Back to Journals » Cancer Management and Research » Volume 11
Secreted TGF-beta-induced protein promotes aggressive progression in bladder cancer cells
Authors Zou J ![]() , Huang R
, Huang R ![]() , Li H, Wang B, Chen Y, Chen S, Ou K, Wang X
, Li H, Wang B, Chen Y, Chen S, Ou K, Wang X
Received 16 March 2019
Accepted for publication 5 July 2019
Published 25 July 2019 Volume 2019:11 Pages 6995—7006
DOI https://doi.org/10.2147/CMAR.S208984
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Video abstract presented by Jun Zou.
Views: 197
Jun Zou,1,* Ruiyan Huang,2,* Huajun Li,1 Bin Wang,3 Yanfei Chen,3 Shuwei Chen,4 Kaifu Ou,4,† Xutao Wang4
1Department of Emergency Surgery, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 2Department of Ultrasonography and Electrocardiograms, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat‑sen University Cancer Center, Guangzhou, Guangdong, People’s Republic of China; 3Department of Urology, Affiliated Cancer Hospital & Institue of Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 4The Third Clinical College of Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
†Kaifu Ou passed away on March 8, 2019.
*These authors contributed equally to this work
Background: Transforming growth factor-beta-induced (TGFBI) is an exocrine protein, which has been found to be able to promote the development of nasopharyngeal carcinoma, glioma, pancreatic cancer, and other tumors. However, there is currently no report concerning the relationship between TGFBI and invasive progression of bladder cancer (BCa).
Methods: IHC staining, qRT-PCR and Western blot were used to analyze TGFBI and EMT markers levels. In vivo tumorigenesis was performed by xenograft tumor model.
Results: In this study, we found that both mRNA and protein levels of TGFBI were significantly up-regulated in muscle invasive bladder cancer (MIBC) tissues compared with non-muscle-invasive bladder cancer (NMIBC) tissues. The high expression level of TGFBI was positively correlated with high histological grade and advanced clinical stage, and BCa patients with high TGFBI levels exhibited poor prognoses. We further confirmed that high expression level of TGFBI can promote proliferation, invasive progression, and epithelial-to-mesenchymal transition (EMT) of BCa cells in vitro, as well as promote tumor growth and EMT in vivo, while silencing of TGFBI inhibited these malignant phenotypes. TGFBI was involved in the up-regulation of EMT by inducing the expression level of Slug, Vimentin, Snail, MMP2, and MMP9 genes, while it down-regulated the expression level of E-cadherin. Moreover, Western blot analysis was carried out to demonstrate that BCa cell lines stably transfected with expression of TGFBI, a secreted protein. Furthermore, conditional medium containing TGFBI protein also resulted in enhanced EMT and malignant phenotype of BCa cells.
Conclusion: Our results indicate that high expression level of TGFBI promotes EMT, proliferation, and invasive progression of BCa cells, and TGFBI is a potential therapeutic target and prognostic marker for BCa.
Keywords: TGFBI, invasive progression, EMT, secreted protein, tumor markers, bladder cancer
Introduction
According to the statistics, 16,900 patients died due to bladder cancer (BCa) in 2014 in the United States, and the rank of cancer-related mortality was eleventh in all tumors.1 There are two types of BCa: non-muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC). A study showed that the 5-year OS of NMIBC was 53.8% and the mean survival time was 54.5 months, while for MIBC, the 5-year OS was 19% and the mean survival time was 25.4 months.2 Although NMIBC has typically shown a good prognosis, however, up to 25% of the progress is invasive diseases.3 A previous research reported that within 75% of the patients with high-grade NMIBC may develop a recurrence at 10 years, while an additional 33% may progress to MIBC.4 Furthermore, among those progressed patients, 40% may die due to BCa.4 Hence, understanding of the possible mechanisms for BCa and identifying effective therapeutic targets are vital.
Epithelial–mesenchymal transition (EMT) is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells; these are multipotent stromal cells that can differentiate into a variety of cell types.5,6 Cancer-associated EMT is a complicated and comprehensive reprogramming, involving in metabolism, epigenetics, and differentiation, through differentiated epithelial cancer cells reversing to an undifferentiated state.7 Tumor cells undergoing EMT may invade the surrounding matrix, and their invasiveness, migration, and anti-apoptosis abilities can be notably enhanced.8 In the process of EMT, some factors, eg, E-cadherin, are responsible for tight junction, while the cell gains mesenchymal markers (eg, Vimentin), and transcription factors, such as Snail, Slug, MMP2, and MMP9 are upregulated during EMT as well.9,10 In recent years, several new oncogenic drivers, including LncDQ, P68, ACK1, PKD1, and LIM have shown playing a pivotal role in the induction of EMT during tumor progression.11–15
Transforming growth factor-beta-induced (TGFBI) protein is a downstream component of the TGF-β signaling pathway.16 Recent studies have indicated that TGFBI acts as an oncogene in pancreatic cancer,17 nasopharyngeal carcinoma,18 and glioma.19 Furthermore, a number of studies reported that it can promote EMT in gastric cancer16 and cholangiocarcinoma.20 However, the relationship between TGFBI and invasive progression of BCa needs to be elucidated. In the present study, we found that overexpression of TGFBI in BCa was important for the acquisition of a poor prognostic phenotype. TGFBI silencing also inhibited EMT, migration, and invasion in vitro, whereas overexpression of TGFBI in BCa could improve these influences and promote tumor growth and EMT in vivo. In addition, we also confirmed that TGFBI was secreted outside the cell to promote EMT and malignant phenotype of BCa cells. This study indicated the functional roles of TGFBI in the development and invasive progression of BCa.
Materials and methods
Ethics statement
All animal experiments were conducted according to the Principles of Laboratory Animal Care (National Society for Medical Research). These experiments were approved by the Internal Research Ethics Board at the Third Affiliated Hospital of Guangzhou Medical University.
Patients and tissue samples
A tissue microarray (TMA) containing 138 effective BCa tissues and 11 adjacent noncancerous bladder tissues was purchased from Alenabio (cat no. BL2081c; Xi’an, China). None of the patients had undergone chemotherapy or radiotherapy prior to surgery.
Immunohistochemistry analysis
Immunohistochemistry analysis of the TMA slice was performed with the DAKO EnVision system (Dako Diagnostics Inc., Zug, Switzerland) on Dako automated immunostaining instruments as previously described.21 The primary antibodies against TGFBI (Anti-TGFBI antibody; 10188-1-AP; Proteintech Group Inc., Rosemont, IL, USA) were used at a dilution of 1:400. Horseradish peroxidase (HRP)-labeled antibodies and alkaline‑phosphatase‑labeled antibodies [dilution, 1:400; UltraSensitive SP (Mouse/Rabbit) IHC kit, Fuzhou Maixin Biotech Co. Ltd., Fuzhou, China] were utilized to detect the specifically bound primary antibodies. Immunostaining was scored by two independent experienced pathologists who were blinded to the clinicopathological data and clinical outcomes of the patients. Those scores were then compared, and any discrepant scores were resolved through re‑examining the staining by the pathologists to achieve a consensus score. The number of stained cells in 10 representative microscopic fields was counted and the percentage of positive cells was calculated using a light microscope (Nikon Corp., Tokyo, Japan). Due to the homogeneity of the staining of the target proteins, tumor specimens were scored in a semi‑quantitative manner. The percentage of immunoreactive cells was separated into five groups as follows: 0, 0%; 1, 1–10%; 2, 11–50%; 3, 51–80%; and 4, >80%. The staining intensity was visually scored and stratified as follows: 0, negative; 1, weak; 2, moderate; and 3, strong. Final immunoreactivity scores were achieved for each case by multiplying the percentage and the intensity score.
Cell lines and cell culture
Human BCa cell lines 5637 and T24 were provided by the American Type Culture Collection (ATCC; Manassas, VA, USA). Fetal bovine serum (FBS) and RPMI 1640 were purchased from Invitrogen (Carlsbad, CA, USA). Besides, 5637 and T24 cells were cultured in RPMI, 1640 containing 10% FBS. The cells were maintained in a humidified atmosphere, containing 5% CO2 at 37°C.
Synthesis and transfection of small interfering RNA (siRNA)
SiRNAs against TGFBI gene sequences were designed: TGFBI #1, sense: 5ʹ-GAUAAGGUCAUCUCCACCATT-3ʹ, anti-sense: 5ʹ-UGGUGGAGAUGACCUUAUCTT-3ʹ; TGFBI #2, sense: 5ʹ-CUUGAAGUCAGCUAUGUGUTT-3ʹ, anti-sense: 5ʹ-ACACAUAGCUGACUUCAAGTT-3ʹ; TGFBI #NC, sense: 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ, anti-sense: 5ʹ-ACGUGACACGUUCGGAGAATT-3ʹ; siRNAs were transfected into 5637 and T24 cells with RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Lentivirus production and construction of stable cell lines
Human TGFBI expression plasmid (pCDH-TGFBI) was constructed with synthetic oligonucleotides and the pCDH vector. Moreover, HEK293T cells were simultaneously transfected with pCDH-TGFBI plasmid, psPAX2 plasmid, and pMD2.G to produce lentivirus. In addition, HEK293T cells were simultaneously transduced with pCDH plasmid, psPAX2 plasmid, and pMD2.G to produce empty lentivirus. For stable expression, 5637 and T24 BCa cells at 80% confluence were infected with empty or TGFBI –expressing lentiviruses using 8 μg/ml polybrene.
RNA extraction and quantitative reverse transcription polymerase chain reaction (Rt-qPCR)
Total RNA of the transfected cells was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RT-qPCR analysis of the concentration and purity of the total RNA was carried out by UV spectrophotometer at 260 nm before the electrophoresis detection. According to the manufacturer’s protocol, total RNA was analyzed by reverse transcription into cDNA by using SuperScript III® (Invitrogen, Carlsbad, CA, USA). The primer sequences used were as follows: forward primers: 5ʹ-AGGAATTTGCTTCGGAACCAC-3ʹ, reverse primers: 5ʹ-GCTGTTCTCAATGCAAAGGCTA-3ʹ; GAPDH was chosen as the internal control gene, and its forward primers: 5ʹ-CCCACTCCTCCACCTTTGAC-3ʹ, reverse primers: 5ʹ-TCTTCCTCTTGTGCTCTTGC-3ʹ. RT-qPCR was conducted using the ABI PRISM 7000 Fluorescent Quantitative PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The mean value in each triplicate was used to calculate the relative amount of TGFBI using 2ˆ-ΔΔCt method. Besides, each experiment was repeated at least three times.
Cell proliferation assay
Here, 1×107 5637 and T24 stably expressing TGFBI cells (5637/TGFBI and T24/TGFBI) or 5637 and T24 BCa cells transfected with the indicated siRNAs for 48 hrs were seeded into 24-well plates. The cells were counted at 24, 48, 72, 96, and 120 hrs. In addition, cell proliferation was also authenticated by EdU incorporation assay using an EdU Kit (C10310-1, RiboBio, China) according to the manufacturer’s instructions. In brief, cells were incubated with 100 μL of 50 μM EdU per well for 2 hrs at 37°C. Afterwards, the cells were fixed for 30 mins at room temperature using 100 μL of fixing buffer (4% paraformaldehyde containing phosphate-buffered saline (PBS)). Subsequently, the cells were incubated with 50 μL of 2 mg/mL glycine for 5 mins followed by washing with PBS (100 μL). After permeabilization with 0.5% TritonX, the cells were reacted with 1× Apollo solution for 30 mins at room temperature in the dark. Next, the cells were incubated with 1× Hoechst 33,342 solution (100 μL) for 30 mins at room temperature in the dark followed by washing with PBS (100 μL). The cells were then visualized under a fluorescence microscope. Experiments were repeated at least three times as well.
Western blot analysis
The cells and tissues were lysed in 1× sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) buffer. The lysates were quantified using BCA Assay Kit (Pierce Biotechnology, Waltham, MA, USA). The lysates were separated by SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membranes. After incubation with primary antibodies specific for TGFB I (1:1000; 10,188-1-AP; Proteintech Group Inc., Rosemont, IL, USA), E-cadherin (1:2000; #14,472; Cell Signaling Technology Danvers, MA, USA), Vimentin (1:2000; #5741; CST, Boston, USA), Slug (1:1000; #9585; CST, Boston, USA), Snail (1:1000; #3879; Cell Signaling Technology, Danvers, MA, USA), MMP9 (1:2000; #15561; Cell Signaling Technology Danvers, MA, USA), and β-actin (1:4000; sc-81178; Santa Cruz Biotechnology, Dallas, TX, USA) at 4°C overnight were indicated by secondary antibodies at room temperature for 1 hr. The blots were detected by an enhanced chemiluminescence (ECL) kit.
Colony formation, migration, and invasion assays
For colony formation assay, 500 5637/TGFBI and T24/TGFBI cells or 5637 and T24 BCa cells transfected with the indicated siRNAs for 48 hrs were seeded into 6-well plates for 12 or 7 days, respectively. For Transwell migration assays, 1×105 treated 5637 or T24 cells were seeded in RPMI 1640 with 0.1% FBS in the upper Transwell chamber (8-µm pore size; BD Biosciences, San Jose, CA, USA), and the lower chamber contained RPMI 1640 with 10% FBS for 36 or 24 hrs, respectively. For Transwell invasion assay, chamber was coated with Matrigel (BD Biosciences, San Jose, CA, USA). In addition, 2×105 treated 5637 or T24 cells were seeded in RPMI 1640 with 0.1% FBS in the upper Transwell chamber (8-µm pore size; BD Biosciences, San Jose, CA, USA), and the lower chamber contained RPMI 1640 with 10% FBS for 96 or 48 hrs, respectively. Clones or cells were fixed with methanol and stained with 0.5% crystal violet and were also counted by Image J software.
Detection of TGFBI protein in cell culture supernatant
After 2×105 5637/NC, 5637/TGFBI, T24/NC, and T24/TGFBI cells were seeded into 6-well plates for 12 hrs, cells were cultured in serum-free RPMI 1640 for 36 hrs. Then, 1 mL cultured serum-free medium was precipitated by 0.15% sodium deoxycholate (Sigma-Aldrich, St. Louis, MO, USA) and 7.2% trichloroacetic acid (Guangzhou Chemical Reagent Factory, Guangzhou, China). After washing with 1 mL acetone (Guangzhou Chemical Reagent Factory, Guangzhou, China) for 3 times, the precipitates were dissolved in 40 µL SDS-PAGE buffer and were then detected by Western blotting.
Animal breeding and treatments
Female BALB/c nude mice (5-week-old) were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). The mice were bred under specific-pathogen-free (SPF) conditions at Guangdong Medical Laboratory Animal Center. A total of 5×106 5637/LV-TGFBI cells (5637 cells stably overexpressing TGFBI) or 5637/LV-control were subcutaneously injected into the right flanks of each mouse (n=5). After three weeks, all mice were euthanized, and the tumors were stripped, photographed, and weighed.
Statistical analysis
Data were presented as mean±standard deviations (SD), which derived from at least three independent experiments and were statistically analyzed using Graph Pad Prism 5.0 software. The survival curve between the groups was described by the Kaplan–Meier analysis and was evaluated using the log-rank test. Two-tailed Student’s t-test (unpaired) was applied for comparing statistical differences between two groups. *P<0.05, **P<0.01, and ***P<0.001 were considered statistically significant.
Results
Upregulation of TGFBI is correlated with poor survival prognosis and invasive progress of BCa
Based on the GSE32584, GSE13507, and GSE 89 gene sets, the mRNA expression levels of TGFBI are upregulated in T2+ tumor grade (MIBC) compared with low-grade T1 and Ta tumors (NMIBC) (Figure 1A–C). To investigate the association of TGFBI level with invasive progress of BCa patients, clinical data collected from The Cancer Genome Atlas (TCGA) database were analyzed, in which the results showed that patients with TGFBI overexpression were at a higher risk of death than those with low expression level of TGFBI (Figure 1D). In the TCGA database, the level of TGFBI turned to be negatively correlated with both disease-free survival (DFS) and overall survival (OS) for BCa patients (Figure 1E and F). Besides, TGFBI expression increased as BCa tumor stage progressed and was at lower levels in normal bladder tissues. (Figure 1G). Additionally, the MIBC showed a higher TGFBI score than the NMIBC and normal tissues (Figure 1H). Then, Chi-square test indicated that TGFBI overexpression was closely associated with clinicopathological features of BCa. As a result, upregulation of TGFBI positively correlated with pathological tumor-staging status (pT status) (P<0.001), pathological lymph node-staging status (pN status) (P=0.007), and clinical stage (P<0.001) (Table 1). In addition, according to the analysis of clinical data obtained from TCGA database, upregulation of TGFBI was significantly associated with tumor progression (Table 2).
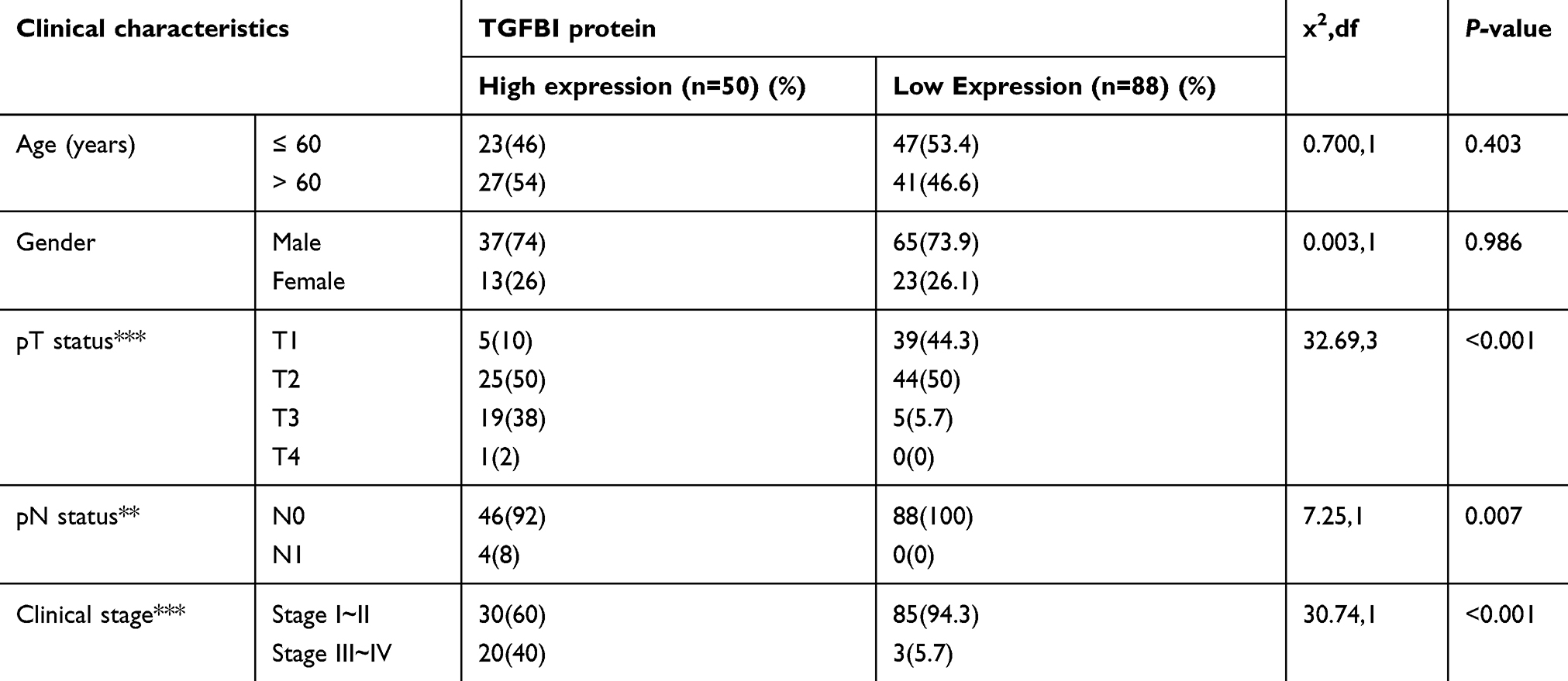 |
Table 1 Correlation between TGFBI expression and clinicopathological characteristics of BCa patients |
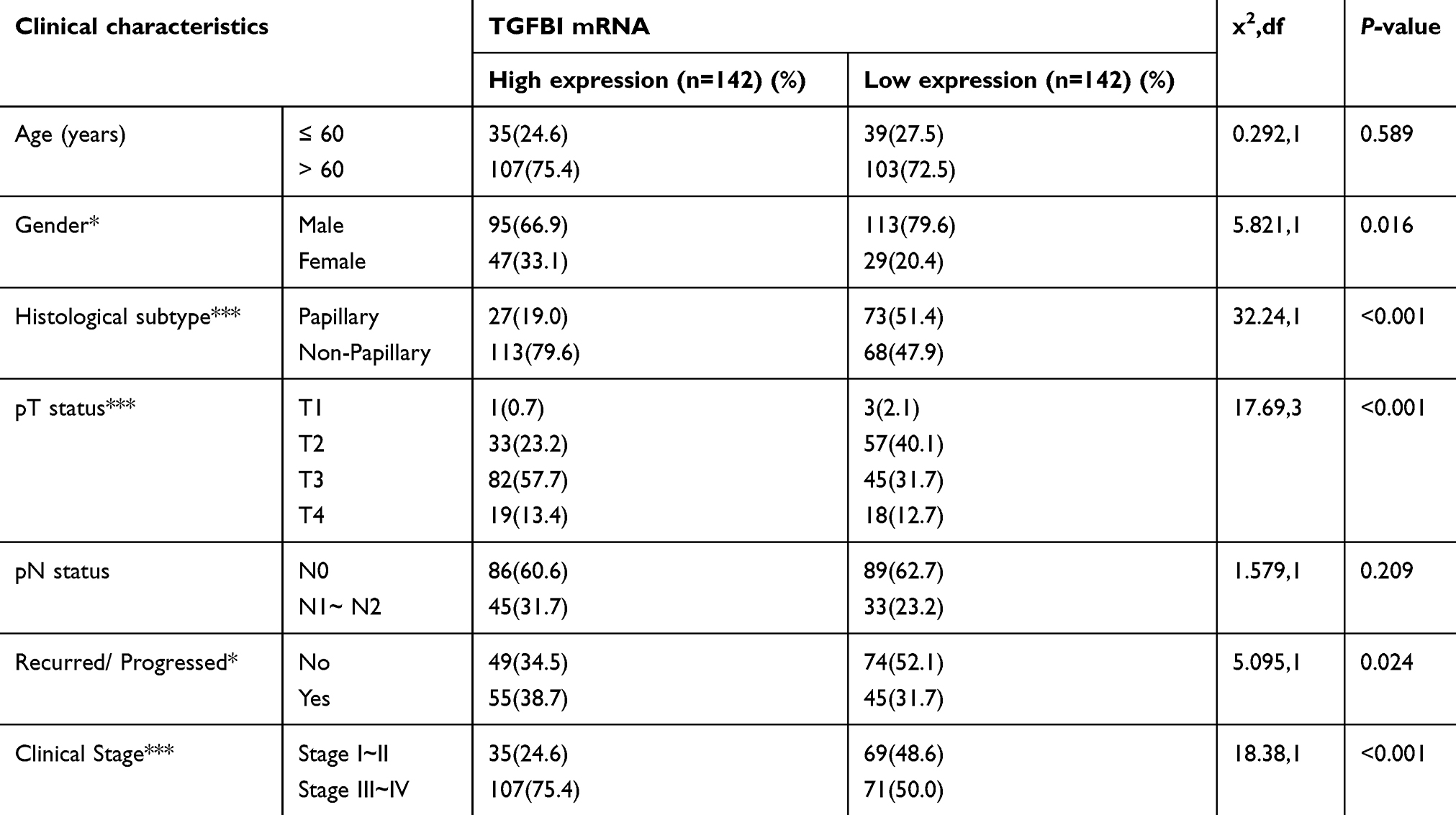 |
Table 2 Comparing clinical features between BCa patients with low and high TGFBI levels in TCGA database |
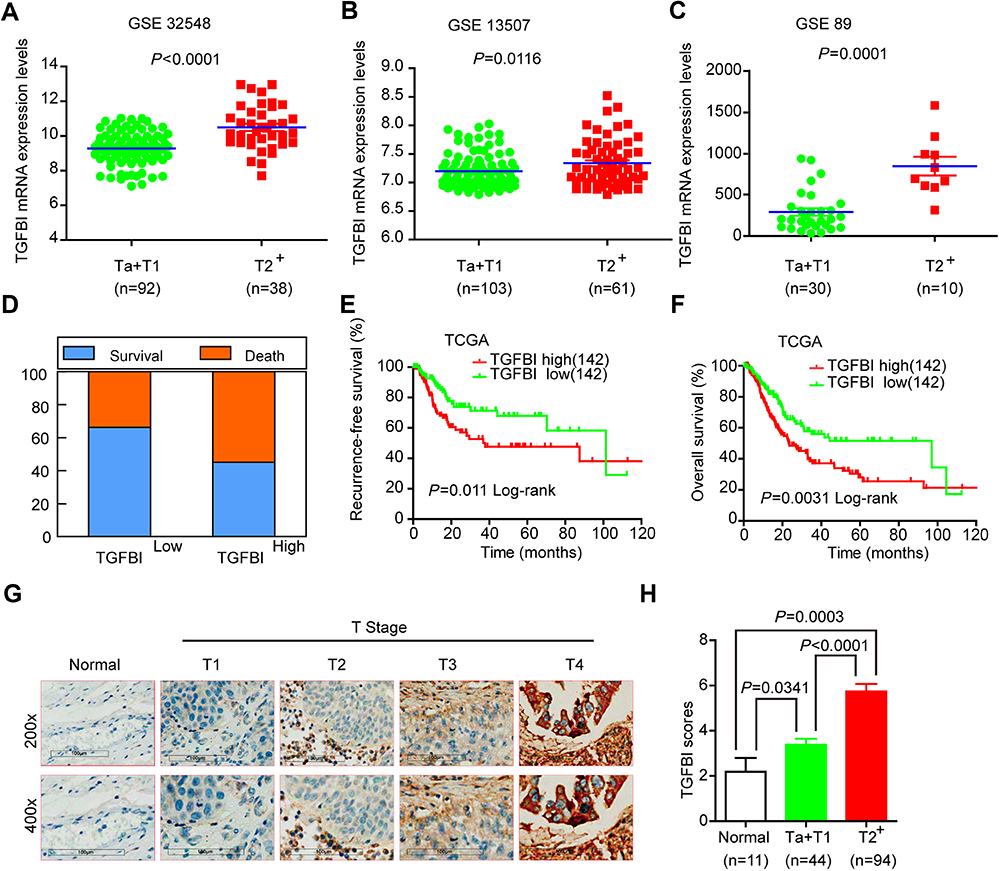 |
Figure 1 Upregulation of TGFBI was correlated with poor survival prognosis and invasive progresses of BCa. (A–C) TGFBI mRNA expression levels were increased in MIBC tissues (≥T2) compared with NMIBC tissues (Ta or T1) obtained from the three publicly accessible BCa datasets (GSE32548, GSE13507, and GSE89). (D–F) From TCGA urothelial cancer specimen cohorts, compared with the patients with low expression level of TGFBI (the lower 35%), the patients with high mRNA expression (the upper 35%) of TGFBI had higher death rates, shorter DFS, and OS. (G) Immunohistochemistry analysis of the expression of TGFBI protein in normal tissues and BCa tissues at different tumor stages. (H) Differences in expression levels of TGFBI protein in normal, NMIBC (Ta or T1), and MIBC tissues (≥T2). |
The silencing of TGFBI inhibits cell proliferation, colony formation, and tumorigenesis in vitro
To investigate the functional roles of TGFBI in BCa tumorigenesis, TGFBI expression was silenced in 5637 and T24 cells, which was confirmed in both mRNA and protein levels (Figure 2A and B). It can be seen that the proliferation of the two former cells was obviously decreased when TGFBI was knocked-down (Figure 2C). With EDU staining, an identical result was presented in cell proliferating ability (Figure 2D). In addition, silencing of TGFBI could restrain the colony formation for 5637 and T24 cell lines, respectively (Figure 2E). To further investigate the action of TGFBI in tumorigenesis in vitro, migration and invasion ability changes of 5637 and T24 cell lines were estimated when TGFBI was knocked-down. Results showed that they were largely hindered with decrease of TGFBI (Figure 2F). Notably, it was found that knockdown of TGFBI depressed the expression of Slug, Vimentin, Snail, MMP2, and MMP9 in 5637 and T24 cells, while augmented the expression of E-cadherin, a classic EMT marker which is inversely associated with tumor cell migration, invasion, and clinical prognosis22 (Figure 2G). Our findings also indicated that TGFBI showed as a key effector on BCa cell progression and its malignant phenotypes.
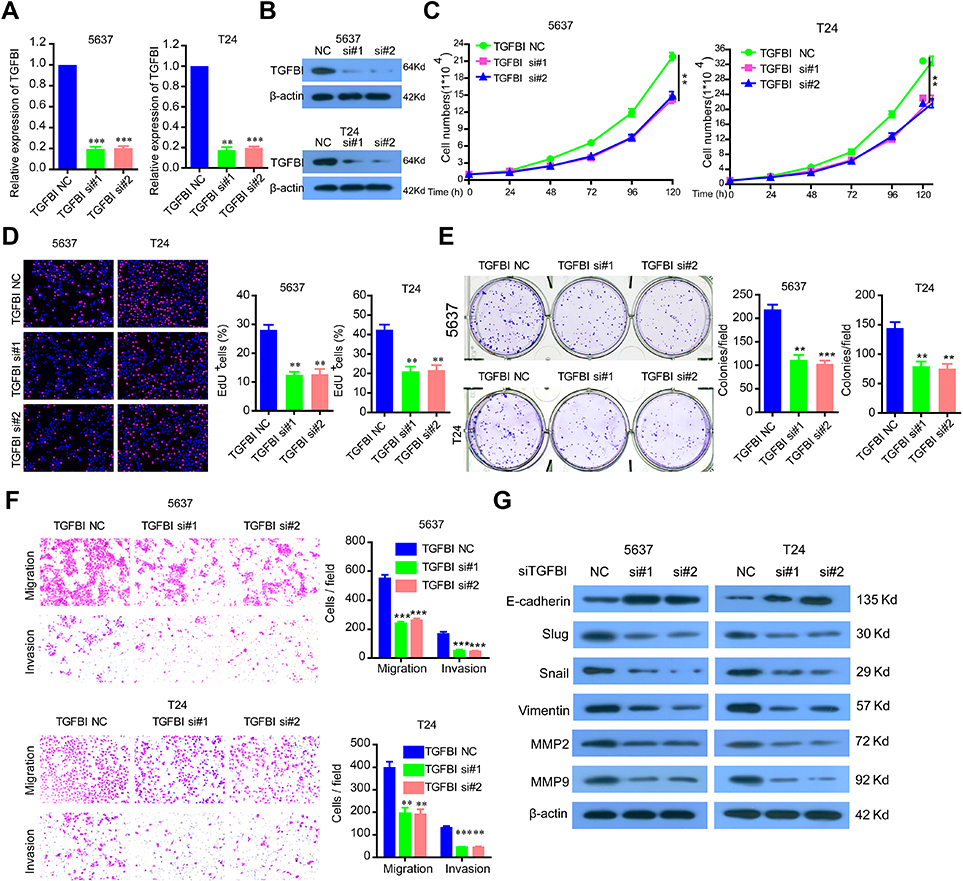 |
Figure 2 The silencing of TGFBI inhibits the malignant phenotypes of BCa cells. (A, B) TGFBI expression was silenced in 5637 and T24 cells, which were detected by RT-q-PCR and Western blotting. (C–F) The effects of TGFBI silencing on 5637 and T24 cell proliferation (C, D), colony formation (E), and migration and invasion (F) were detected. (G) After TGFBI silencing, EMT markers were detected by Western blotting. **P<0.01 and ***P<0.001 were considered statistically significant. |
Overexpression of TGFBI promotes the malignant phenotypes of tumor cells in vitro
For further assessment, TGFBI was stably overexpressed in 5637 and T24 cells (Figure 3A and B). As expected, overexpression of TGFBI stimulated cell growth (Figure 3C and D), colony formation (Figure 3E), migration and invasion (Figure 3F) in 5637 and T24 cells. Given insights into the molecular mechanism underlying the pro-tumorigenic role of TGFBI, overexpression of TGFBI and EMT correlative factors were similarly detected by Western blotting in 5637 and T24 cells. In contrast to the TGFBI silencing results, we found that overexpression of TGFBI promotes EMT of BCa cells (Figure 3G). Taken together, we deduced that TGFBI had a pivotal role, facilitating the advancement of BCa cells to malignant phenotypes, that resulted in the poor prognosis in BCa patients.
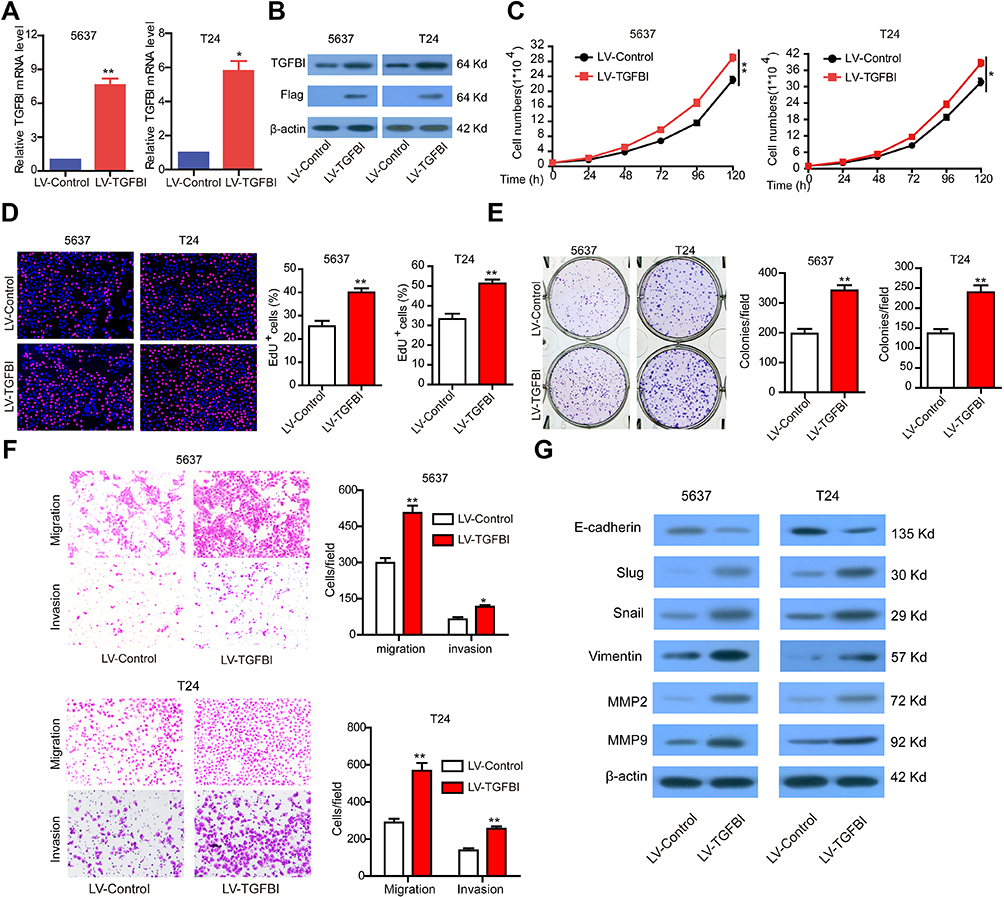 |
Figure 3 The stable overexpression of TGFBI promotes the malignant phenotypes of BCa cells. (A, B) The stable overexpression of TGFBI was detected by RT-q-PCR and Western blotting in 5637 and T24 cells. (C–F) The effects of stable overexpression of TGFBI on cell proliferation (C, D), colony formation (E), and migration and invasion (F) were detected. (H) After stable overexpression of TGFBI, EMT markers were detected by Western blotting in 5637 and T24 cells. *P<0.05 and **P<0.01 were considered statistically significant. |
TGFBI in the supernatant promotes the malignant phenotypes of tumor cells in vitro
TGFBI, as a secreted protein,23 was efficiently secreted to cell culture medium (Figure 4A). To evaluate the potential of TGFBI as a new prognostic factor and therapeutic target for BCa, we examined the effects of TGFBI in the supernatant on BCa cells. Then, supernatants of two BCa cell lines stably expressing TGFBI were collected from the conditional media and used to treat their own non-stable expression cell lines. Accordingly, identical results with the intracellular TGFBI overexpression approach were obtained. The conditional medium derived from 5637/LV-TGFBI and T24/LV-TGFBI cells significantly facilitated cell proliferation (Figure 4B), migration and invasion (Figure 4C), respectively, compared with the conditional medium derived from 5637/LV-control and T24/LV-control cells. The levels of EMT markers (eg, Slug, Vimentin, Snail, MMP2, and MMP9) were all promoted except for the E-cadherin, which was restrained partly (Figure 4D). Taken together, TGFBI may become a promising prognostic marker and target.
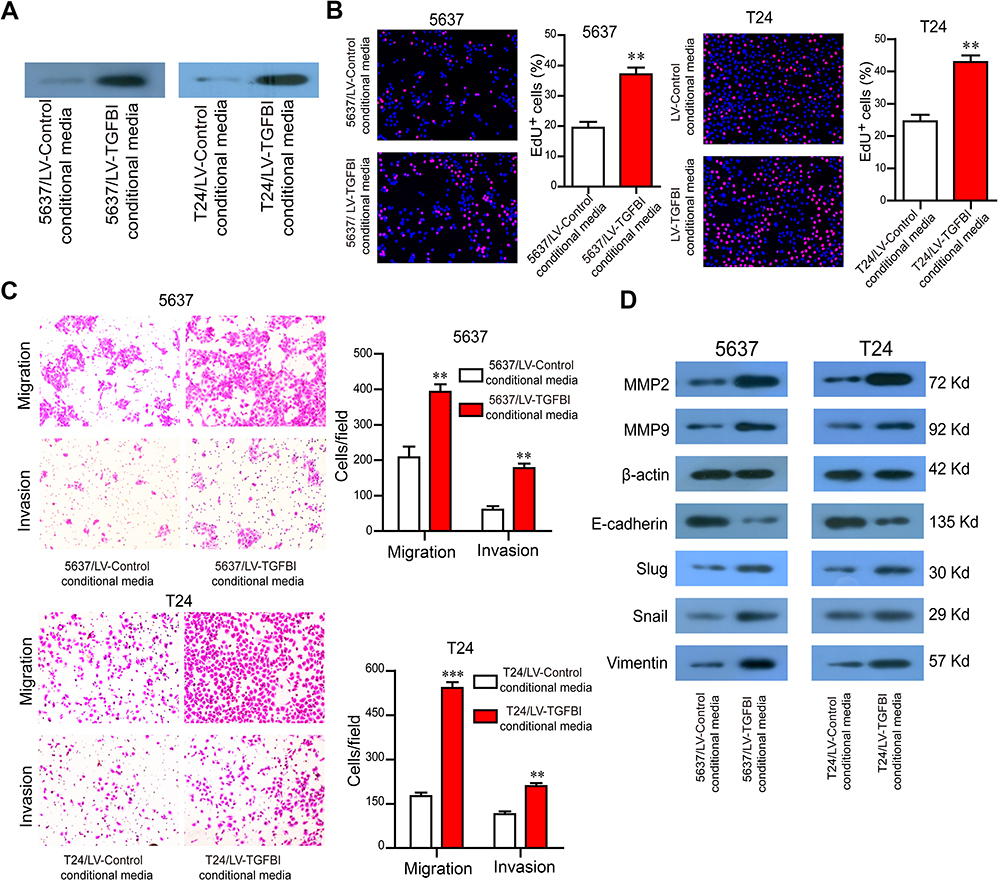 |
Figure 4 TGFBI in the supernatant promotes the malignant phenotypes of BCa cells. (A) TGFBI was efficiently secreted to cell culture medium. (B–D) The conditional medium derived from 5637/LV-TGFBI and T24/LV-TGFBI cells was treated to 5637 and T24 cells, respectively. The effects of TGFBI in the supernatant on cell proliferation (B) migration and invasion (C), and EMT markers (D) were detected. **P<0.01 and ***P<0.001 were considered statistically significant. |
Increase of tumor growth by upregulated TGFBI in vivo
Our results showed that TGFBI was a potential target for the treatment of BCa and prognostic markers. To further assess the effects of high-level TGFBI on the malignant phenotypes of tumor cells in vivo, 5637 cell lines with or without TGFBI overexpression were transplanted into nude mice. As a result, upregulation of TGFBI significantly increased the growth of tumor cells in size and weight (Figure 5A and B). In addition, expression levels of tumor tissues were detected by immunohistochemistry using antibodies against the following six markers, including TGFBI, E-cadherin, Vimentin, MMP2, MMP9, and Ki-67. The outcomes showed that the levels of all checked proteins in 5637/TGFBI tumors were increased except for the E-cadherin compared with 5637/control tumors (Figure 5C). Western blot analysis showed that the expression of Slug, Vimentin, Snail, MMP2, and MMP9 was increased in 5637/TGFBI tumors, while the expression of E-cadherin was decreased (Figure 5D). These results suggested that TGFBI reliably aggravated tumor progression in vivo, due to expediting the growth of tumor cells and alteration of the level of EMT-relevant proteins. Hence, these evidences indicated that TGFBI plays a key role in the tumorigenesis and progression of Bca. However, from another point of view, of the abovementioned results related to TGFBI may show it as a positive target, facilitating the clinical prognosis and treatment of BCa.
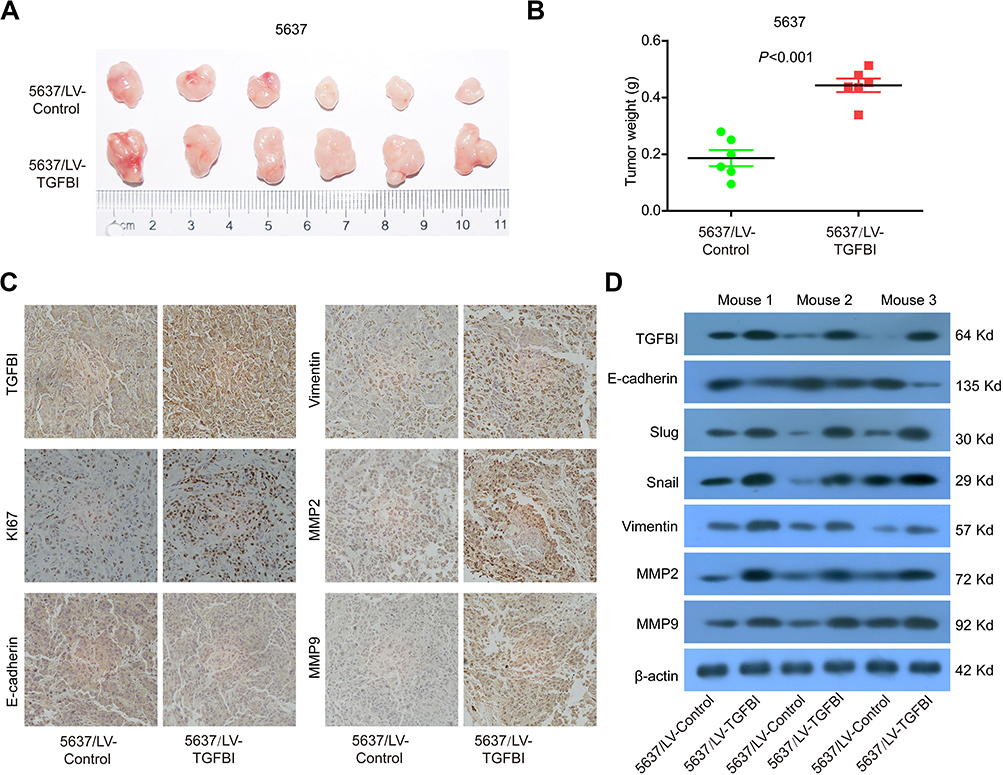 |
Figure 5 TGFBI promoted the tumorigenicity of BCa cells in vivo. (A) Representative images of tumors in5637/LV-control and 5637/LV-TGFBI groups in nude mice. (B) Weights of the xenograft tumors are presented (n=6). (C) Representative images of TGFBI, KI67, E-cadherin, Vimentin, MMP2, and MMP9 in tumors formed by the indicated cells. (D) Tumors were lysed, and the total levels of TGFBI, EMT markers, and GAPDH were detected by Western blotting. |
Discussion
At present, it is extremely necessary to identify novel and efficient biomarkers that can discriminate MIBC and NMIBC, so that patients with low risk of BCa may highly benefit from avoiding disease progression.24 Previous studies have shown that TGF signaling pathway plays a key role in cell proliferation, apoptosis, differentiation, cytokine secretion, extracellular matrix modification, tumor metastasis, and other cellular biological activities.25,26 TGFBI, a downstream gene of TGF-beta pathway, may be relevant to the EMT process.20 TGFBI protein consists of 683 amino acids, containing an amino-terminal secretory sequence and a C-terminal Arg-Gly-Asp (RGD) sequence that can serve as a ligand recognition site for several integrins.27 TGFBI also contained short amino acid regions being homologous to similar regions in Drosophila fasciclin-I and four homologous internal domains, which can be folded into a potential bivalent structure and could act as a bridge between cells expressing appropriate ligand.27 A study showed that TGFBI acts as a tumor suppressor,28 while several scholars believe that it is a tumor promoter.29–31
In this study, we found, for the first time, that the expression of TGFBI at gene and protein levels was significantly higher in MIBC patients than that in NMIBC patients. We analyzed TCGA data and found that TGFBI mRNA was negatively correlated with OS and DFS in patients with BCa. Besides, statistical analysis showed that TGFBI mRNA was significantly associated with pT status, pN status, and clinical stage. Clinical data also suggest that TGFBI mRNA promotes the progression of BCa from NMIBC to MIBC, leading to worse prognosis. The results of immunohistochemistry also confirmed that the expression level of TGFBI in BCa was higher than that in benign bladder tissue and that was significantly associated with pathological grade and TNM stage. Besides, the protein expression level of TGFBI in NMIBC (≤T1) was significantly lower than that in MIBC (≥T2), which was consistent with the mRNA expression level of TGFBI. In vitro cell experiments also confirmed that overexpression of TGFBI promotes the malignant phenotype (proliferation, invasion, migration, and cloning) of BCa, and the expression of Slug, Vimentin, Snail, MMP2, and MMP9 increased, while the expression of E-cadherin decreased. After inhibiting the expression level of this gene, an opposite result was observed, demonstrating that TGFBI promotes the malignant phenotype and EMC of BCa cells. In vivo tumorigenesis experiments in nude mice confirmed this finding as well. These results demonstrate that TGFBI is a potential therapeutic target and prognostic marker.
Previous studies have shown that TGFBI is an exocrine protein.32,33 Our immunohistochemical results also confirmed that TGFBI protein was stained in BCa cells and intercellular substances. In addition, it was revealed that exocrine TGFBI also promoted proliferation, invasion, metastasis, and EMT of tumor cells. To our knowledge, the urinary bladder is a hollow muscular organ in humans and some other animals that collects and stores urine from the kidneys before disposal by urination. Hence, detection of exfoliated cells and tumor markers of BCa is of great significance for clinical diagnosis of BCa,33 while infusion of chemotherapeutic drugs into the bladder is an important approach to treat bladder without muscle invasion.33 Our results suggested that it may help diagnose the disease severity of BCa patients by detecting the expression level of TGFBI in his/her urine. Simultaneously, TGFBI may also be a promising indicator for the efficacy of intravesical chemotherapy. Additionally, TGF-β acts as a tumor suppressor in premalignant tumor development and as a tumor promoter in advanced tumors, specifically during invasion and metastasis.34,35 In previous studies, TGFBI has been taken as a downstream effector protein of TGF-β into account;35 therefore, searching for upstream regulatory genes can be a hot topic research. Our results suggest that TGFBI may act as an upstream element to regulate the downstream genes and promote the occurrence, proliferation, metastasis, and EMT of BCa.
Ethics approval and consent to participate
These procedures were approved by the Research Ethics Committee of The Third Affiliated Hospital of Guangzhou Medical University (Guangzhou, China).
Consent for publication
Not applicable.
Data sharing statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
TGFBI, transforming growth factor-beta-induced; BCa, bladder cancer; MIBC, muscle-invasive bladder cancer; NMIBC, non-muscle-invasive bladder cancer; EMT, epithelial-to-mesenchymal transition; pT status, pathological T-staging status; pN status, pathological N-staging status.
Acknowledgments
The present study was financially supported by the Natural Science Foundation of Guangdong Province (grant no. 2018A0303130327), Guangzhou Health Science and Technology General Guidance Project (Health Science and Technology [2017] 6), and The Third Affiliated Hospital of Guangzhou Medical University (grant no. 2018Q10).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Mokdad AH, Dwyer-Lindgren L, Fitzmaurice C, et al. Trends and patterns of disparities in cancer mortality among US counties, 1980-2014. JAMA. 2017;317(4):388–406. doi:10.1001/jama.2016.20324
2. Supit W, Mochtar CA, Sugiono M, Umbas R. Survival of patients with transitional cell carcinoma of the urinary bladder in indonesia: a single institution review. Asian Pac J Cancer P. 2011;12(2):549–553.
3. van Rhijn BWG, Burger M, Lotan Y, et al. Recurrence and progression of disease in non-muscle-invasive bladder cancer: from epidemiology to treatment strategy. Eur Urol. 2009;56(3):430–442. doi:10.1016/j.eururo.2009.06.028
4. Chamie K, Litwin MS, Bassett JC, et al. Recurrence of high-risk bladder cancer: A population-based analysis. Cancer-Am Cancer Soc. 2013;119(17):3219–3227.
5. Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 Promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154(1):61–74. doi:10.1016/j.cell.2013.06.005
6. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. doi:10.1038/nm.3336
7. Li LN, Li WL. Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Therapeut. 2015;150:33–46. doi:10.1016/j.pharmthera.2015.01.004
8. Iwatsuki M, Mimori K, Yokobori T, et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101(2):293–299. doi:10.1111/j.1349-7006.2009.01419.x
9. Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Resp Res. 2018;19(1):136–146. doi:10.1186/s12931-018-0834-8.
10. Mrkvicova A, Chmelarova M, Peterova E, et al. The effect of sodium butyrate and cisplatin on expression of EMT markers. PLoS One. 2019;14(1):e0210889. doi:10.1371/journal.pone.0210889
11. Zeng B, Lin ZW, Ye HL, et al. Upregulation of LncDQ is associated with poor prognosis and promotes tumor progression via epigenetic regulation of the EMT pathway in HCC. Cell Physiol Biochem. 2018;46(3):1122–1133. doi:10.1159/000488841
12. Li MY, Liu JQ, Chen DP, et al. p68 prompts the epithelial-mesenchymal transition in cervical cancer cells by transcriptionally activating the TGF-β1 signaling pathway. Oncol Lett. 2018;15(2):2111–2116. doi:10.3892/ol.2017.7552
13. Xu SH, Huang JZ, Xu ML, et al. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. 2015;236(2):175–185. doi:10.1002/path.4515
14. Zheng H, Shen M, Zha YL, et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26(3):358–373. doi:10.1016/j.ccr.2014.07.022
15. Hui W, Jiaolong S, Yuhao L, et al. LIM and SH3 protein 1 induces TGFβ-mediated epithelial-mesenchymal transition in human colorectal cancer by regulating S100A4 expression. Clin Cancer Res. 2014;20(22):5835–5847. doi:10.1158/1078-0432.CCR-14-0485
16. Yokobori T, Nishiyama M. TGF-Î2 signaling in gastrointestinal cancers: progress in basic and clinical research. J Clin Med Res. 2017;6(1):11. doi:10.3390/jcm6010011
17. Sato T, Muramatsu T, Tanabe M, Inazawa J. Identification and characterization of transforming growth factor beta-induced in circulating tumor cell subline from pancreatic cancer cell line. Cancer Sci. 2018;109(11):3623–3633. doi:10.1111/cas.13783
18. Bissey PA, Law JH, Bruce JP, et al. Dysregulation of the MiR-449b target TGFBI alters the TGFβ pathway to induce cisplatin resistance in nasopharyngeal carcinoma. Oncogenesis. 2018;7(5):40. doi:10.1038/s41389-018-0050-x
19. Guo SK, Shen MF, Yao HW, Liu YS. Enhanced expression of TGFBI promotes the proliferation and migration of glioma cells. Cell Physiol Biochem. 2018;49(3):1138–1150. doi:10.1159/000493293
20. Yoo HJ, Yun BR, Kwon JH, et al. Genetic and expression alterations in association with the sarcomatous change of cholangiocarcinoma cells. Exp Mol Med. 2009;41(2):102–115. doi:10.3858/emm.2009.41.2.013
21. Zou J, Huang RY, Jiang FN, et al. Overexpression of TPX2 is associated with progression and prognosis of prostate cancer. Oncol Lett. 2018;16(3):2823–2832. doi:10.3892/ol.2018.9016
22. Bronsert P, Enderle-Ammour K, Bader M, et al. Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer-host interface. J Pathol. 2014;234(3):410–422. doi:10.1002/path.4416
23. Skonier J, Bennett K, Rothwell V, et al. beta ig-h3: a transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol. 1994;13(6):571–584. doi:10.1089/dna.1994.13.571
24. Zhang ZL, Yu CP, Li YH, Jiang LJ, Zhou FJ. Utility of SAM68 in the progression and prognosis for bladder cancer. BMC Cancer. 2015;15:364
25. Joan M. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi:10.1038/nrm3434
26. Heldina CH, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012;586(14):1959–1970. doi:10.1016/j.febslet.2012.02.037
27. Skonier J, Neubauer M, Madisen L, Bennett K, GD P, AF P. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992;11(7):511–522. doi:10.1089/dna.1992.11.511
28. Zhang Y, Wen GG, Wang C, et al. TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res. 2009;69(1):37–44. doi:10.1158/0008-5472.CAN-08-1648
29. Han B, Cai H, Chen Y, et al. The role of TGFBI (βig-H3) in gastrointestinal tract tumorigenesis. Mol Cancer. 2015;14(1):64. doi:10.1186/s12943-014-0278-9
30. Andrei T, Davide M, Yinghong W, et al. Identification of novel accessible proteins bearing diagnostic and therapeutic potential in human pancreatic ductal adenocarcinoma. J Proteome Res. 2011;10(9):4302–4313. doi:10.1021/pr200527z
31. Buckhaults P, Rago C, St Croix B, et al. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 2001;61(19):6996–7001.
32. Choi SI, Kim BY, Dadakhujaev S, et al. Impaired autophagy and delayed autophagic clearance of transforming growth factor β-induced protein (TGFBI) in granular corneal dystrophy type 2. Autophagy. 2012;8(12):1782–1797. doi:10.4161/auto.22067
33. Dominik A, Mirjam B, Daniel Stephan E, Hans-Peter S. Therapeutic options for intractable hematuria in advanced bladder cancer. Int J Urol. 2013;20(7):651–660. doi:10.1111/iju.12113
34. Isabel F, Joan F, Jessica M, Patricia S. TGF-beta signaling in cancer treatment. Curr Pharm Des. 2014;20(17):2934–2947. doi:10.2174/13816128113199990591
35. Ikushima H, Miyazono K. TGF-β signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-β. Cell Tissue Res. 2012;347(1):37–49. doi:10.1007/s00441-011-1179-5
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.