (A) The expression of de novo lipogenic proteins Fas, Acc1, Srebp1c, Dgat1, and Elovl6 in the liver from vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. β-actin was used as a loading control. (B) Quantitation of protein expression by ImageJ. Data are presented as the means ± sem, n=3. *P < 0.05; **P < 0.01; by unpaired t-test with Welch’s correction. Abbreviations: ns, not significant; Fas, fatty acid synthase; Acc1, acetyl-CoA carboxylase 1; Srebp1c, sterol response element-binding protein-1c; Dgat1, Acyl-CoA, diacylglycerol acyltransferase 1; Elovl6, elongation of very-long-chain fatty acids protein 6. |
The Low Dose of BVT.2733 Affects Fat Deposition and Reduces Lipolysis in the Adipose Tissue
Lipolysis in adipose tissue is an important source of FFAs flux to the liver. Hence, we assessed fat distribution in mice treated with BVT.2733. Results showed that BVT.2733 treatment mildly, although without statistical significance, reduced total fat in mice fed with HFD (Table 3). This is consistent with the finding that administration of BVT.2733 slightly reduced the body weight of mice fed with HFD but not mice fed with LFD (Figure 2C). Interestingly, BVT.2733 treatment significantly reduced the amount of perirenal adipose tissue in HFD fed mice (Table 3). We further assessed the expression of lipogenic and lipolytic genes in perirenal adipose tissue. We found that a high-fat diet significantly decreased the expression of lipogenic related proteins including Fas, Acc1, and Srebp1c in perirenal adipose tissue, which is consistent with previous studies.28,29 The low dose of BVT.2733 treatment reduced the protein expression of Fas and Srebp1c in perirenal adipose tissue of mice fed with LFD, but does not affect perirenal adipose tissue of mice fed with HFD (Figure 5A and B).
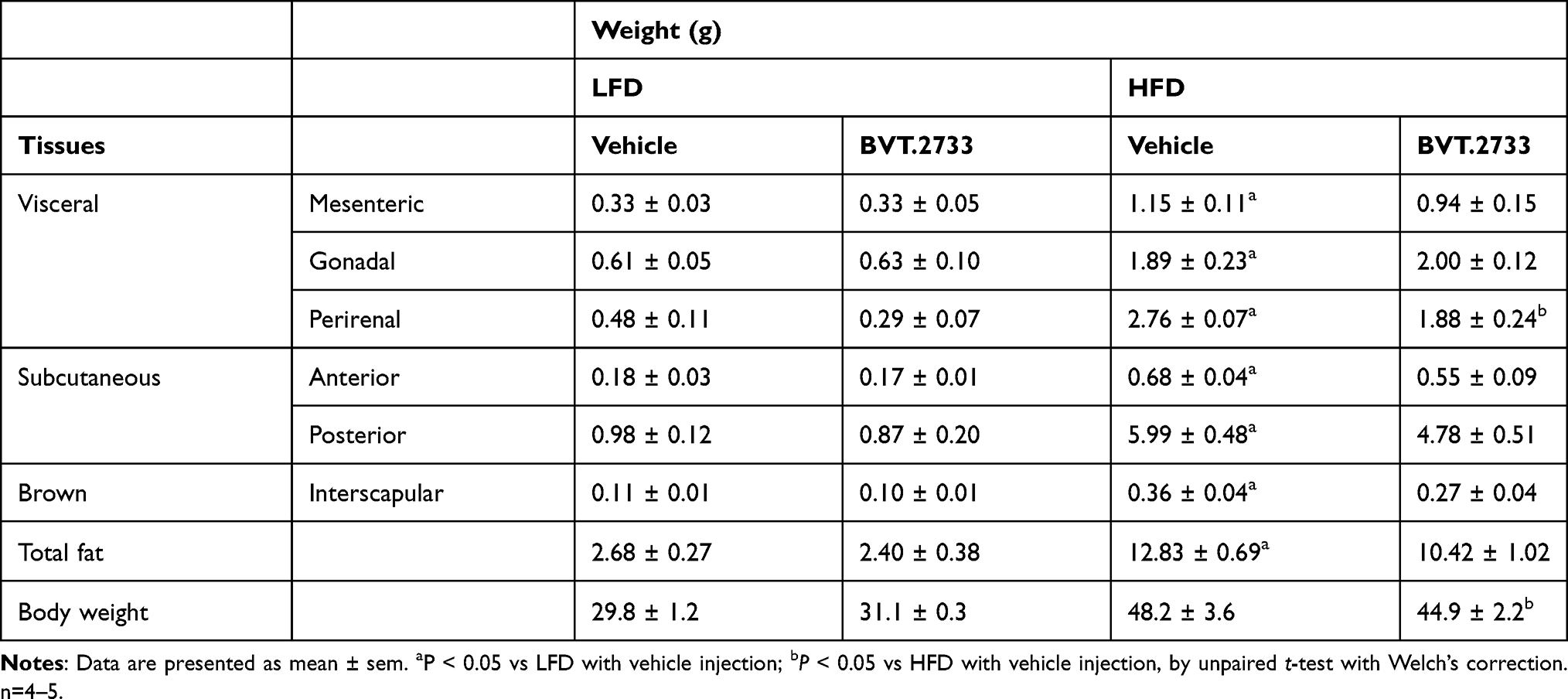 |
Table 3 The Effect of BVT.2733 on Weight of Adipose Tissues |
 |
Figure 5 BVT.2733 reduces the expression of the lipolytic gene in adipose tissue. (A) The expression of de novo lipogenic proteins Fas, Acc1, and Srebp1c, in perirenal adipose tissue from vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. (B) Quantitation of protein expression by ImageJ. Data are presented as the means ± sem, n=3. (C) The relative mRNA expression of lipolysis genes Lxrα, Lxrβ, Atgl, Lipe in perirenal adipose tissue. Data are presented as means ± sem, n=4–5. *P < 0.05; **P < 0.01; by unpaired t-test with Welch’s correction. Abbreviations: ns, not significant; Lxrα, liver X receptor α; Lxrβ, liver X receptor β; Atgl, adipose triglyceride lipase; lipase E (Lipe) or hormone-sensitive lipase (HSL). |
In contrast, HFD increased the expression of lipolytic genes Lxrα, Lxrβ, Atgl, and Lipe (or hormone-sensitive lipase, HSL) in perirenal adipose tissue, and the expression of Lxrα and Lxrβ was attenuated by BVT.2733 treatment (Figure 5C). These results suggested that the low dose of BVT.2733 inhibited lipolysis in adipose tissue of mice fed with HFD.
The Low Dose of BVT.2733 Reduces Hepatic FFAs and Serum Corticosterone Levels
To determine whether loss of fat tissue and decreased lipolysis in adipose tissues directly affected FFAs level, we measured FFAs levels both in the plasma and liver tissue of mice treated with the low dose of BVT.2733. Administration of BVT.2733 significantly reduced the level of FFAs in the liver (Figure 6A). The reduction in FFAs could not be explained by impaired hepatic FFA absorption since the expression of Cd36, a transporter of FFAs, was not reduced (Figure 6B). Given that 11β-HSD1 converts inactive 11-dehydrocortisterone into corticosterone, we measured the level of plasma corticosterone. Administration of a low dose of BVT.2733 did not change the plasma level of corticosterone in mice fed with LFD, but significantly ameliorated the increase of corticosterone level in HFD fed mice (Figure 6C). Besides, administration of the low dose of BVT.2733 did not alter the expression of 11β-HSD1 and Nr3c1 genes, which encode glucocorticoid receptor (GR) in the liver of LFD or HFD fed mice (Figure 6D).
 |
Figure 6 BVT.2733 reduces the concentration of plasma free fatty acids and corticosterone in NAFLD mice. (A) The concentration of plasma and liver FFAs in mice serum and liver were measured in the liver from vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. (B) The relative mRNA expression of Cd36 in vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. (C) The concentration of plasma corticosterone from vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. (D) The relative mRNA expression of 11β-HSD1 and Nr3c1 in the liver from vehicle or BVT.2733 (50 mg/kg/day) groups of mice fed with either LFD or HFD. Data are presented as means ± sem. *P < 0.05; **P < 0.01; by unpaired t-test with Welch’s correction, n=4–5. Abbreviation: ns, not significant. |
Discussion
NAFLD has been extensively studied because of its pronounced negative impact on people’s health. Currently, NAFLD is treated by: 1) reducing the source of fatty acids in the liver; 2) diverting hepatic metabolic substrates (fatty acids and glucose) to oxidative tissues for disposal or thermogenesis; and 3) inhibiting cell stress and apoptosis pathway, and fibrosis to block the NASH progression. For instance, miRNA mimics or anti-miRNAs were used as therapeutic approaches for the treatment of NAFLD and liver fibrosis.30 Targeted delivery of siRNA/peptide nucleic acid (PNA) hybrid nanocomplex effectively reversed rat liver fibrosis.31 Despite tremendous efforts to identify therapeutic targets and drug development, unmet challenges remain.
Obesity is one of the major risk factors of NAFLD. Nearly 80% of patients with obesity have NAFLD. In contrast, the prevalence of NAFLD in individuals with normal body mass index (BMI) and no metabolic risk factors is only 16%.32,33 Visceral fat has been found to be correlated with NAFLD.34 The cross-talk between adipose tissue and the liver may lead to the pathogenesis and progression of NAFLD.6,35 Studies have confirmed that during visceral obesity, hypertrophic adipocytes release several factors, including FFAs, inflammatory factors, and adipo-secretory factors.36 FFAs transported to the liver directly increase hepatic FFAs load. In the substrate-overload liver injury model, FFAs overload in the liver was implicated in the pathogenesis of NAFLD. We have previously found that inhibition of 11β-HSD1 by BVT.2733 reduces body weight and adipose tissue inflammation in diet-induced obese mice.17,18 The purpose of our study was to reverse hepatic steatosis in obese mice by BVT.2733 treatment. We successfully established a diet-induced obese mice model. Mice fed with 60% HFD for 28 weeks showed increased body weight and liver weight, accompanied by hepatic steatosis, the typical characteristics of NAFLD. Obese mice then received BVT.2733 injection to determine whether BVT.2733 had the protective effect on hepatic steatosis. The low dose of BVT.2733 dramatically reduced the body weight, liver weight, serum cholesterol, LDL, and HDL in diet-induced obese mice. It also attenuated hepatic steatosis, reduced serum levels of ALT and hepatic FFAs. We speculate that treatment of the low dose of BVT.2733 before mice were obese will help to protect against obesity and have a protective effect on hepatic steatosis. Previous studies showed that food intake was not affected by the BVT.2733 treatment (167 mg/kg/day) in spontaneously hyperglycaemic KKAy mice.37 Similarly, treatment of a selective 11β-HSD1 inhibitor, AZD6925, did not affect body weight or food intake in control or db/db mice.20 In our study, we treated mice with a low dose of BVT.2733 (50 mg/kg/day). Thus, we speculate that BVT.2733 should not influence the energy consumption of mice.
11β-HSD1 is a key enzyme that regulates peripheral glucocorticoid metabolism by converting cortisone to cortisol in humans (11-dehydrocorticosterone to corticosterone in rodents). It enhances the action of glucocorticoids in local tissues, acting as a local amplifier. Corticosterone in the liver and adipose tissue are key drivers of NAFLD development. A clinical study showed that 11β-HSD1 expression was markedly increased in adipose tissue of obese individuals and with a higher level in visceral fat than subcutaneous fat.38 The low dose of BVT.2733 significantly reduced the protein expression of Acc1 in liver tissues of mice fed with LFD and HFD, indicating that BVT.2733 inhibits lipogenesis in the liver. Additionally, the low dose of BVT.2733 protects against high-fat diet-induced hepatic steatosis by inhibiting lipolysis in fatty tissues. We showed that the increased expression of Lxrα and Lxrβ in adipose tissue of HFD fed mice was attenuated by BVT.2733 treatment. It has been reported that corticosterone treatment increased expression of lipase HSL and Atgl and knockdown of 11β-HSD1 by shRNA attenuated corticosterone-induced lipolysis.39 Our results are strongly supported by the knockout mice study. Mice with the specific knockout of 11β-HSD1 in adipose tissue are protected from the development of 11-DHC-induced hepatic steatosis and significantly protected from CORT- and 11-DHC-induced increased adipose expression of HSL, Atgl, Lxrα, and Lxrβ.40 In fact, AMP-activated protein kinase (AMPK) has been emerging as a negative regulator of HSL, and phosphorylation of AMPK at Thr172 is a critical event.41 Therefore, it could be very interesting to investigate whether BVT.2733 inhibits lipolysis of adipose tissue by regulating AMPK phosphorylation.
Transgenic mice with high expression of 11β-HSD1 in adipose tissue showed significantly increased adipose glucocorticoid levels and a 3-fold elevation of glucocorticoid transport to the liver via the portal system.15 When fed with a high-fat diet, 11β-HSD1 deficient mice exhibited improved insulin sensation and loss of visceral adipose,42,43 however, mice with liver 11β-HSD1 knockout did not prevent the gain of body weight, adipose tissue weight, and liver weight.44 Therefore, 11β-HSD1 in adipose tissue appeared to more important than in the liver on the development of NAFLD. Besides, deficiency or inhibition of 11β-HSD1 may lead to a negative influence on hepatic steatosis. Liver-specific loss of 11β-HSD1 did not protect from hepatic steatosis but enhanced hepatic inflammation in mice fed with fructose and trans-fats.45 In our study, inhibition of 11β-HSD1 by the low dose of BVT.2733 reduced body weight and liver weight in mice fed on a high-fat diet. This effect was more pronounced in the liver since the weights of visceral fat, except perirenal fat, subcutaneous fat, and brown fat were not significantly altered. Thus, adipose tissue 11β-HSD1 is the key factor leading to NAFLD formation and the development of hepatic steatosis. Inhibition of 11β-HSD1 activity in adipose tissue not only reduces visceral obesity and improves adipose tissue function, but also reduces glucocorticoids and FFAs levels in the hepatocytes, thereby effectively preventing hepatic steatosis. Our results were strongly supported by a recent study that showed that adipose-specific, but not liver-specific 11β-HSD1 knockout mice, prevented glucocorticoids-induced hepatic steatosis.40 This suggests that activation of 11β-HSD1 in adipose tissue promotes the development of hepatic steatosis.
11β-HSD1 inhibition in diabetic and obese mice models prevented weight gain.37 11β-HSD1 is expressed in white adipose tissue, where it functions as the predominant reductase amplifying glucocorticoids. Glucocorticoids drive the pathogenesis of metabolic syndrome in adipose tissue. Glucocorticoids have been reported to stimulate lipolysis via induction of hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL) desnutrin, increasing the release of adipocyte fatty acid and production of metabolic fuel.40,41,46 Consistent with this, our study showed that low dose BVT.2733 significantly attenuated the HFD-induced upregulation of lipolytic genes Lxrα and Lxrβ expression in adipose tissue. Lipolysis in adipose tissue is the major contributor to hepatic FFAs flux. A high level of glucocorticoids has been reported to promote adipogenesis and reduce lipid oxidation in the liver.47,48
Chronic endoplasmic reticulum (ER) stress induces numerous intracellular pathways that can lead to NAFLD development and progression, including hepatic steatosis, systemic inflammation, and hepatocyte cell death.49,50 A recent study showed that over-expression of 11β‑HSD1 promoted endoplasmic reticulum stress.51 Based on these findings, we speculate that inhibition of 11β‑HSD1 by BVT.2733 may directly affect the function of the endoplasmic reticulum. There are some limitations to this study. Recently, 11β-HSD1 has been reported to be involved in liver fibrosis. A recent study showed that in a mouse model of chemical liver injury, 11β-HSD1 deficiency promoted fibrosis by enhancing the activation of myofibroblasts.52 Therefore, the use of BVT.2733 may carry a risk of promoting hepatic fibrosis, which will cause irreversible damage to the liver. Another study showed that, in a chemical liver injury mice model, 11β-HSD1 deficiency promotes fibrosis by enhancing myofibroblast activation.52 The small number of animals is another limitation in our research. Although the measurements were statistically significant, a larger number of animals will convince our conclusion strongly.
In conclusion, our study shows that the high-fat diet may activate 11β-HSD1 and induce glucocorticoids and FFAs from adipose tissues to the liver, thus leads to the development of NAFLD. Meanwhile, the activation of 11β-HSD1 increases lipogenesis in the liver of mice fed with HFD (Figure 7A). Inhibition of 11β-HSD1 by the low dose of BVT.2733 protects against high-fat diet-induced hepatic steatosis by inhibiting lipolysis in fatty tissues and lipogenesis in the liver, and therefore attenuates delivery of glucocorticoids and FFAs to the liver where they are re-esterified to promote hepatic lipid accumulation (Figure 7B).
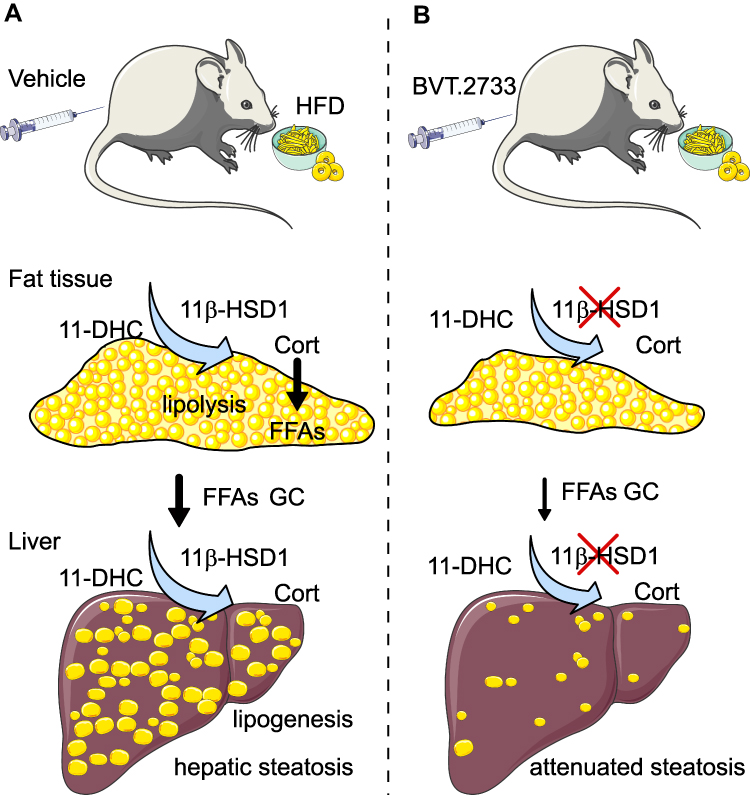 |
Figure 7 An illustration of the effect of 11β-HSD1 inhibition by the low dose of BVT.2733 on NAFLD. (A) The activation of 11β-HSD1 is elevated in both adipose tissue and liver in diet-induced obesity. This stimulates lipolysis in the adipose tissue, and increases glucocorticoid and free fatty acids transport to the liver through the portal vein. Meanwhile, the activation of 11β-HSD1 increases lipogenesis in the liver. (B) Inhibition of 11β-HSD1 by BVT.2733 reduces adipose tissue mass and portal glucocorticoid and free fatty acids levels, and reverses lipogenesis in the liver. Therefore, inhibition of 11β-HSD1 attenuates hepatic steatosis. Abbreviations: 11-DHC, 11-dehydrocorticosterone; Cort, corticosterone. |
Abbreviations
11β-HSD1, 11β-hydroxysteroid dehydrogenase type1; Acc1, Acetyl-coa carboxylase 1; Acc2, Acetyl-coa carboxylase 2; ALP, Alkaline phosphatase; ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; ATGL, Adipose triglyceride lipase; Atgl, Adipose triglyceride lipase; Dgat1, Acyl-coa: diacylglycerol acyltransferase 1; DNL, De novo lipogenesis; Elovl6, Elongation of very-long-chain fatty acids protein 6; ER, Endoplasmic reticulum; Fas, Fatty acid synthase; FFAs, Free fatty acids; GR, Glucocorticoid receptor; GTT, Glucose tolerance test; HDL, Lipoprotein; HFD, High-fat diet; HSL, Hormone-sensitive lipase; ITT, Insulin tolerance test; LDL, Low-density cholesterol; LFD, Low-fat diet; Lipe, Lipase E, hormone-sensitive type; Lxrα, Liver X receptor α; Lxrβ, Liver X receptor β; NAFLD, Nonalcoholic fatty liver disease; PBS, Phosphate-buffered saline; PFA, Paraformaldehyde; qRT-PCR, Quantitative Real-time PCR; Srebp1c, Sterol response element-binding protein-1c; TC, Total cholesterol; TG, Triglyceride.
Acknowledgment
Outstanding Youth Foundation of Jiangsu Province (BK20190043), The National Natural Science Foundation of China (31971062, 31900326, and 81200620), The Natural Science Foundation of Jiangsu Province (BK20180838), The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (20KJA180003, 19KJB320003), The Livelihood and Technology Program of Suzhou City (SYS2020100, SYS2019030), The Open Fund of State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University (KF-GN-202004), and The Research Innovation Program for College Graduates of Jiangsu Province (KYCX19-1981). This work is also supported by the International Joint Research Center for Genomic Resources (2017B01012) and the Tang Scholar of Soochow University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
There are no conflicts of interest.
References
1. Younossi ZM, Blissett D, Blissett R, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016;64(5):1577–1586. doi:10.1002/hep.28785
2. Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism. 2016;65(8):1017–1025. doi:10.1016/j.metabol.2016.01.012
3. Farrell GC, Wong VW, Chitturi S. NAFLD in Asia – as common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10(5):307–318. doi:10.1038/nrgastro.2013.34
4. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592–1609. doi:10.1053/j.gastro.2012.04.001
5. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):
6. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. doi:10.1038/s41591-018-0104-9
7. Monetti M, Levin MC, Watt MJ, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6(1):69–78. doi:10.1016/j.cmet.2007.05.005
8. Liao W, Hui TY, Young SG, Davis RA. Blocking microsomal triglyceride transfer protein interferes with apoB secretion without causing retention or stress in the ER. J Lipid Res. 2003;44(5):978–985. doi:10.1194/jlr.M300020-JLR200
9. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142(4):711–725. e716. doi:10.1053/j.gastro.2012.02.003
10. Bril F, Lomonaco R, Orsak B, et al. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology. 2014;59(6):2178–2187. doi:10.1002/hep.26988
11. Stojsavljevic S, Palcic MG, Jukic LV, Duvnjak LS, Duvnjak M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(48):18070–18091. doi:10.3748/wjg.v20.i48.18070
12. Odermatt A, Arnold P, Stauffer A, Frey BM, Frey FJ. The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem. 1999;274(40):28762–28770. doi:10.1074/jbc.274.40.28762
13. Rahimi L, Rajpal A, Ismail-Beigi F. Glucocorticoid-induced fatty liver disease. Diabetes Metab Syndr Obes. 2020;13:1133–1145. doi:10.2147/dmso.s247379
14. Paterson JM, Morton NM, Fievet C, et al. Metabolic syndrome without obesity: hepatic overexpression of 11 beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A. 2004;101(18):7088–7093. doi:10.1073/pnas.0305524101
15. Masuzaki H, Paterson J, Shinyama H, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294(5549):2166–2170. doi:10.1126/science.1066285
16. Candia R, Riquelme A, Baudrand R, et al. Overexpression of 11 beta-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue and portal hypercortisolism in non-alcoholic fatty liver disease. Liver Int. 2012;32(3):392–399. doi:10.1111/j.1478-3231.2011.02685.x
17. Wang L, Liu J, Zhang A, et al. BVT.2733, a selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, attenuates obesity and inflammation in diet-induced obese mice. PLoS One. 2012;7(7):e40056. doi:10.1371/journal.pone.0040056
18. Huang M, Liu J, Sheng Y, et al. 11β-hydroxysteroid dehydrogenase type 1 inhibitor attenuates high-fat diet induced cardiomyopathy. J Mol Cell Cardiol. 2018;125:106–116. doi:10.1016/j.yjmcc.2018.10.002
19. Berthiaume M, Laplante M, Festuccia WT, Berger JP, Thieringer R, Deshaies Y. Additive action of 11beta-HSD1 inhibition and PPAR-gamma agonism on hepatic steatosis and triglyceridemia in diet-induced obese rats. Int J Obes (Lond). 2009;33(5):601–604. doi:10.1038/ijo.2009.33
20. Koh EH, Kim AR, Kim H, et al. 11β-HSD1 reduces metabolic efficacy and adiponectin synthesis in hypertrophic adipocytes. J Endocrinol. 2015;225(3):147–158. doi:10.1530/joe-15-0117
21. Yuan X, Li H, Bai H, et al. The 11β-hydroxysteroid dehydrogenase type 1 inhibitor protects against the insulin resistance and hepatic steatosis in db/db mice. Eur J Pharmacol. 2016;788:140–151. doi:10.1016/j.ejphar.2016.05.034
22. Stefan N, Ramsauer M, Jordan P, et al. Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):406–416. doi:10.1016/s2213-8587(13)70170-0
23. Harno E, Cottrell EC, Yu A, et al. 11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors still improve metabolic phenotype in male 11β-HSD1 knockout mice suggesting off-target mechanisms. Endocrinology. 2013;154(12):4580–4593. doi:10.1210/en.2013-1613
24. Zhao P, Wong KI, Sun X, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172(4):731–743.e712. doi:10.1016/j.cell.2018.01.007
25. Barf T, Vallgårda J, Emond R, et al. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11β-hydroxysteroid dehydrogenase type 1. J Med Chem. 2002;45(18):3813–3815. doi:10.1021/jm025530f
26. Wang SJ, Birtles S, de Schoolmeester J, et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 reduces food intake and weight gain but maintains energy expenditure in diet-induced obese mice. Diabetologia. 2006;49(6):1333–1337. doi:10.1007/s00125-006-0239-y
27. Li G, Hernandez-Ono A, Crooke RM, Graham MJ, Ginsberg HN. Effects of antisense-mediated inhibition of 11β-hydroxysteroid dehydrogenase type 1 on hepatic lipid metabolism. J Lipid Res. 2011;52(5):971–981. doi:10.1194/jlr.M013748
28. Figarola JL, Singhal P, Rahbar S, Gugiu BG, Awasthi S, Singhal SS. COH-SR4 reduces body weight, improves glycemic control and prevents hepatic steatosis in high fat diet-induced obese mice. PLoS One. 2013;8(12):e83801. doi:10.1371/journal.pone.0083801
29. Gaidhu MP, Anthony NM, Patel P, Hawke TJ, Ceddia RB. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of ATGL, HSL, and AMPK. Am J Physiol Cell Physiol. 2010;298(4):C961–C971. doi:10.1152/ajpcell.00547.2009
30. Su Q, Kumar V, Sud N, Mahato RI. MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis. Adv Drug Deliv Rev. 2018;129:54–63. doi:10.1016/j.addr.2018.01.009
31. Jain A, Barve A, Zhao Z, et al. Targeted delivery of an siRNA/PNA hybrid nanocomplex reverses carbon tetrachloride-induced liver fibrosis. Adv Ther (Weinh). 2019;2(8):1900046. doi:10.1002/adtp.201900046
32. Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–131. doi:10.1053/j.gastro.2010.09.038
33. Bellentani S, Saccoccio G, Masutti F, et al. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. 2000;132(2):112–117. doi:10.7326/0003-4819-132-2-200001180-00004
34. Thamer C, Machann J, Haap M, et al. Intrahepatic lipids are predicted by visceral adipose tissue mass in healthy subjects. Diabetes Care. 2004;27(11):2726–2729. doi:10.2337/diacare.27.11.2726
35. Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell. 2018;9(2):164–177. doi:10.1007/s13238-017-0436-0
36. van der Poorten D, Milner KL, Hui J, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48(2):449–457. doi:10.1002/hep.22350
37. Alberts P, Engblom L, Edling N, et al. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia. 2002;45(11):1528–1532. doi:10.1007/s00125-002-0959-6
38. Veilleux A, Rhéaume C, Daris M, Tchernof A. Omental adipose tissue 11β-HSD1 oxoreductase activity, body fat distribution and metabolic alterations in women. J Clin Endocrinol Metab. 2009;94(9):3550–3557. doi:10.1210/jc.2008-2011
39. Bujalska IJ, Gathercole LL, Tomlinson JW, et al. A novel selective 11beta-hydroxysteroid dehydrogenase type 1 inhibitor prevents human adipogenesis. J Endocrinol. 2008;197(2):297–307. doi:10.1677/joe-08-0050
40. Morgan SA, McCabe EL, Gathercole LL, et al. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci U S A. 2014;111(24):E2482–2491. doi:10.1073/pnas.1323681111
41. Wang Y, Yan C, Liu L, et al. 11β-Hydroxysteroid dehydrogenase type 1 shRNA ameliorates glucocorticoid-induced insulin resistance and lipolysis in mouse abdominal adipose tissue. Am J Physiol Endocrinol Metab. 2015;308(1):E84–95. doi:10.1152/ajpendo.00205.2014
42. Morton NM, Paterson JM, Masuzaki H, et al. Novel adipose tissue–mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1–deficient mice. Diabetes. 2004;53(4):931–938. doi:10.2337/diabetes.53.4.931
43. Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2010;11(1):11–18. doi:10.1111/j.1467-789X.2009.00623.x
44. Lavery GG, Zielinska AE, Gathercole LL, et al. Lack of significant metabolic abnormalities in mice with liver-specific disruption of 11β-hydroxysteroid dehydrogenase type 1. Endocrinology. 2012;153(7):3236–3248. doi:10.1210/en.2012-1019
45. Larner DP, Morgan SA, Gathercole LL, et al. Male 11β-HSD1 knockout mice fed trans-fats and fructose are not protected from metabolic syndrome or nonalcoholic fatty liver disease. Endocrinology. 2016;157(9):3493–3504. doi:10.1210/en.2016-1357
46. Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem. 2004;279(45):47066–47075. doi:10.1074/jbc.M403855200
47. Macfarlane DP, Forbes S, Walker BR. Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol. 2008;197(2):189–204. doi:10.1677/JOE-08-0054
48. Campbell JE, Peckett AJ, D’Souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am J Physiol Cell Physiol. 2011;300(1):C198–C209. doi:10.1152/ajpcell.00045.2010
49. Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(7):1768–1776. doi:10.3748/wjg.v20.i7.1768
50. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–947. doi:10.1016/j.jhep.2018.06.008
51. Li C, Xia J, Zhu W, et al. Systemic overexpression of the 11β‑HSD1 promotes endoplasmic reticulum stress in multiple tissues and the development of metabolic syndrome in mice. Mol Med Rep. 2017;16(5):7738–7744. doi:10.3892/mmr.2017.7530
52. Zou X, Ramachandran P, Kendall TJ, et al. 11Beta-hydroxysteroid dehydrogenase-1 deficiency or inhibition enhances hepatic myofibroblast activation in murine liver fibrosis. Hepatology (Baltimore, Md). 2018;67(6):2167–2181. doi:10.1002/hep.29734
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.