")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Schisandra chinensis regulates drug metabolizing enzymes and drug transporters via activation of Nrf2-mediated signaling pathway
Authors He J, Zhou Z, Yin J, He C, Zhou S, Yu Y
Received 28 May 2014
Accepted for publication 17 July 2014
Published 17 December 2014 Volume 2015:9 Pages 127—146
DOI https://doi.org/10.2147/DDDT.S68501
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jin-Lian He,1 Zhi-Wei Zhou,2,3 Juan-Juan Yin,2 Chang-Qiang He,1 Shu-Feng Zhou,2,3 Yang Yu1
1College of Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, Guizhou, People’s Republic of China
Abstract: Drug metabolizing enzymes (DMEs) and drug transporters are regulated via epigenetic, transcriptional, posttranscriptional, and translational and posttranslational modifications. Phase I and II DMEs and drug transporters play an important role in the disposition and detoxification of a large number of endogenous and exogenous compounds. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a critical regulator of a variety of important cytoprotective genes that are involved in disposition and detoxification of xenobiotics. Schisandra chinensis (SC) is a commonly used traditional Chinese herbal medicine that has been primarily used to protect the liver because of its potent antioxidative and anti-inflammatory activities. SC can modulate some DMEs and drug transporters, but the underlying mechanisms are unclear. In this study, we aimed to explore the role of Nrf2 in the regulatory effect of SC extract (SCE) on selected DMEs and drug transporters in human hepatocellular liver carcinoma cell line (HepG2) cells. The results showed that SCE, schisandrin A, and schisandrin B significantly increased the expression of NAD(P)H: Nicotinamide Adenine Dinucleotide Phosphate-oxidase or:quinone oxidoreductase 1, heme oxygenase-1, glutamate–cysteine ligase, and glutathione S-transferase A4 at both transcriptional and posttranscriptional levels. Incubation of HepG2 cells with SCE resulted in a significant increase in the intracellular level of glutathione and total glutathione S-transferase content. SCE significantly elevated the messenger ribonucleic acid and protein levels of P-glycoprotein and multidrug resistance-associated protein 2 and 4, whereas the expression of organic anion transporting peptide 1A2 and 1B1 was significantly downregulated by SCE. Knockdown of Nrf2 by small interfering ribonucleic acid attenuated the regulatory effect of SCE on these DMEs and drug transporters. SCE significantly upregulated Nrf2 and promoted the translocation of Nrf2 from cytoplasm to the nuclei. Additionally, SCE significantly suppressed the expression of cytosolic Kelch-like ECH-associated protein 1 (the repressor of Nrf2) and remarkably increased Nrf2 stability in HepG2 cells. Taken together, our findings suggest that the hepatoprotective effects of SCE may be partially ascribed to the modulation of DMEs and drug transporters via Nrf2-mediated signaling pathway. SCE may alter the pharmacokinetics of other coadministered drugs that are substrates of these DMEs and transporters and thus cause unfavorable herb–drug interactions.
Keywords: Nrf2, Keap1, HepG2 cell, drug metabolizing enzyme, drug transporter, P-gp, MRP, OATP, Schisandra chinensis
Introduction
There is an increasing popularity of natural products including Chinese herbal medicines that are used for the management of various ailments and improvement of body health, although the clinical evidence for their application is poor or lacking.1 An estimated 25% of adults in developed countries and more than 80% of the population in developing countries consume herbal medicines for disease treatment. The 2002 National Health Interview Survey of the US (n=5,456) indicated that 18.9% of adults used herbal supplements within the past 12 months and 57.3% (n=3,315) of these people used herbs to treat a specific health condition.2 Based on the 2002 National Health Interview Survey, an estimated ~38.2 million adults in the US took herbal products and supplements in 2002.3 In a study including 61,587 individuals aged 50–76 years in the US, one-third of the population stated that they used herbal supplements.4 However, there are limited data on the pharmacokinetics, mechanisms of action, and safety profiles for most Chinese herbal medicines.5,6 There are minimal regulatory requirements for safety and efficacy of herbal medicines in Western countries. Many herbal medicines and phytochemicals are traditionally used as organ protectants or chemopreventive agents, since they can induce antioxidant response and the expression of drug metabolizing enzymes (DMEs) and drug transporters that are responsible for the disposition and excretion of a large number of xenobiotics.7 On the other hand, many Chinese herbal medicines have been reported to act as inducers or inhibitors of important DMEs and drug transporters, and this has raised a safety concern due to potential harmful herb–drug interactions.8–15 However, the underlying mechanisms for herbal medicine-induced alterations in the expression of DMEs and drug transporters are not fully understood. A number of DMEs and drug transporters are tightly regulated via epigenetic, transcriptional, posttranscriptional, translational, and posttranslational modifications. To sense and respond to xenobiotic exposure, DMEs and drug transporters are regulated by a panel of ligand-activated transcriptional factors, namely nuclear receptors, including pregnane X receptor (PXR/NR1I2), constitutive androstane/activated receptor (CAR/NR1I3), farnesoid X receptor (FXR/NR1H4), liver X receptor-α (LXRα/NR1H3), and hepatocyte nuclear factor 4α (HNF4α/NR2A1).16–19 These nuclear receptors serve as xenobiotic sensors to upregulate important DMEs and drug transporters that participate in the metabolism and excretion of xenobiotics.
The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a ubiquitous transcriptional factor encoded by the NFE2L2 gene in humans and plays a critical role in the maintenance of cellular homeostasis.20–22 Nrf2-mediated antioxidant response pathway is the primary cellular defense mechanism against the cytotoxic effects of oxidative stress caused by xenobiotic exposure and other factors and carcinogen-induced carcinogenesis.20,23 Under physiological conditions, Nrf2 is localized in the cytosol and associated with its negative regulator, the Kelch-like ECH-associated protein 1 (Keap1, also known as the Nrf2 repressor).20 The Nrf2 protein, upon synthesis, is rapidly degraded by the 26S proteasome exclusively in the cytoplasm of cells, with a half-life of 15-20 minutes.24,25 Keap1 is crucial for the rapid turnover of Nrf2 and functions as an adaptor for Nrf2 ubiquitination at the lysine residues of the Neh2 domain by a Cullin-3-dependent ubiquitin–ligase complex.26,27 Upon exposure to xenobiotics, Nrf2 dissociates from Keap1 and translocates into the nucleus where it heterodimerizes with other leucine zipper proteins such as Maf or other partners and binds to a cis-acting deoxyribonucleic acid (DNA) regulatory element, namely the antioxidant response element (ARE) in the upstream promoter region, and initiates the transcription of a wide array of cytoprotective target genes that participate in xenobiotic disposition and clearance from the body and maintain essential cellular defensive functions against myriad toxic products, including metalloids, chemical carcinogens, and radiation.23,28 Nrf2 deficiency or inactivation of Nrf2-mediated pathway can lead to pathological changes such as abnormal cellular survival and apoptosis and prolonged inflammation recovery. Nrf2 knockout mice have showed significantly increased susceptibility to cancer, neurodegeneration, premature senescence, lung injury and fibrosis, and inflammation.29–37 Nrf2 also plays a key role in the regulation of many DMEs and drug transporters that contribute to the disposition, detoxification, and clearance of a large number of xenobiotics. Most DMEs and drug transporters have a critical role in the detoxification of xenobiotics and endogenous compounds. For example, NAD(P)H:quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), uridine 5′-diphospho-glucuronosyltransferase–glucuronosyltransferases, glutamate–cysteine ligase, modifier subunit (GCLM), and glutathione (GSH) S-transferases (GSTs) are important contributors to xenobiotic disposition and detoxification. Drug transporters such as P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRPs/ABCCs), and organic anion transporting polypeptides (OATPs) are critical for drug distribution, disposition, and excretion.38 As such, pharmacological activation of Nrf2 has been widely advocated as a useful strategy for cancer prevention and treatment of several diseases. Induction or inhibition of DMEs and drug transporters may cause considerable alteration in the profiles of absorption, distribution, metabolism, and excretion of drugs.39 Therefore, it is important to identify the modulatory factors and signaling pathways involved in the regulation of DMEs and drug transporters.
Schisandra chinensis (SC, Wuweizi in Chinese) is a traditional Chinese herbal medicine that has been used for thousands of years. It has a wide range of pharmacological activities, including antioxidant, hepatoprotective, antibacterial, anticancer, cardioprotective, and anti-inflammatory effects.40 SC is widely and mainly used to protect the liver in clinical settings in Asian countries, including the People’s Republic of China, Japan, and Korea.40 SC extract (SCE) contains a mixture of various bioactive compounds, and the lignanoids are the main bioactive components, including schisandrin (Sch); pseudo Sch; SchA (ie, deoxyschisandrin), SchB, and SchC; γ-Sch; schisandrols A and B; Wuweizi ethers A and B; and schisanhenol.41,42 Among them, SchB is the most abundant dibenzocyclooctadiene lignanoid present in SCE. Previous preclinical studies have found that SCE and its major active lignanoids protected the liver in various liver-injured models, with different mechanisms involved in its hepatoprotective activities.43–59 These include antioxidation, inhibition of cytokine production, inhibition of apoptosis, activation of Nrf2, and other possible mechanisms.51–59 However, the mechanism is not fully understood, and the role of Nrf2 in the hepatoprotection of SCE is not elucidated. In this study, we aimed to examine the role of Nrf2 in the modulating effects of SCE on important DMEs and drug transporters in human hepatocellular liver carcinoma (HepG2) cells.
Materials and methods
Chemicals and reagents
SC was purchased from Daxiang Pharmaceuticals Inc. (Guangzhou, Guangdong, People’s Republic of China). SchA (purity =98.0%, relative molecular mass =416.51 Dal) and SchB (purity =98.0%, Mr =400.46 Dal) were bought from the National Institute for Food and Drug Control, Beijing, People’s Republic of China. tert-Butylhydroquinone (tBHQ, purity =97%, used as a known Nrf2 activator60,61), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), cycloheximide (CHX, a known protein synthesis inhibitor), and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich Inc. (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and 0.25% trypsin were purchased from Gibco Inc. (Grand Island, NY, USA). TRIzol reagent and real-time polymerase chain reaction (RT-PCR) kit were purchased from TaKaRa Inc. (Dalian, Liaoning, People’s Republic of China). Radio immunoprecipitation assay buffer (Beyotime Bio Inc., Shanghai, People’s Republic of China) and super signal-enhanced chemiluminescence detection system were purchased from Amersham Biosciences Inc. (Piscataway, NJ, USA). The bicinchoninic acid kit for protein concentration measurement was bought from Beyotime Bio Inc. (Shanghai, People’s Republic of China). Primary antibodies against human HO-1 and GCLM were purchased from Epitomics Inc. (Burlingame, CA, USA), and primary antibodies against Nrf2, MRP1, MRP2, OATP1A2, GADPH and Keap1 were bought from Abcam Inc. (Cambridge, MA, USA). ARE–luciferase plasmids and pEZX–PG04 control plasmids were purchased from GeneCopoeia Inc. (Rockville, MD, USA).
Cell culture
The HepG2 cell line was obtained from American Tissue Collection Center (ATCC, Manassas, VA, USA). Cells were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere. Medium was refreshed every 2–3 days.
SCE preparation
To prepare SCE, the raw materials were dissolved in 95% ethanol at 70°C with fresh solvent changed after each 180 minutes three times, followed by macroporous resin (AB-8) purification using 75% ethanol as the eluting solvent. To quantify the contents of SchA and SchB in SCE, a quota of SCE (20 μL) was subject to high-performance liquid chromatography (HPLC, LC-2010C) (Simadzu, Kyoto, Japan) equipped with an SPD-100MAVP detector and a Discovery® C-18 HPLC column (4.6×25 mm, 5 μm, Penomenex Nucleosil, Torrance, CA, USA). The column was eluted using H2O:methanol (v/v, 30:70) at a flow rate of 1.0 mL/minute for 35 minutes. The absorbance at 254 nm was monitored. To examine the total content of lignanoids of SCE, a quota of SCE (60 μL) was examined at 570 nm on an ultra-violet (UV)-6100S scanning spectrophotometer (Shanghai Precision instrument Co. Ltd, Shanghai, People’s Republic of China).
MTT assay
The effect of SCE on the viability of HepG2 cells was evaluated using the MTT assay. Cells were seeded into a 96-well plate (4×103 cells/well) and treated with various concentrations of SCE, SchA, SchB, and tBHQ for 24 hours. Following the treatment, the cells were incubated with 10 μL (5 mg/mL) MTT for 4 hours at 37°C. The medium was removed and the formazan crystals produced by viable cells were dissolved in 150 μL DMSO. The absorbance was detected using a microplate reader (THERMO Multiskan FC, MIT, USA) at wavelength of 570/630 nm.
NQO1–ARE luciferase reporter assay
HepG2 cells in a 96-well plate were transfected with a mixture of 0.5 μg of ARE–luciferase plasmids, 0.05 μg of pEZX–PG04 control plasmids (both from GeneCopoeia Inc.), and Lipofectamine 2000 reagent (Invitrogen). After 6 hours, the transfection mixture was replaced with fresh complete DMEM. Cells were treated with various concentrations of SCE for 24 hours. The Renilla and Firefly luciferase activities were measured using the Dual Luciferase Assay System (GeneCopoeia Inc.) with a microplate reader (THERMO Multiskan FC).
Total ribonucleic acid isolation and RT-PCR analysis
Total ribonucleic acid (RNA) was extracted from HepG2 cells using RNAiso Plus reagent according to the manufacturer’s instruction. The concentration of total RNA was examined by spectroscope at 260/280 nm. Following the RNA extraction, 1 μg of total RNA was used to synthesize complementary deoxyribonucleic acid (cDNA). The cDNA was reverse transcripted using the Super Script III First-Strand Synthesis System (TaKaRa, Tokyo, Japan). The resultant cDNA was subject to RT-PCR analysis using Power SYBR Green PCR Master Mix (TaKaRa) in ABI7500 Real-Time PCR system (Applied Biosystems Inc., Grand Island, NY, USA). The sequences of primers sequences are listed in Table 1.

|
Table 1 Sequences of primers designed for real-time polymerase chain reaction analysis |
Western blotting assay
Cells were lysed with the RIPA buffer containing the protease inhibitor cocktail. The protein concentration was measured by bicinchoninic acid assay, and protein samples were mixed with 4× Laemmli loading buffer containing dithiothreitol. The mixture was denatured at 95°C for 5 minutes. An equal amount of protein (20 μg) was separated by a criterion 8% Tris–HCl gel (Bio-Rad, Hercules, CA, USA), and subsequently transferred onto a polyvinylidene difluoride membrane using a Trans-Blot semidry transfer cell (Bio-Rad). The membrane was blocked with 5% skim milk for 2 hours at room temperature and then probed with specific primary antibodies at 4°C overnight. Following that, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 hours. Subsequently, the blots were visualized using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech., Piscataway, NJ, USA).
RNA interference
RNA interference experiments were performed using a specific pool of oligonucleotides against human Nrf2 (On-Targetplus SMART pool human NFE2L2; RIBOBIO Inc., Guangzhou, People’s Republic of China). According to the manufacturer’s instructions, HepG2 cells were transfected with 25 nM Nrf2 small interfering RNA (siRNA) (siNrf2) using Lipofectamine 2000 (Invitrogen) for 24 hours. The negative control was performed by transfection with a nontargeting silencing pool (On-Targetplus siGAPDH; RIBOBIO Inc.) at the same concentration as siNrf2. The silencing efficiency was evaluated after 24 hours by analyzing the gene expression level of target of interest.
Measurement of intracellular GSH level and total GST content
HepG2 cells were seeded in a six-well plate at a density of 5.0×105 cells/well. After 24 hours, cells were treated with serum-free medium containing different concentrations of drugs for 24 hours. Then, cells were washed with phosphate-buffered saline twice and harvested using the lysis buffer. Cell lysate (20 μL) was used for protein concentration measurement and 6.5% (wt/vol) sulfosalicylic acid was added to samples at a ratio of 1:4 (v/v) for protein precipitation. Samples were incubated on ice for 10 minutes and then centrifuged at 1,000× g for 10 minutes. The GSH level was immediately detected using a microplate reader at 405 nm, and the total GST content was measured at 412 nm.
Statistical analysis
Data are presented as the mean ± standard deviation from at least three independent experiments. Statistical analyses were performed by SPSS 16.0 software (SPSS Inc., Chicago, IL, USA) using one-way analysis of variance. P<0.05 is considered as statistical significance.
Results
Determination of major components of SCE
As is shown in Figure 1, both SchA and SchB were eluted by the mobile phase in the HPLC system. The retention time for SchA and SchB was 19.17 and 26.34 minutes, respectively.
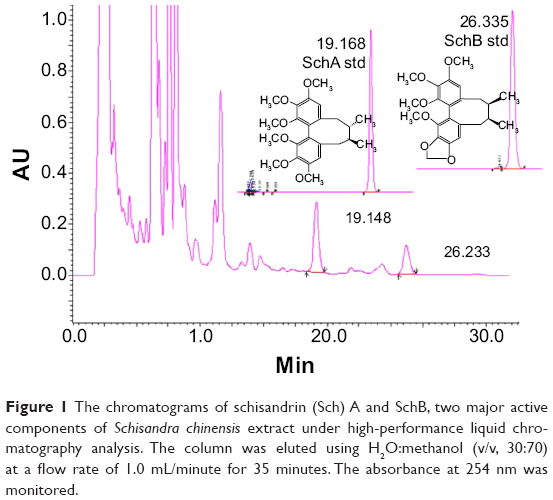
|
Figure 1 The chromatograms of schisandrin (Sch) A and SchB, two major active components of Schisandra chinensis extract under high-performance liquid chromatography analysis. The column was eluted using H2O:methanol (v/v, 30:70) at a flow rate of 1.0 mL/minute for 35 minutes. The absorbance at 254 nm was monitored. |
The linearity for SchA and SchB determination by HPLC was found in the range of 0.012-0.240 mg/mL (r=0.9999) and 0.0125–0.2500 mg/mL (r=0.9999), respectively. The content of SchA in SCE was 1.577 mg/g crude extracts and 0.763 mg/g crude extracts for SchB. A UV spectrophotometer was used to determine the total lignanoids of SCE with SchB used as the reference compound. The linearity was in the range of 4.84–24.20 mg/L (r=0.9948) and the total lignanoid content in SCE prepared was 39.77 mg/mL.
Effect of SCE, SchA, and SchB on HepG2 cell viability
First, in order to evaluate the cytotoxic effect of SCE and its major active lignanoids on HepG2 cells, the cell viability was examined using the MTT assay. Cells were incubated with SCE, SchA, SchB, and tBHQ for 24 hours, which resulted in a decrease in cell viability in a concentration-dependent manner (Figure 2). The cell viability was 96.1%±10.3%, 75.8%±10.3%, 63.4%±2.4%, 53.1%±3.9%, 28.4%±14.4%, and 15.25%±7.0% when HepG2 cells were treated with SCE at 15, 30, 60, 120, 240, and 480 μg/mL for 24 hours, respectively (Figure 2A). The cell viability was 93.3%±17.4%, 83.1%±19.7%, 72.3%±2.5%, 62.0%±2.5%, 40.3%±17.1%, and 28.3%±9.7% with the incubation of SchA at 3.125, 6.25, 12.5, 25, 50, and 100 μM for 24 hours, respectively (Figure 2B). After cells were treated with SchB at 12.5, 25, 50, 100, 200, and 400 μM, the cell viability was 98.4%±27.2%, 82.7%±6.2%, 83.5%±27.7%, 72.7%±34.1%, 45.8%±22.9%, and 30.2%±10.8%, respectively. When the cells were treated with tBHQ at 3.125, 6.25, 12.5, 25, 50, and 100 μM, the cell viability was 97.0%±11.8%, 87.3%±36.0%, 74.9%±11.0%, 62.1%±24.6%, 45.3%±11.1%, and 16.5%±10.0%, respectively (Figure 2B). The half maximal inhibitory concentration values for SCE, SchA, SchB, and tBHQ were 306.1 μg/mL, 70.6 μM, 266.4 μM, and 50.7 μM, respectively. These compounds only showed a low cytotoxicity toward HepG2 cells.
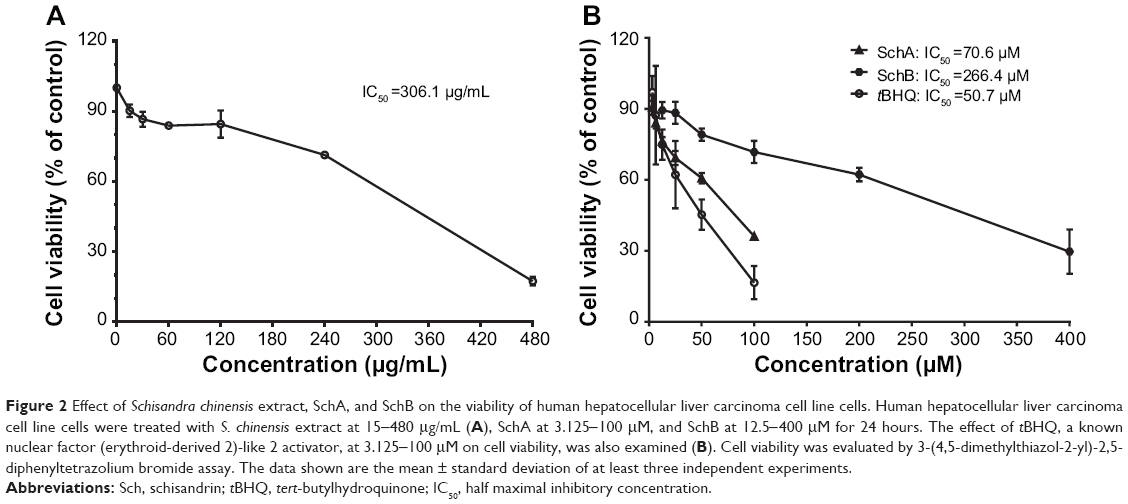
|
Figure 2 Effect of Schisandra chinensis extract, SchA, and SchB on the viability of human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with S. chinensis extract at 15-480 μg/mL (A), SchA at 3.125-100 μM, and SchB at 12.5-400 μM for 24 hours. The effect of tBHQ, a known nuclear factor (erythroid-derived 2)-like 2 activator, at 3.125-100 μM on cell viability, was also examined (B). Cell viability was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The data shown are the mean ± standard deviation of at least three independent experiments. |
SCE, SchA, and SchB induce the expression of selected DMEs in HepG2 cells
SCE has been reported to be a potent antioxidant that is primarily used to treat liver diseases.40 Herein, the effect of SCE on the expression of several important DMEs was examined at transcriptional and posttranscriptional levels. HepG2 cells were incubated with SCE at 100, 150, 200, 250, and 300 μg/mL for 24 hours; the gene expression level of NQO1 was increased 1.9-, 2.6-, 1.6-, 2.5-, and 3.2-fold, respectively (P<0.05) (Figure 3A); and the gene expression level of GCLM was elevated 1.4-, 4.2-, 1.4-, 2.2-, and 3.5-fold, respectively (P<0.05) (Figure 3B). Treatment of HepG2 cells with 250 and 300 μg/mL SCE resulted in a 2.1- and 3.9-, 1.5- and 1.3-fold increase (P<0.01) in the gene expression level of HO-1 (Figure 3C) and GSTA4 (Figure 3D), respectively. Moreover, there was a significant increase in the gene expression of NQO1, GCLM, and HO-1 when cells were incubated with SchA at 20 μM for 24 hours. Treating cells with 200 μM SchB also significantly induced the gene expression of NQO1 and GSTA4. On the other hand, the protein expression level of HO-1 and GCLM was also examined in order to evaluate the effect of SCE on the posttranscriptional regulation of DMEs. Incubation of HepG2 cells with SCE at 100, 150, 200, 250, and 300 μg/mL for 24 hours concentration dependently increased the expression levels of HO-1 and GCLM (Figure 4). SchA at 20 μM and SchB at 200 μM also significantly increased the expression levels of HO-1 and GCLM in HepG2 cells (Figure 4).
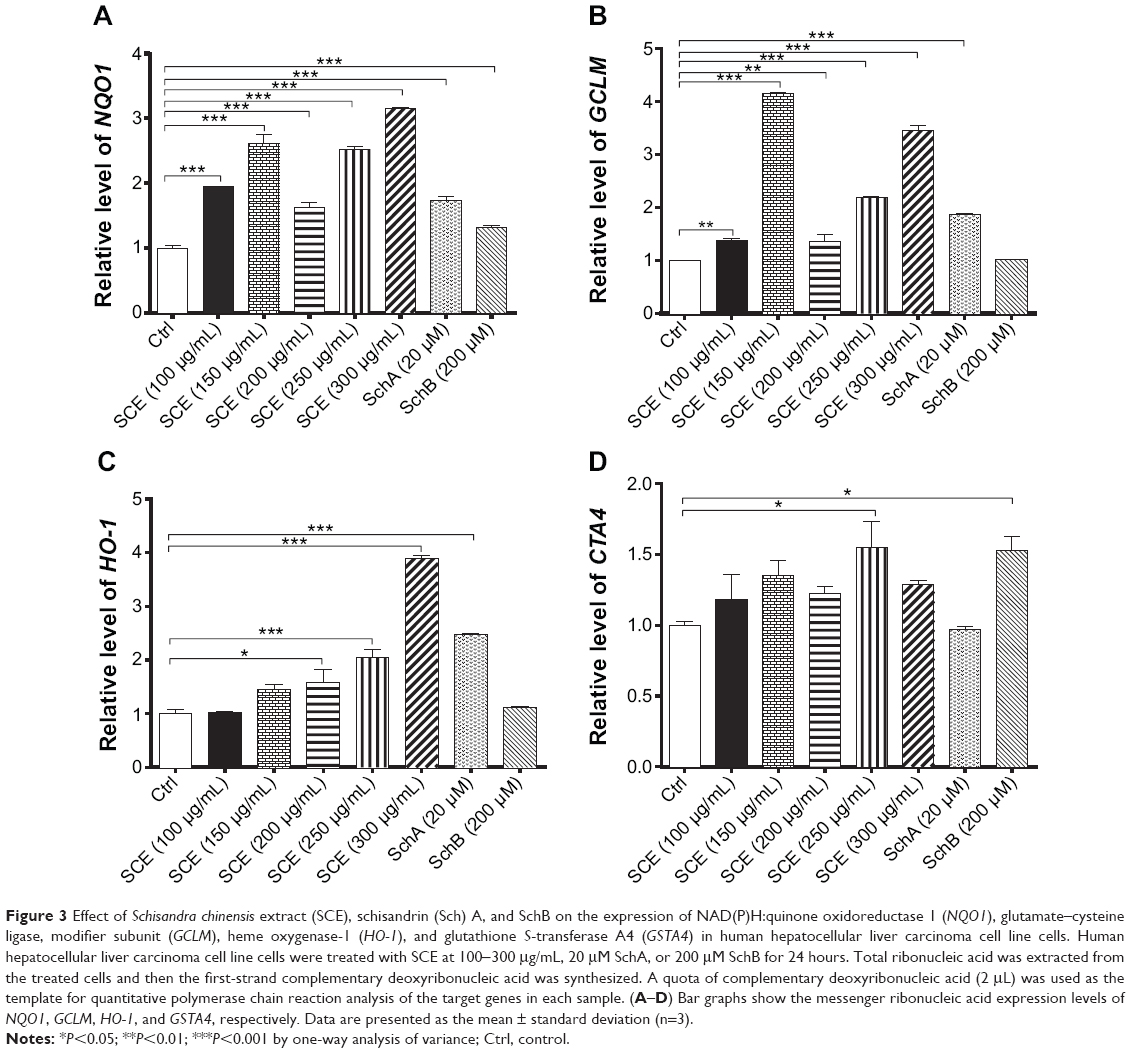
|
Figure 3 Effect of Schisandra chinensis extract (SCE), schisandrin (Sch) A, and SchB on the expression of NAD(P)H:quinone oxidoreductase 1 (NQO1), glutamate–cysteine ligase, modifier subunit (GCLM), heme oxygenase-1 (HO-1), and glutathione S-transferase A4 (GSTA4) in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. Total ribonucleic acid was extracted from the treated cells and then the first-strand complementary deoxyribonucleic acid was synthesized. A quota of complementary deoxyribonucleic acid (2 μL) was used as the template for quantitative polymerase chain reaction analysis of the target genes in each sample. (A–D) Bar graphs show the messenger ribonucleic acid expression levels of NQO1, GCLM, HO-1, and GSTA4, respectively. Data are presented as the mean ± standard deviation (n=3). |
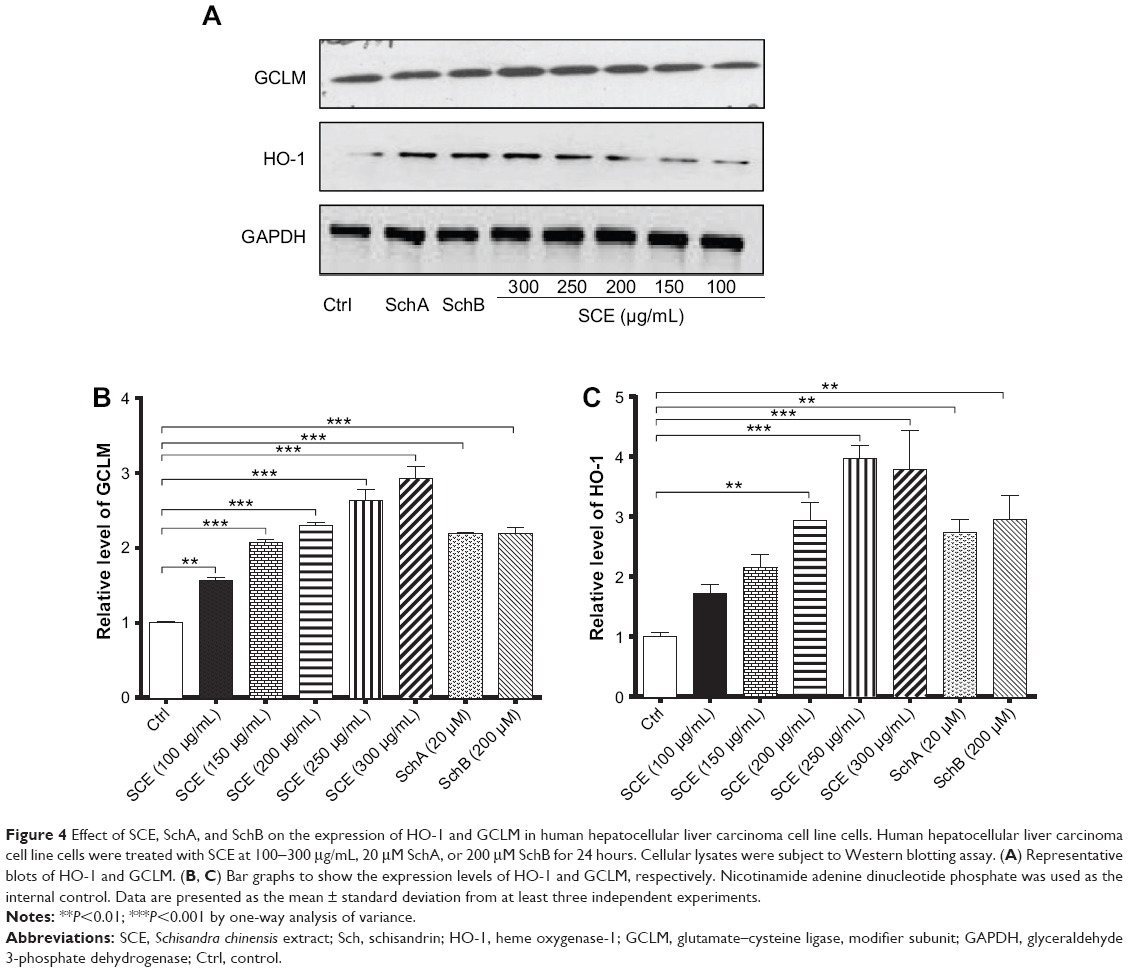
|
Figure 4 Effect of SCE, SchA, and SchB on the expression of HO-1 and GCLM in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. Cellular lysates were subject to Western blotting assay. (A) Representative blots of HO-1 and GCLM. (B, C) Bar graphs to show the expression levels of HO-1 and GCLM, respectively. Nicotinamide adenine dinucleotide phosphate was used as the internal control. Data are presented as the mean ± standard deviation from at least three independent experiments. |
SCE and SchB differentially modulate the expression levels of selected drug transporters in HepG2 cells
Drug transporters play an important role in the excretion of xenobiotics and thus protect important organs from their toxicities.38 On the other hand, the regulatory effect of xenobiotics, including drugs and herbal medicines, on drug transporters may have substantial impact on the pharmacokinetic and pharmacodynamic profiles of a coadministered drug. As such, the effect of SCE on selected important drug transporters was evaluated at both transcriptional and posttranscriptional levels. Incubation of HepG2 cells with SCE significantly increased the gene expression level of P-gp, MRP2, and MRP4 (Figure 5). There was a 2.0- and 1.5-fold increase in the gene expression level of P-gp in HepG2 cells treated with 200 or 250 μg/mL of SCE, respectively (Figure 5C). Treatment of HepG2 cells with SCE at 100, 150, 200, 250, and 300 μg/mL increased 1.5-, 1.8-, 1.8-, 2.5-, and 2.0-fold in MRP2 gene expression level (Figure 5A) and elevated 1.9-, 1.8-, 2.2-, and 1.7-fold in MRP4 gene expression level, respectively (Figure 5B). SchB at 200 μM significantly increased the gene expression level of MRP2 and MRP4. In addition, tBHQ (a known Nrf2 agonist/activator) significantly increased the gene expression level of P-gp and MRP2. There was no significant effect of SchA at 20 μM on the gene expression level of P-gp, MRP2, and MRP4 (Figure 5).
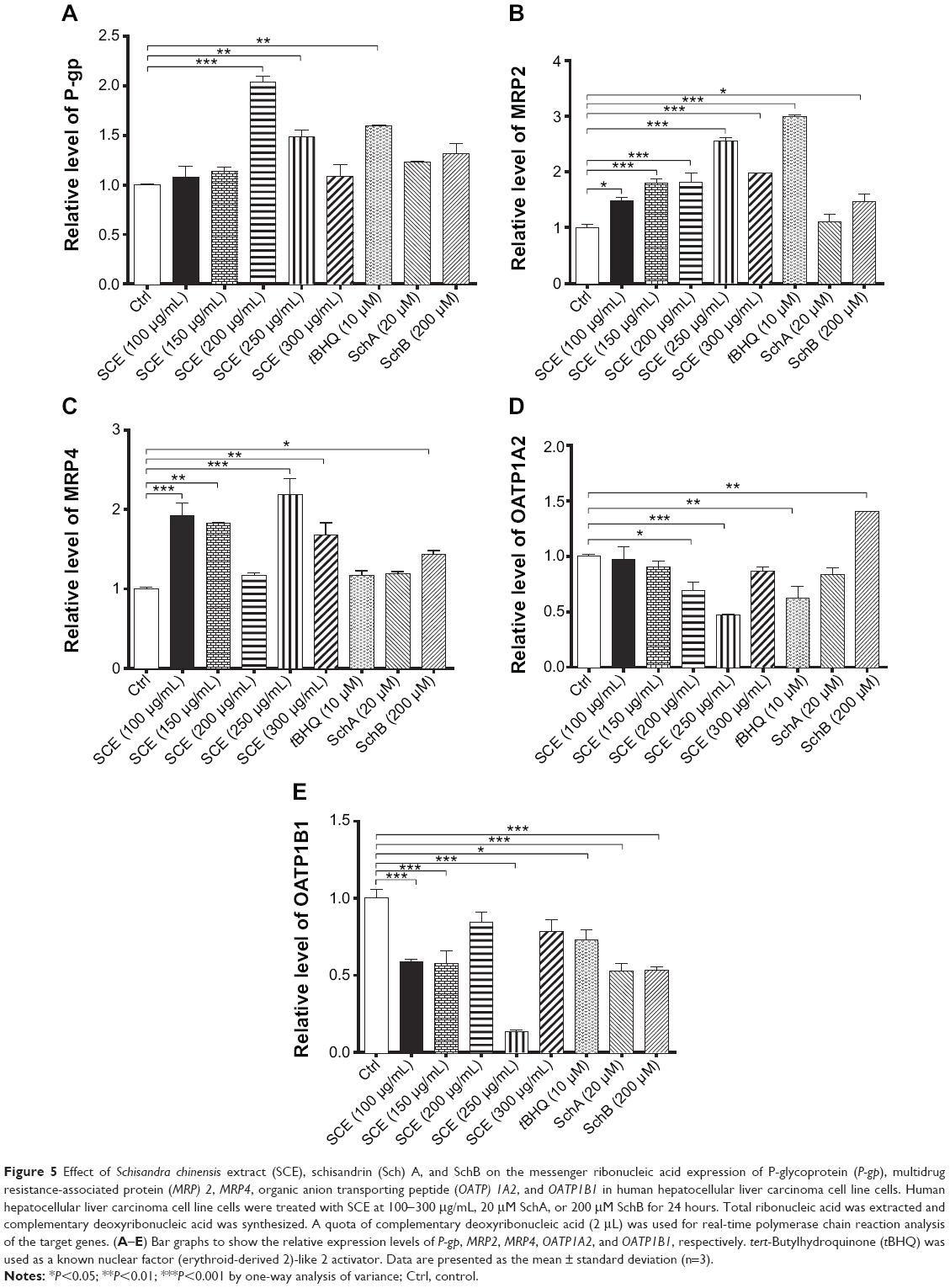
|
Figure 5 Effect of Schisandra chinensis extract (SCE), schisandrin (Sch) A, and SchB on the messenger ribonucleic acid expression of P-glycoprotein (P-gp), multidrug resistance-associated protein (MRP) 2, MRP4, organic anion transporting peptide (OATP) 1A2, and OATP1B1 in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. Total ribonucleic acid was extracted and complementary deoxyribonucleic acid was synthesized. A quota of complementary deoxyribonucleic acid (2 μL) was used for real-time polymerase chain reaction analysis of the target genes. (A–E) Bar graphs to show the relative expression levels of P-gp, MRP2, MRP4, OATP1A2, and OATP1B1, respectively. tert-Butylhydroquinone (tBHQ) was used as a known nuclear factor (erythroid-derived 2)-like 2 activator. Data are presented as the mean ± standard deviation (n=3). |
On the contrary, incubation of HepG2 cells with SCE significantly reduced the gene expression level of OATP1A2 and OATP1B1. There was a 30.9% and 53.1% reduction in the gene expression of OATP1A2 when cells were treated with SCE at 200 and 300 μg/mL, respectively (Figure 5D), and there was a 41.6%, 42.8%, and 86.4% decrease in the gene expression of OATP1B1 when cells were incubated with SCE at 100, 150, and 250 μg/mL, respectively (Figure 5E). However, SchB at 200 μM increased 1.4-fold in the gene expression level of OATP1A2 (P<0.05). There was no significant effect of SchA at 20 μM on the gene expression level of OATP1A2 and OATP1B1 (Figure 5). Notably, incubation of HepG2 cells with tBHQ significantly decreased 37.9% and 27.3% of the messenger RNA (mRNA) levels of OATP1A2 and OATP1B1, respectively (Figure 5).
In addition, the protein expression levels of P-gp, MRP2, and OATP1A2 were examined in HepG2 cells treated with SCE, SchA, or SchB. As shown in Figure 6, incubation of HepG2 cells with 250 and 200 μg/mL SCE significantly increased 1.9- and 1.4-fold in the levels of P-gp and MRP2, respectively, whereas there was an 85.7% reduction in OATP1A2 level at a concentration of 250 μg/mL SCE. Treatment of cells with 10 μM tBHQ increased 2.3-fold in the expression level of MRP2. In addition, SchA at 20 μM significantly enhanced the expression of P-gp and MRP2 but decreased the expression level of OATP1A2. SchB at 200 μM significantly increased the expression level of P-pg but reduced the expression of MRP2 and OATP1A2 (Figure 6). These findings demonstrate that SCE, SchA, and SchB at different concentrations have distinct effects on the expression of various drug transporters; SchA and SchB are the major modulators of these drug transporters; and Nrf2 appears to play an important role in the regulation of these drug transporters.
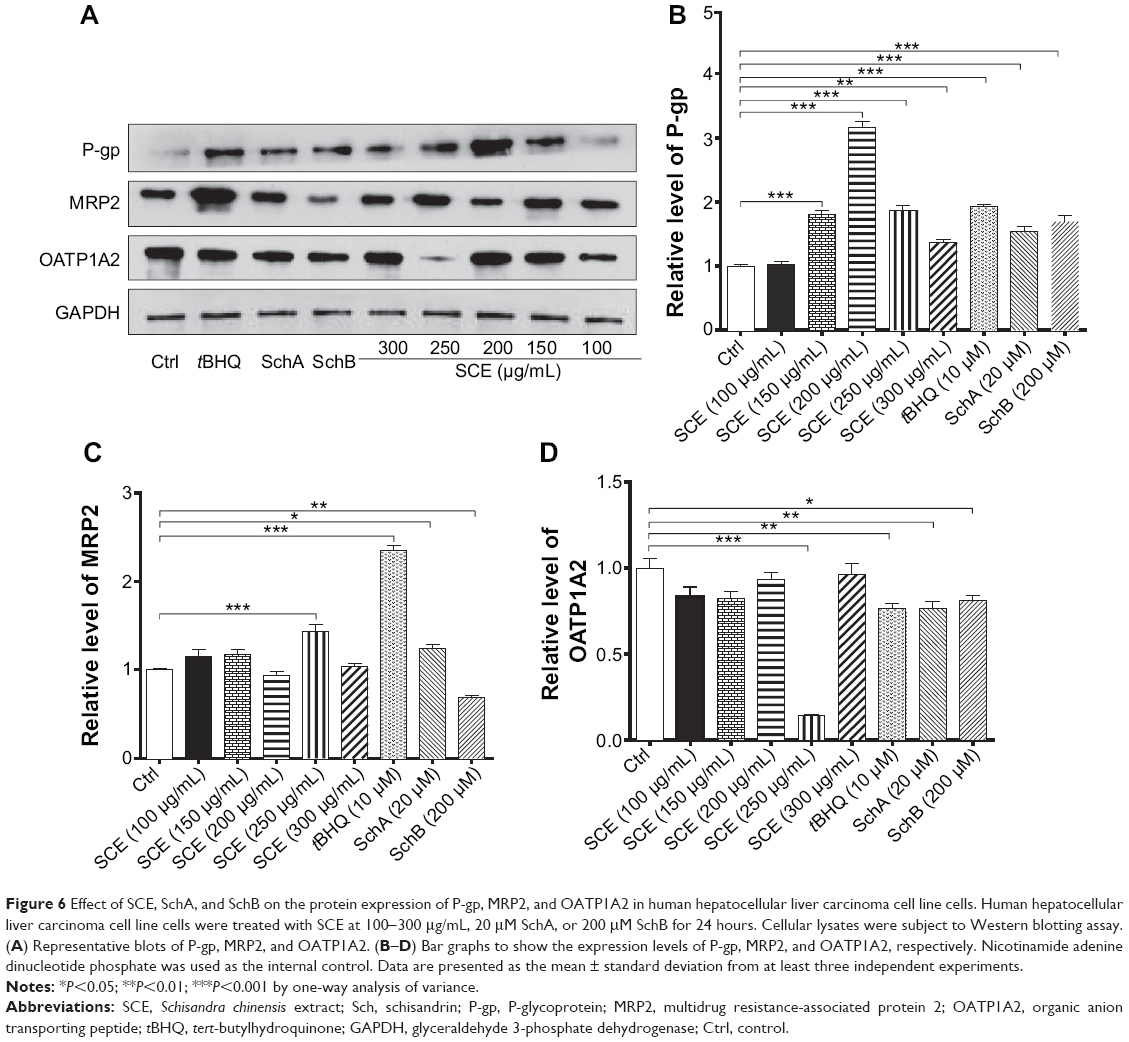
|
Figure 6 Effect of SCE, SchA, and SchB on the protein expression of P-gp, MRP2, and OATP1A2 in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. Cellular lysates were subject to Western blotting assay. (A) Representative blots of P-gp, MRP2, and OATP1A2. (B–D) Bar graphs to show the expression levels of P-gp, MRP2, and OATP1A2, respectively. Nicotinamide adenine dinucleotide phosphate was used as the internal control. Data are presented as the mean ± standard deviation from at least three independent experiments. |
SCE, SchA, and SchB increase intracellular GSH levels and total GST contents
Owing to their potent antioxidant activity, GSH and GSTs have an important role in maintaining essential cellular function against the cytotoxic effects of xenobiotics. Herein, the effect of SCE on intracellular levels of GSH and total GST content was examined in HepG2 cells. SCE remarkably elevated the intracellular level of GSH and the total content of GSTs in HepG2 cells. There was a 2.6-, 8.2-, 6.3-, and 4.0-fold increase in the intracellular level of GSH when HepG2 cells were treated with 150, 200, 250, and 300 μg/mL SCE, respectively (Figure 7A). Similarly, there was a 2.7-, 4.9-, 3.6-, and 2.5-fold increase in the total content of GSTs when cells were incubated with 150, 200, 250, and 300 μg/mL SCE, respectively (Figure 7B). SchA at 20 μM and SchB at 200 μM showed a comparable promoting effect on the intracellular level of GSH and the total content of GSTs as SCE in HepG2 cells.
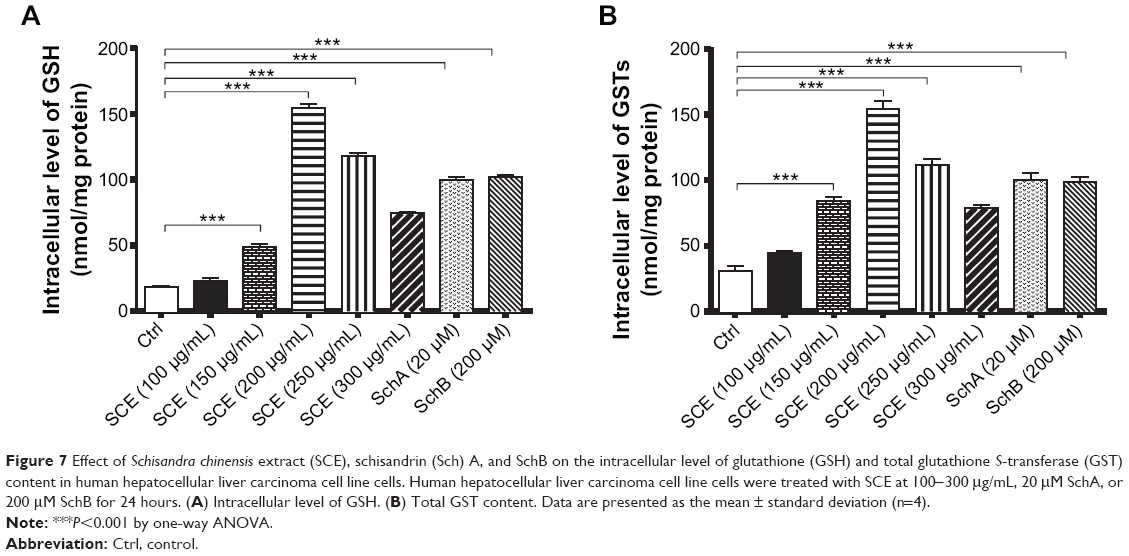
|
Figure 7 Effect of Schisandra chinensis extract (SCE), schisandrin (Sch) A, and SchB on the intracellular level of glutathione (GSH) and total glutathione S-transferase (GST) content in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. (A) Intracellular level of GSH. (B) Total GST content. Data are presented as the mean ± standard deviation (n=4). |
SCE activates Nrf2 in HepG2 cells as indicated by the NQO1–ARE luciferase assay
Following the observations of the regulatory effect of SCE on selected DMEs, drug transporters, and redox status in HepG2 cells, we further investigated the role of Nrf2 in the regulatory effect of SCE and its major components SchA and SchB on DMEs and drug transporters. It has been recognized that Nrf2 is a critical nuclear transcriptional factor regulating the expression of Phase II cytoprotective genes in response to xenobiotic exposure and other external stimuli.20,22 Herein, we examined the effect of SCE on the expression/activity of Nrf2 by testing the activity of NQO1–ARE luciferase, which indicates the activity of Nrf2. Incubation of HepG2 cells with SCE significantly enhanced the activity of Nrf2. There was a 2.2-, 3-, 6-, 6.1-, 5.9-, and 3.2-fold increase in the level of NQO1–ARE luciferin when cells were treated with SCE at 50, 100, 150, 200, 250, and 300 μg/mL, respectively. tBHQ at 10 μM, SchA at 20 μM, and SchB at 200 μM increased 4.4-, 4.0-, and 4.3-fold in the level of NQO1–ARE luciferin, respectively (P<0.01) (Figure 8). These results suggest that the SCE acts as an Nrf2 activator in HepG2 cells.
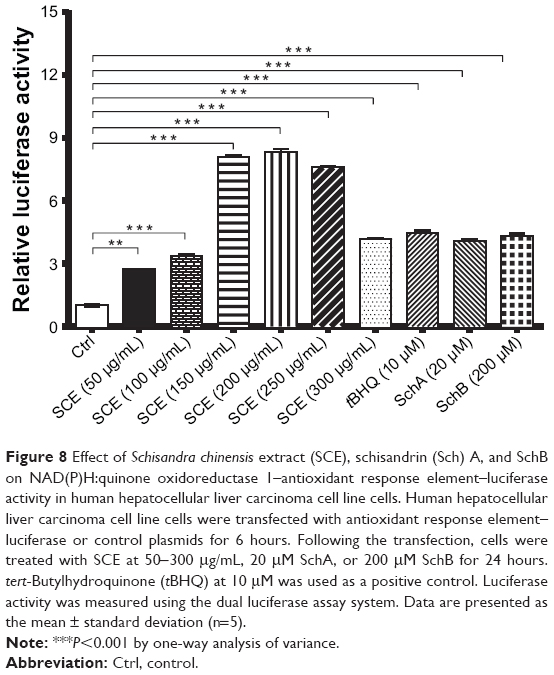
|
Figure 8 Effect of Schisandra chinensis extract (SCE), schisandrin (Sch) A, and SchB on NAD(P)H:quinone oxidoreductase 1–antioxidant response element–luciferase activity in human hepatocellular liver carcinoma cell line cells. Human hepatocellular liver carcinoma cell line cells were transfected with antioxidant response element–luciferase or control plasmids for 6 hours. Following the transfection, cells were treated with SCE at 50-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. tert-Butylhydroquinone (tBHQ) at 10 μM was used as a positive control. Luciferase activity was measured using the dual luciferase assay system. Data are presented as the mean ± standard deviation (n=5). |
Nrf2 knockdown attenuates the induction of DMEs and efflux drug transporters
Nrf2 is a key transcriptional factor that regulates DMEs and drug transporters.20,22 To clarify the role of Nrf2 in the regulation of DMEs and drug transporters by SCE, RNA interference was performed to knock down Nrf2 in HepG2 cells. In comparison with the control group (si-Ncontrol), incubation of HepG2 cells with Nrf2 siRNA significantly decreased 50% and 48% in the gene expression of NQO1 (Figure 9C) and MRP2 (Figure 9E), respectively. There was a 30% reduction in the gene expression of HO-1 when treated with Nrf2 siRNA (P<0.05) (Figure 9D). Furthermore, there was a 60% decrease in the expression level of Nrf2 compared with the control, and the inducing effect of SCE on the expression of Nrf2 was attenuated (Figure 9A and B). These results indicate that Nrf2-mediated signaling pathway is involved in the regulatory effect of SCE on DMEs and drug transporters in HepG2 cells.
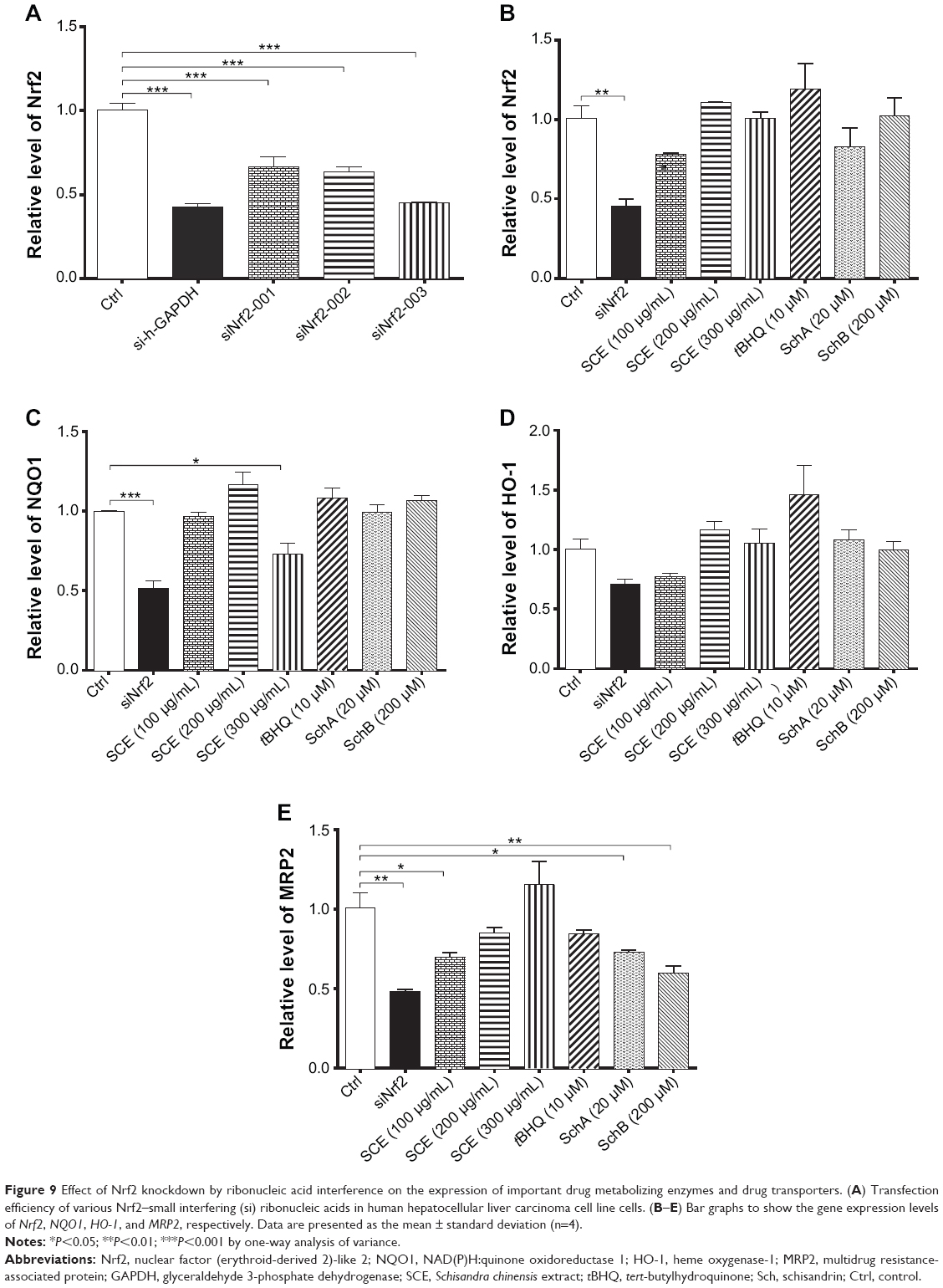
|
Figure 9 Effect of Nrf2 knockdown by ribonucleic acid interference on the expression of important drug metabolizing enzymes and drug transporters. (A) Transfection efficiency of various Nrf2–small interfering (si) ribonucleic acids in human hepatocellular liver carcinoma cell line cells. (B–E) Bar graphs to show the gene expression levels of Nrf2, NQO1, HO-1, and MRP2, respectively. Data are presented as the mean ± standard deviation (n=4). |
SCE, SchA, and SchB suppress Keap1 expression and facilitate nuclear translocation of Nrf2 in HepG2 cells
Keap1 is known as an Nrf2 repressor that binds Nrf2 with cellular actin cytoskeleton in cytoplasm.20–22 We have observed that Nrf2 plays an important role in the regulatory effect of SCE on DMEs and drug transporters. Next, we further examined the effect of SCE, SchA, and SchB on Keap1 expression and the nuclear translocation of Nrf2 to clarify the regulatory effect of SCE on the interaction between Keap1 and Nrf2. Incubation of HepG2 cells with SCE concentration dependently decreased the cytosolic level of Keap1 (P<0.01) (Figure 10). Treatment of HepG2 cells with SCE at 250 and 300 μg/mL significantly decreased the cytosolic level of Keap1 by 45.85% and 70.68%, respectively (P<0.001). SchA at 20 μM and SchB at 200 μM significantly decreased the cytosolic level of Keap1 by 32.94% and 35.90%, respectively (P<0.05). We also examined the effect of SCE on Nrf2 protein expression in the nucleus and cytoplasm. The results showed an increase in Nrf2 at both total and nuclear protein levels, which indicates that SCE facilitates the nuclear translocation of Nrf2 (Figure 10).
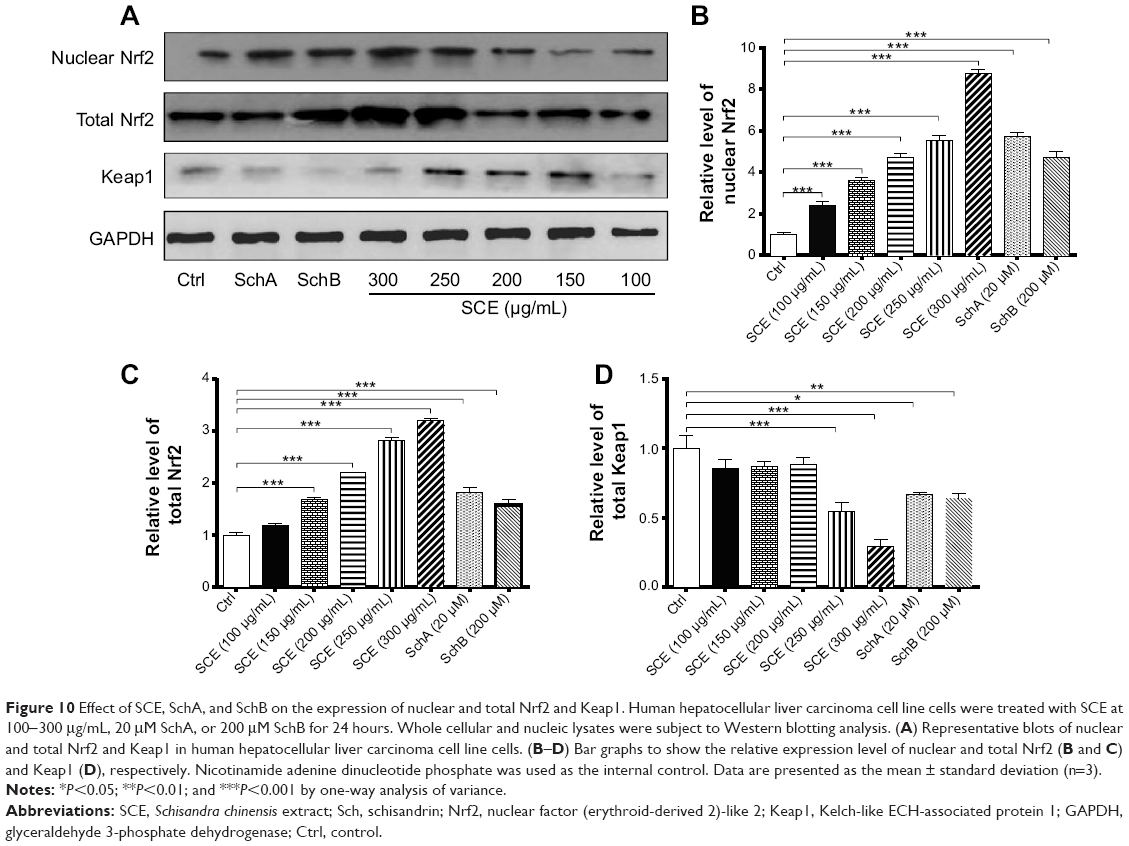
|
Figure 10 Effect of SCE, SchA, and SchB on the expression of nuclear and total Nrf2 and Keap1. Human hepatocellular liver carcinoma cell line cells were treated with SCE at 100-300 μg/mL, 20 μM SchA, or 200 μM SchB for 24 hours. Whole cellular and nucleic lysates were subject to Western blotting analysis. (A) Representative blots of nuclear and total Nrf2 and Keap1 in human hepatocellular liver carcinoma cell line cells. (B–D) Bar graphs to show the relative expression level of nuclear and total Nrf2 (B and C) and Keap1 (D), respectively. Nicotinamide adenine dinucleotide phosphate was used as the internal control. Data are presented as the mean ± standard deviation (n=3). |
SCE increases Nrf2 protein stability
In addition, to further elucidate how SCE regulated DMEs and drug transporters through Nrf2 activation, the effect of SCE on Nrf2 protein stability was examined. HepG2 cells were treated with the protein synthesis inhibitor CHX at indicated times with or without pretreatment of SCE, SchA, and SchB. At the basal level, the half-life of Nrf2 protein was 13.07±0.50 minutes. Pretreatment of cells with SCE at 200 μg/mL, SchA at 20 μM, and SchB at 200 μM significantly prolonged the half-lives of Nrf2 protein (P<0.001), which were 26.02±0.46, 24.14±0.80, and 19.41±1.96 minutes, respectively (Figure 11). These results indicate that SCE, SchA, and SchB remarkably increase Nrf2 stability primarily through inhibition of protein degradation. Since SCE, SchA, and SchB downregulated Keap1, this would contribute to the prolonged half-lives of Nrf2 in HepG2 cells.
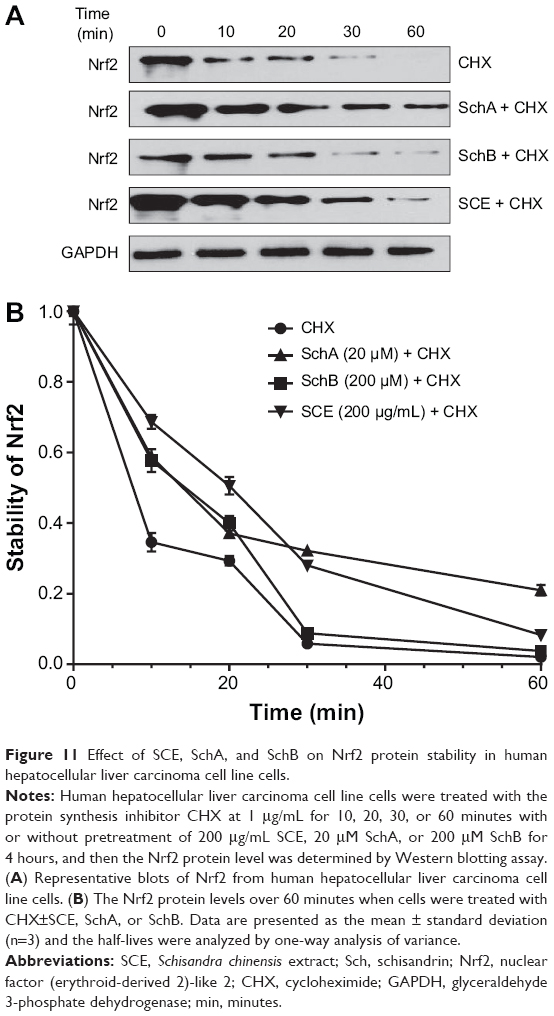 |
Figure 11 Effect of SCE, SchA, and SchB on Nrf2 protein stability in human hepatocellular liver carcinoma cell line cells. |
Discussion
SCE (Wuweizi), dried mature fruit from SC, is one of the most commonly used Chinese herbal medicines as a liver protectant that has been documented in Sheng Nong’s Herbal Classic.62,63 To date, it has been shown that SCE has remarkable protective effects on the liver, digestive system, central nervous system, and cardiovascular system.64 These beneficial effects can be ascribed to its antioxidative and anti-inflammatory activities. In the present study, we have observed that SCE upregulates important DMEs and drug transporters that participate in xenobiotic detoxification with the involvement of Nrf2-mediated signaling pathway in HeG2 cells.
The choice of SCE concentrations (150-300 μg/mL) in this study is based on the dosage of SCE used in patients who are administered 1.0–6.0 g of SCE per day. This equals 20.0–120.0 μg/mL of SCE, assuming a standard adult contains 50 kg of water and SCE is evenly dissolved in the body’s water. Our prepared SCE at 200 μg/mL contains 0.76 μM SchA and 0.39 μM SchB. The concentrations of SchA and SchB used in this study were significantly higher than those in SCE but did not elicit remarkable cytotoxicity toward HepG2 cells. Notably, we tested the effects of low concentrations of SchA and SchB on the expression of DMEs and drug transporters in HepG2 cells in our pilot experiment, but we did not see any effects.
The liver is the primary organ in the biotransformation and detoxification of endogenous and exogenous compounds by a large number of Phase I and II DMEs.65 Of great importance, Phase II conjugating DMEs have an important role in the detoxification of drugs and environmental toxicants and the maintenance of liver function.66 Recently, studies have shown that Phase I and II detoxifying and antioxidant enzymes such as GSTs, GCLM, NQO1, and HO-1 are critical in the maintenance of redox balance in the liver.65,66 NQO1 is an flavin adenine dinucleotide-binding cytosolic protein that forms homodimers and reduces quinones to hydroquinones, inhibiting the production of radical species.67 NQO1 also has anti-inflammatory effects, scavenges superoxide anion radicals, and stabilizes p53 and other tumor suppressors. Mutations in the NQO1 gene have been associated with tardive dyskinesia, an increased risk of hematotoxicity after exposure to benzene, and susceptibility to cancer, Alzheimer’s disease, and cardiovascular diseases.68 HO-1 cleaves the heme ring at the α-methene bridge to form either biliverdin or verdoglobin when the heme is still attached to a globin, with the release of CO and free iron.69 Biliverdin is subsequently converted to bilirubin by biliverdin reductase. Unlike HO-2, HO-1 is an inducible isoform in response to stress such as oxidative stress, hypoxia, heavy metals, and cytokines. GCL is the first rate-limiting enzyme of GSH synthesis.70 Human GSTs contribute to the GSH conjugation or GSH-dependent biotransformation of xenobiotics, and many catalyze GSH peroxidase or thiol transferase reactions. GSTs also catalyze GSH-dependent isomerization reactions required for the synthesis of several prostaglandins and steroid hormones and the catabolism of tyrosine. Furthermore, GSTs serve as significant regulators of pathways determining cell proliferation and survival and as regulators of ryanodine receptors that are essential for muscle function.71 Dysregulation of NQO1, GCLM, GSTs, and HO-1 results in disruption of redox homeostasis in the liver or other organs when the level of reactive oxygen species outweighs the antioxidants under oxidative stress.72 Consequently, disrupted redox homeostasis may cause liver and organ damage. In the present study, we have observed that SCE significantly induced the expression of NQO1, GCLM, GSTA4, and HO-1 at both transcriptional and translational levels in HepG2 cells. These results can, at least in part, explain the antioxidative and hepatoprotective effects of SCE.
GSH is a ubiquitous intracellular tripeptide with diverse functions including detoxification, antioxidant defense, maintenance of thiol status, and modulation of cell proliferation.73 GSH is synthesized in the cytosol of mammalian cells in a tightly regulated manner. The major determinants of GSH synthesis are the availability of cysteine, the sulfur amino acid precursor, and the activity of the rate-limiting enzyme GCL, which is composed of a catalytic (GCLC) and modifier (GCLM) subunit.73 GSH has an important role in maintaining liver function via the regulation of redox homeostasis, and inadequate GSH levels have a significant association with toxicant-induced hepatotoxicity.73 Restoration of the ratio of GSH to its oxidized form can ameliorate toxicant-induced hepatotoxicity. In our study, we found that incubation of HepG2 cells with SCE remarkably elevated the intercellular level of GSH, which could partially explain the hepatoprotective effect of SCE. Moreover, we also observed that SCE significantly increased the total GST content that plays a critical role in the detoxification of a large number of xenobiotics. Taken together, the antioxidative effect of SCE can be ascribed, at least in part, to the upregulation of antioxidative enzymes and the elevation of intracellular levels of GSH. Given the effects of SCE on GSH and GSTs, SCE shows a coordinated regulatory effect in response to oxidative stress or chemical stimuli to exert its liver protective effects.
Drug transporters have an important role in cellular protection because of their capability of removing xenobiotics and endogenous substrates that can accumulate in cells and lead to toxicity.74,75 The ATP-binding cassette (ABC) transporters, such as P-gp (MDR1), MRP2, and MRP4, are responsible for the efflux of a number of endogenous and exogenous compounds.76–78 ABC transporters are divided into seven distinct subfamilies, including ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, and white. P-gp is located on the apical surface of intestinal epithelial cells, bile canaliculi, renal tubular cells, and placenta and the luminal surface of capillary endothelial cells in the brain and testes. MRP2 is generally localized at the apical membrane, and MRP4 is at the sinusoidal membrane of hepatocytes.76–78 P-gp transports various substrates across the cell membrane, including drugs such as colchicine, tacrolimus, and quinidine; chemotherapeutic agents such as etoposide, doxorubicin, and vinblastine; lipids; steroids; peptides; bilirubin; digoxin; immunosuppressive agents; and environmental compounds.76 MRP2/ABCC2 is an efflux transporter that aides in biliary excretion and renal excretion of drugs and also prevents xenobiotics from penetrating or accumulating in tissues such as lungs and from crossing the placenta.79 MRP2 has broad substrate specificity and can transport organic anions such as glucuronate, GSH conjugates of lipophilic substances, compounds without anionic conjugate residues (eg, methotrexate and bromosulfophthalein), reduced and oxidized GSH, nucleotide analogs, and anticancer drugs. MRP2 transports conjugated endogenous and xenobiotic substances, including toxins and carcinogens into bile (from hepatocytes), urine (from renal proximal tubular cells), and the intestinal lumen (from enterocytes).79 MRP4 is unique among ABCC transporters due to its dual localization in polarized cells.80 In the liver, choroid plexus, and prostatic acinar cells, MRP4 is expressed at the basolateral membrane, while in brain capillary endothelium and renal proximal tubule cells, MRP4 is expressed at the apical membrane. MRP4 mediates the transport of signaling molecules (such as cyclic adenosine monophosphate and cyclic guanosine monophosphate), uric acid, eicosanoids, anticancer drugs, and antiviral agents (eg, topotecan, tenofovir, adefovir) as well as nucleoside analogs (eg, 6-mercaptopurine).77,80 MRP4 transports bile acids in the presence of GSH and serves as a backup system for eliminating bile acids from hepatocytes. Both MRP2 and MRP4 transport their substrates in the presence of GSH. Adequate GSH level facilitates the transport by maintaining the integrity of cell membrane, protecting cells against oxidative stress, recovering electron transfer, and stabilizing mitochondria membrane potential.77,78 In the present study, we found that SCE upregulated the expression of P-gp, MRP2, and MRP4 at both transcriptional and translational levels. The regulatory effect of SCE appeared more potent than SchA and SchB alone, probably due to the presence of multiple components in SCE and potential synergistic effect. Notably, the transcriptional and posttranscriptional levels of P-gp were significantly enhanced by SchA and SchB. Both SchA and SchB are substrates of P-gp.81,82 The results indicate that SCE can modulate both the activity and expression of P-gp. SchA and SchB are inhibitors for P-gp and MRP1.83,84
On the other hand, OATP1A2 and OATP1B1 are members of the OATP/SLCO21 family that belongs to uptake transporters localized in the hepatic sinusoidal membrane.85–89 In the intestine, OATP1A2 is localized to the brush border membrane of enterocytes, cholangiocytes (cells lining the bile ducts), the apical membrane of the distal nephron, and the luminal membrane of the endothelial cells of brain capillaries. OATP1A2 transports a range of organic anionic, neutral, and cationic pharmacological drugs and xenobiotics, including bile acids, conjugated sex steroids, linear and cyclic peptides, mushroom toxins, prostaglandin E2, the thyroid hormones T3 and T4, BQ-123 (an endothelin receptor antagonist), CRC-220 (a thrombin inhibitor), magnetic resonance imaging contrast agents, fexofenadine, methotrexate, ouabain, statins, and the cyanobacterial toxin microcystin.88 OATP1B1 exhibits broad substrate selectivity that includes anionic (eg, statins such as pravastatin, pitavastatin, and rosuvastatin), zwitterionic (eg, rifampicin), and neutral lipophilic (eg, paclitaxel) drugs.88 OATP1B1 also transports endogenous substances such as bile acids, thyroid hormones, steroid sulfates, glucuronide conjugates, and peptides. Our results showed that SCE significantly suppressed the expression of OATP1A2 and OATP1B1 at transcriptional and posttranscriptional levels. In contrast to SCE, SchB at 200 μM upregulated the expression of OATP1A2. Nrf2 may contribute to this effect of SchB, but the involvement of other regulators and related signaling pathways such as FXR and protein kinase C cannot be excluded.
In our study, we further revealed the role of Nrf2-mediated signaling pathway in the regulatory effect of SCE and its main components on DMEs and drug transporters. This is consistent with the reports from several previously published studies where Nrf2 was found to play a role in the antioxidative and anti-inflammatory effects of SCE and its main active compounds.90–92 Activation of Nrf2-mediated pathway by pharmacologic approach serves to stimulate coordinated induction of hepatic DMEs such as GSTs and efflux transporters such as P-gp, MRP2, MRP3, and MRP4 upon binding to the ARE at the proximal region of the promoter in the target genes.
Nrf2 signaling is known as a mastery pathway that regulates the expression of a number of DMEs and transporters.20,22 Besides, Nrf2-mediated signaling pathway is widely involved in cell protection in cancer chemoprevention and anti-inflammatory process. Nrf2 belongs to the basic Lucien zipper nuclear transcriptional factor family, which shares regions of homology with the Cap ‘n’ Collar (CNC) structure. Nrf2 contains six highly conserved domains known as Nrf2–ECH homology (Neh) domains.20–22 The Neh1 domain is a CNC–bZIP domain that allows Nrf2 to heterodimerize with small Maf proteins; the Neh2 domain allows for binding of Nrf2 to its cytosolic repressor Keap1; the Neh3 domain plays a role in Nrf2 protein stability and may act as a transactivation domain, interacting with components of the transcriptional apparatus; the Neh4 and Neh5 domains also act as transactivation domains but bind to a different protein called cAMP response element binding protein, which possesses intrinsic histone acetyltransferase activity; and the Neh6 domain may contain a degron that is involved in the degradation of Nrf2.35,44,45 Nrf2 is considered to play an important role in the cellular defense mechanisms against xenobiotic toxicity and carcinogenicity.20,22 Activation of Nrf2 signaling pathway can increase the expression of the rate-limiting enzymes in GSH synthesis, thus enhancing cells’ capability against oxidative stress. It is evident that many small natural or synthetic molecules can activate Nrf2 signaling pathway leading to upregulation of cytoprotective genes such as detoxification and GSH biosynthetic enzymes. Under physiological condition, Nrf2 locates in cytoplasm, forming a protein complex with its cytosolic repressor Keap1.20,22 When cells are stimulated by oxidative stress and/or electrophiles, Nrf2 is disassociated from Keap1, followed by nuclear translocation. In the nucleus, Nrf2 forms a protein complex dimer with small Maf protein and other cofactors to control the expression of its target genes. In other words, translocation of Nrf2 can enhance the transcription of numerous antioxidative and Phase II detoxifying genes such as HO-1, NQO1, and GSTs.20,22 Thus, activation of Nrf2 signal can protect cells from challenges of drugs, toxicants, and carcinogens. Our results showed that HO-1, NQO1, and GCLM were upregulated by SCE at a transcriptional level in a concentration-dependent manner. In addition, SCE remarkably increased the total content of GSTs. Moreover, the SCE-mediated upregulation of HO-1 and NQO1 was abolished by Nrf2 knockdown. These results suggest that SCE modulated DMEs and drug transporters via the activation of the Nrf2 signaling pathway (Figure 12).
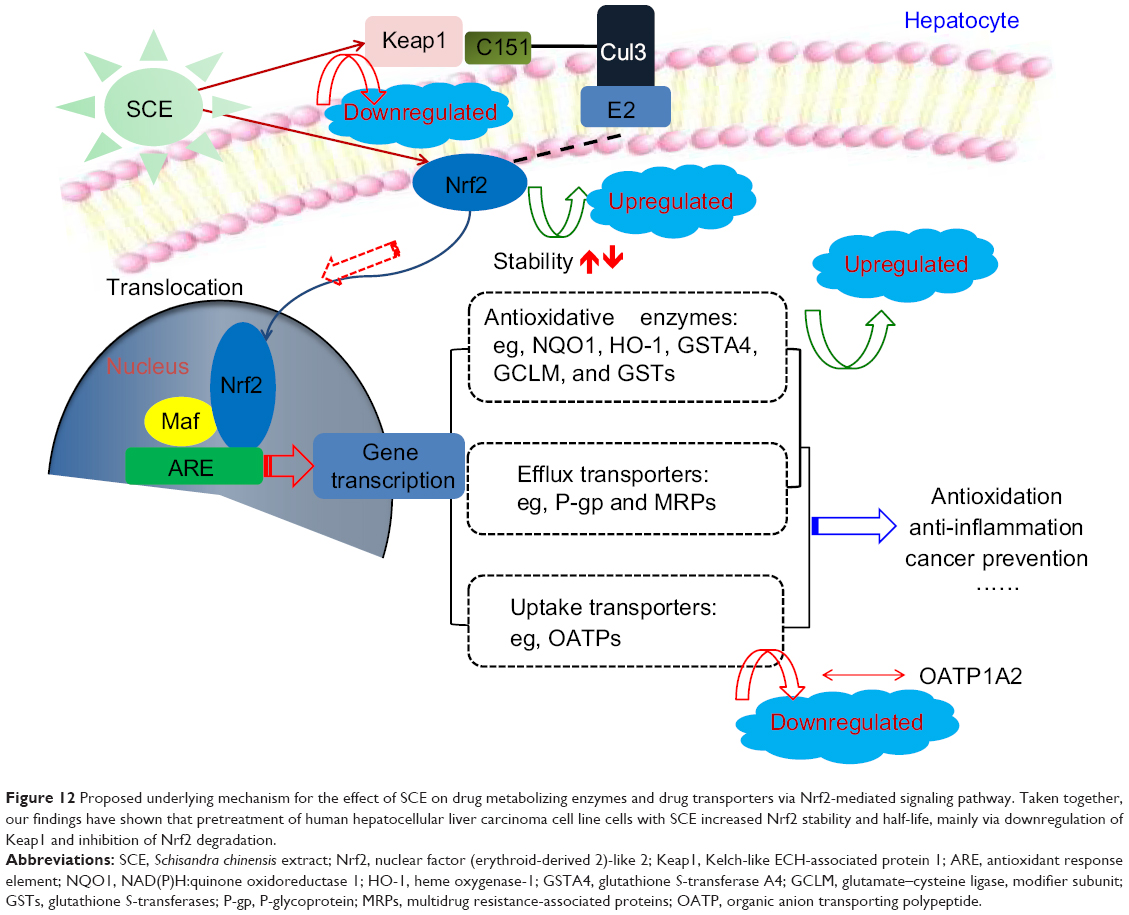
|
Figure 12 Proposed underlying mechanism for the effect of SCE on drug metabolizing enzymes and drug transporters via Nrf2-mediated signaling pathway. Taken together, our findings have shown that pretreatment of human hepatocellular liver carcinoma cell line cells with SCE increased Nrf2 stability and half-life, mainly via downregulation of Keap1 and inhibition of Nrf2 degradation. |
Disassociation of Nrf2–Keap1 complex and translocation of Nrf2 into the nucleus for ARE binding in the promoter regions of target genes are the prerequisites of transcriptional activation of DME and transporter genes.20,22 In our experiments, we found that SCE concentration dependently enhanced Nrf2 translocation as well as increased total Nrf2 expression level. Our results also showed that SCE significantly inhibited Keap1 expression in a concentration-dependent manner. Previous studies have shown that Nrf2 activators might stabilize Nrf2 by increasing the ubiquitination and proteasomal degradation of Keap1 and consequently increased the half-life of Nrf2.93,94 In agreement with these findings, our findings showed that pretreatment of HepG2 cells with SCE increased Nrf2 stability and half-life mainly via downregulation of Keap1 and inhibition of Nrf2 degradation.
In this study, we found that both SchA and SchB induced the gene expression of HO-1 and NQO1 and prolonged the half-life of Nrf2 in HepG2 cells. However, SchA and SchB showed different potency and regulatory effects of some selected genes. In the SCE we prepared, its total lignanoid concentration was 44.97 mg/g crude extracts. In 300 μg/mL SCE used for cell treatment, the total lignanoid concentration is only 13.15 μg/mL, and thus the concentration of SchA and SchB in SCE was much lower than that we used for SchA at 20 μM and SchB at 200 μM. Our results demonstrate that the major components in SCE are lignanoids, and interaction of these lignanoid components may enhance the biological activities of SCE.
SCE at 100–300 μg/mL considerably up- or downregulated the expression of DMEs and drug transporters in HepG2 cells, but there is no clear dose response relationship, while maximum effect was often observed at 200 μg/mL SCE. The reason for this is unknown, but we suspect that gene regulation by SCE in HepG2 cells may be saturated when SCE concentration achieved 200 μg/mL, and the presence of multiple active components in SCE may antagonize and synergize depending on individual component concentrations. It has been reported that a low concentration of SCE exerts an antioxidative effect, whereas a high concentration of SCE exhibits an opposite effect.47
In the present study, we observed that SCE, SchA, and SchB induced the expression of NQO1, HO-1, and MRP2 in HepG2 cells, but this effect could not be fully abolished using Nrf2 siRNA. This suggests that there may be other related signaling pathways involved in the regulatory effect of SCE on the expression of DMEs and drug transporters in HepG2 cells. Recently, it has been reported that inhibition of c-Jun N-terminal kinase signaling pathway is involved in the liver protective effect of SCE in mice with acetaminophen-induced liver injury.59 Moreover, SCE or its components can inhibit nuclear transcription factor-κB and mitogen-activated protein kinase signaling pathways, contributing to its beneficial effects.80,83,84 Moreover, the RNA interfering efficiency may change, contributing to these varying effects.
Conclusion
In summary, our study has shown that SCE upregulates the expression of NQO1, HO-1, GCL, GSTA4, P-gp, and MRPs but downregulates the expression of OATPs primarily via the activation of Nrf2-mediated signaling pathway in HepG2 cells. Our findings may provide better understanding of the molecular mechanism for the hepatoprotective action of SCE in clinical practice. On the other hand, SCE may have potential to interact with other drugs, and this should be taken into consideration in polypharmacy. More mechanistic studies are needed to dissect how SCE protects the liver through regulation of cytoprotective DMEs and drug transporters.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No: 81102548) and the special funds from Central Finance of China in Support of the Development of Local Colleges and University [Educational finance grant No: 276(2014)]. The authors would like to thank the Institute of Biomedicine and Guangdong Provincial Key Laboratory of Bioengineering, Guangzhou, People’s Republic of China, for technical assistance.
Author contributions
YY and SFZ designed and coordinated the study. JLH, ZWZ, JJY, and CQH conducted the experiments and data analysis. JLH and ZWZ drafted the manuscript. All authors critically reviewed the manuscript and approved the final version for publication.
Disclosure
The authors have no financial interests that might be perceived to affect the results or the discussion reported in this article.
References
Qiu J. Traditional medicine: a culture in the balance. Nature. 2007;448(7150):126–128. | ||
Mehta DH, Gardiner PM, Phillips RS, McCarthy EP. Herbal and dietary supplement disclosure to health care providers by individuals with chronic conditions. J Altern Complement Med. 2008;14(10):1263–1269. | ||
Kennedy J. Herb and supplement use in the US adult population. Clin Ther. 2005;27(11):1847–1858. | ||
Gunther S, Patterson RE, Kristal AR, Stratton KL, White E. Demographic and health-related correlates of herbal and specialty supplement use. J Am Diet Assoc. 2004;104(1):27–34. | ||
He SM, Chan E, Zhou SF. ADME properties of herbal medicines in humans: evidence, challenges and strategies. Curr Pharm Des. 2011;17(4):357–407. | ||
Chen XW, Sneed KB, Zhou SF. Pharmacokinetic profiles of anticancer herbal medicines in humans and the clinical implications. Curr Med Chem. 2011;18(21):3190–3210. | ||
Hun Lee J, Shu L, Fuentes F, Su ZY, Kong TAN. Cancer chemoprevention by traditional chinese herbal medicine and dietary phytochemicals: targeting Nrf2-mediated oxidative stress/anti-inflammatory responses, epigenetics, and cancer stem cells. J Tradit Complement Med. 2013;3(1):69–79. | ||
Fugh-Berman A. Herb-drug interactions. Lancet. 2000;355(9198):134–138. | ||
Yang XX, Hu ZP, Duan W, Zhu YZ, Zhou SF. Drug-herb interactions: eliminating toxicity with hard drug design. Curr Pharm Des. 2006;12(35):4649–4664. | ||
Zhou SF, Zhou ZW, Li CG, et al. Identification of drugs that interact with herbs in drug development. Drug Discov Today. 2007;12(15–16):664–673. | ||
Di YM, Li CG, Xue CC, Zhou SF. Clinical drugs that interact with St John’s wort and implication in drug development. Curr Pharm Des. 2008;14(17):1723–1742. | ||
Yang AK, He SM, Liu L, Liu JP, Wei MQ, Zhou SF. Herbal interactions with anticancer drugs: mechanistic and clinical considerations. Curr Med Chem. 2010;17(16):1635–1678. | ||
He SM, Yang AK, Li XT, Du YM, Zhou SF. Effects of herbal products on the metabolism and transport of anticancer agents. Expert Opin Drug Metab Toxicol. 2010;6(10):1195–1213. | ||
Zhou S, Gao Y, Jiang W, Huang M, Xu A, Paxton JW. Interactions of herbs with cytochrome P450. Drug Metab Rev. 2003;35(1):35–98. | ||
Zhou S, Lim LY, Chowbay B. Herbal modulation of P-glycoprotein. Drug Metab Rev. 2004;36(1):57–104. | ||
Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62(13):1238–1249. | ||
Aleksunes LM, Klaassen CD. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARa-, and Nrf2-null mice. Drug Metab Dispos. 2012;40(7):1366–1379. | ||
Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8(7):803–817. | ||
Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol. 2007;47(5):566–578. | ||
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. | ||
Keum YS, Choi BY. Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules. 2014;19(7):10074–10089. | ||
Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012;64(4):1055–1081. | ||
Shen G, Kong AN. Nrf2 plays an important role in coordinated regulation of Phase II drug metabolism enzymes and Phase III drug transporters. Biopharm Drug Dispos. 2009;30(7):345–355. | ||
Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278(4):2396–2402. | ||
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8(4):379–391. | ||
Takaya K, Suzuki T, Motohashi H, et al. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med. 2012;53(4):817–827. | ||
Baird L, Lleres D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci U S A. 2013;110(38):15259–15264. | ||
Stewart JD, Hengstler JG, Bolt HM. Control of oxidative stress by the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85(4):239. | ||
Iida K, Itoh K, Kumagai Y, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64(18):6424–6431. | ||
Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27(6):1094–1100. | ||
Kobayashi A, Tsukide T, Miyasaka T, et al. Central nervous system-specific deletion of transcription factor Nrf1 causes progressive motor neuronal dysfunction. Genes Cells. 2011;16(6):692–703. | ||
Aw Yeang HX, Hamdam JM, Al-Huseini LM, et al. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem. 2012;287(13):10556–10564. | ||
Weerachayaphorn J, Mennone A, Soroka CJ, et al. Nuclear factor-E2-related factor 2 is a major determinant of bile acid homeostasis in the liver and intestine. Am J Physiol Gastrointest Liver Physiol. 2012;302(9):G925–G936. | ||
Cho HY, van Houten B, Wang X, et al. Targeted deletion of Nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid Redox Signal. 2012;17(8):1066–1082. | ||
Volonte D, Liu Z, Musille PM, et al. Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol Biol Cell. 2013;24(12):1852–1862. | ||
Al-Huseini LM, Aw Yeang HX, Sethu S, et al. Nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) modulates dendritic cell immune function through regulation of p38 MAPK-cAMP-responsive element binding protein/activating transcription factor 1 signaling. J Biol Chem. 2013;288(31):22281–22288. | ||
Muramatsu H, Katsuoka F, Toide K, Shimizu Y, Furusako S, Yamamoto M. Nrf2 deficiency leads to behavioral, neurochemical and transcriptional changes in mice. Genes Cells. 2013;18(10):899–908. | ||
Gu X, Manautou JE. Regulation of hepatic ABCC transporters by xenobiotics and in disease states. Drug Metab Rev. 2010;42(3):482–538. | ||
Wu TY, Khor TO, Lee JH, et al. Pharmacogenetics, pharmacogenomics and epigenetics of Nrf2-regulated xenobiotic-metabolizing enzymes and transporters by dietary phytochemical and cancer chemoprevention. Curr Drug Metab. 2013;14(6):688–694. | ||
Chan SW. Panax ginseng, Rhodiola rosea and Schisandra chinensis. Int J Food Sci Nutr. 2012;63 Suppl 1:75–81. | ||
Wang MC, Lai YC, Chang CL. High throughput screening and antioxidant assay of dibenzo[a,c]cyclooctadiene lignans in modified-ultrasonic and supercritical fluid extracts of Schisandra chinensis Baill by liquid chromatography-mass spectrometry and a free radical-scavenging method. J Sep Sci. 2008;31(8):1322–1332. | ||
Lu Y, Chen DF. Analysis of Schisandra chinensis and Schisandra sphenanthera. J Chromatogr A. 2009;1216(11):1980–1990. | ||
Ip SP, Mak DH, Li PC, Poon MK, Ko KM. Effect of a lignan-enriched extract of Schisandra chinensis on aflatoxin B1 and cadmium chloride-induced hepatotoxicity in rats. Pharmacol Toxicol. 1996;78(6):413–416. | ||
Zhu M, Lin KF, Yeung RY, Li RC. Evaluation of the protective effects of Schisandra chinensis on Phase I drug metabolism using a CCl4 intoxication model. J Ethnopharmacol. 1999;67(1):61–68. | ||
Ip SP, Yiu HY, Ko KM. Differential effect of schisandrin B and dimethyl diphenyl bicarboxylate (DDB) on hepatic mitochondrial glutathione redox status in carbon tetrachloride intoxicated mice. Mol Cell Biochem. 2000;205(1–2):111–114. | ||
Zhu M, Yeung RY, Lin KF, Li RC. Improvement of phase I drug metabolism with Schisandra chinensis against CCl4 hepatotoxicity in a rat model. Planta Med. 2000;66(6):521–525. | ||
Chiu PY, Mak DH, Poon MK, Ko KM. In vivo antioxidant action of a lignan-enriched extract of Schisandra fruit and an anthraquinone-containing extract of Polygonum root in comparison with schisandrin B and emodin. Planta Med. 2002;68(11):951–956. | ||
Chang HF, Lin YH, Chu CC, Wu SJ, Tsai YH, Chao JC. Protective effects of Ginkgo biloba, Panax ginseng, and Schizandra chinensis extract on liver injury in rats. Am J Chin Med. 2007;35(6):995–1009. | ||
Chen N, Chiu PY, Ko KM. Schisandrin B enhances cerebral mitochondrial antioxidant status and structural integrity, and protects against cerebral ischemia/reperfusion injury in rats. Biol Pharm Bull. 2008;31(7):1387–1391. | ||
Yan F, Zhang QY, Jiao L, et al. Synergistic hepatoprotective effect of Schisandrae lignans with Astragalus polysaccharides on chronic liver injury in rats. Phytomedicine. 2009;16(9):805–813. | ||
Stacchiotti A, Li Volti G, Lavazza A, Rezzani R, Rodella LF. Schisandrin B stimulates a cytoprotective response in rat liver exposed to mercuric chloride. Food Chem Toxicol. 2009;47(11):2834–2840. | ||
Kim KJ, Yoon KY, Hong HD, Lee BY. Schisandra chinensis prevents hepatic lipid peroxidation and oxidative stress in rats subjected to heat environmental stress. Phytother Res. 2012;26(11):1674–1680. | ||
Kang JW, Kim SJ, Kim HY, et al. Protective effects of HV-P411 complex against D-galactosamine-induced hepatotoxicity in rats. Am J Chin Med. 2012;40(3):467–480. | ||
Pan SY, Yu Q, Zhang Y, et al. Dietary Fructus Schisandrae extracts and fenofibrate regulate the serum/hepatic lipid-profile in normal and hypercholesterolemic mice, with attention to hepatotoxicity. Lipids Health Dis. 2012;11:120. | ||
Cheng N, Ren N, Gao H, Lei X, Zheng J, Cao W. Antioxidant and hepatoprotective effects of Schisandra chinensis pollen extract on CCl4-induced acute liver damage in mice. Food Chem Toxicol. 2013;55:234–240. | ||
Hwang IS, Kim JE, Lee YJ, et al. Protective effects of gomisin A isolated from Schisandra chinensis against CCl4-induced hepatic and renal injury. Int J Mol Med. 2013;31(4):888–898. | ||
Li L, Zhang T, Zhou L, et al. Schisandrin B attenuates acetaminophen-induced hepatic injury through heat-shock protein 27 and 70 in mice. J Gastroenterol Hepatol. 2014;29(3):640–647. | ||
Park HJ, Lee SJ, Song Y, et al. Schisandra chinensis prevents alcohol-induced fatty liver disease in rats. J Med Food. 2014;17(1):103–110. | ||
Wang KP, Bai Y, Wang J, Zhang JZ. Inhibitory effects of Schisandra chinensis on acetaminophen-induced hepatotoxicity. Mol Med Rep. 2014;9(5):1813–1819. | ||
Cheung KL, Yu S, Pan Z, Ma J, Wu TY, Kong AN. tBHQ-induced HO-1 expression is mediated by calcium through regulation of Nrf2 binding to enhancer and polymerase II to promoter region of HO-1. Chem Res Toxicol. 2011;24(5):670–676. | ||
Park JS, Kim HS. Regulation of hemeoxygenase-1 gene expression by Nrf2 and c-Jun in tertiary butylhydroquinone-stimulated rat primary astrocytes. Biochem Biophys Res Commun. 2014;447(4):672–677. | ||
Koncic MZ, Tomczyk M. New insights into dietary supplements used in sport: active substances, pharmacological and side effects. Curr Drug Targets. 2013;14(9):1079–1092. | ||
Panossian A, Wikman G. Pharmacology of Schisandra chinensis Bail: an overview of Russian research and uses in medicine. J Ethnopharmacol. 2008;118(2):183–212. | ||
Li YJ, Takizawa H, Azuma A, et al. Disruption of Nrf2 enhances susceptibility to airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clinical Immunology. 2008;128(3):366–373. | ||
Iyanagi T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: implications for detoxification. Int Rev Cytol. 2007;260:35–112. | ||
Zhang Q, Pi J, Woods CG, Andersen ME. Phase I to II cross-induction of xenobiotic metabolizing enzymes: a feedforward control mechanism for potential hormetic responses. Toxicol Appl Pharmacol. 2009;237(3):345–356. | ||
Siegel D, Yan C, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem Pharmacol. 2012;83(8):1033–1040. | ||
Zhu H, Li Y. NAD(P)H:quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc Toxicol. 2012;12(1):39–45. | ||
Wilks A, Heinzl G. Heme oxygenation and the widening paradigm of heme degradation. Arch Biochem Biophys. 2014;544:87–95. | ||
Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005;280(40):33766–33774. | ||
Board PG, Menon D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim Biophys Acta. 2013;1830(5):3267–3288. | ||
Wang Z, Dou X, Li S, et al. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology. 2014;59(4):1381–1392. | ||
Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1–2):42–59. | ||
Wong K, Ma J, Rothnie A, Biggin PC, Kerr ID. Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem Sci. 2014;39(1):8–16. | ||
Sahoo S, Aurich MK, Jonsson JJ, Thiele I. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front Physiol. 2014;5:91. | ||
Zhou SF. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica. 2008;38(7–8):802–832. | ||
Zhou SF, Wang LL, Di YM, et al. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15(20):1981–2039. | ||
Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of P-gp and MRP1 to control multidrug resistance (MDR). Curr Drug Target. 2006;7(7):893–909. | ||
Nies AT, Keppler D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 2007;453(5):643–659. | ||
Russel FG, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008;29(4):200–207. | ||
Yoo HH, Lee M, Lee MW, Lim SY, Shin J, Kim DH. Effects of Schisandra lignans on P-glycoprotein-mediated drug efflux in human intestinal Caco-2. Planta Med. 2007;73(5):444–450. | ||
Madgula VL, Avula B, Choi YW, et al. Transport of Schisandra chinensis extract and its biologically-active constituents across Caco-2 cell monolayers – an in-vitro model of intestinal transport. J Pharm Pharmacol. 2008;60(3):363–370. | ||
Sun M, Xu X, Lu Q, Pan Q, Hu X. Schisandrin B: a dual inhibitor of P-glycoprotein and multidrug resistance-associated protein 1. Cancer Lett. 2007;246(1–2):300–307. | ||
Qiangrong P, Wang T, Lu Q, Hu X. Schisandrin B – a novel inhibitor of P-glycoprotein. Biochem Biophys Res Commun. 2005;335(2):406–411. | ||
Svoboda M, Riha J, Wlcek K, Jaeger W, Thalhammer T. Organic anion transporting polypeptides (OATPs): regulation of expression and function. Curr Drug Metab. 2011;12(2):139–153. | ||
Obaidat A, Roth M, Hagenbuch B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol. 2012;52:135–151. | ||
Iusuf D, van de Steeg E, Schinkel AH. Functions of OATP1A and 1B transporters in vivo: insights from mouse models. Trends Pharmacol Sci. 2012;33(2):100–108. | ||
Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos. 2013;34(1):45–78. | ||
Tamai I, Nakanishi T. OATP transporter-mediated drug absorption and interaction. Curr Opin Pharmacol. 2013;13(6):859–863. | ||
Lee SB, Kim CY, Lee HJ, Yun JH, Nho CW. Induction of the phase II detoxification enzyme NQO1 in hepatocarcinoma cells by lignans from the fruit of Schisandra chinensis through nuclear accumulation of Nrf2. Planta Med. 2009;75(12):1314–1318. | ||
Park SY, Park da J, Kim YH, Kim Y, Choi YW, Lee SJ. Schisandra chinensis a-iso-cubebenol induces heme oxygenase-1 expression through PI3K/Akt and Nrf2 signaling and has anti-inflammatory activity in Porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. Int Immunopharmacol. 2011;11(11):1907–1915. | ||
Checker R, Patwardhan RS, Sharma D, et al. Schisandrin B exhibits anti-inflammatory activity through modulation of the redox-sensitive transcription factors Nrf2 and NF-kappaB. Free Radic Biol Med. 2012;53(7):1421–1430. | ||
Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280(36):31768–31775. | ||
Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278(7):4536–4541. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.