Back to Journals » Drug Design, Development and Therapy » Volume 14
Scalable in silico Simulation of Transdermal Drug Permeability: Application of BIOiSIM Platform
Authors Maharao N ![]() , Antontsev V, Hou H, Walsh J, Varshney J
, Antontsev V, Hou H, Walsh J, Varshney J ![]()
Received 10 March 2020
Accepted for publication 7 May 2020
Published 11 June 2020 Volume 2020:14 Pages 2307—2317
DOI https://doi.org/10.2147/DDDT.S253064
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Neha Maharao,* Victor Antontsev,* Hypatia Hou, Jason Walsh, Jyotika Varshney
VeriSIM Life Inc., San Francisco, CA, USA
*These authors contributed equally to this work
Correspondence: Jyotika Varshney
VeriSIM Life Inc, 315 Montgomery St,10th Floor, San Francisco, CA 94104 USA
Email [email protected]
Introduction: Transdermal drug delivery is gaining popularity as an alternative to traditional routes of administration. It can increase patient compliance because of its painless and noninvasive nature, aid compounds in bypassing presystemic metabolic effects, and reduce the likelihood of adverse effects through decreased systemic exposure. In silico physiological modeling is critical to predicting dermal exposure for a therapeutic and assessing the impact of different formulations on transdermal disposition.
Methods: The present study aimed at developing a physiologically based transdermal platform, “BIOiSIM”, that could be globally applied to a wide variety of compounds to predict their transdermal disposition. The platform integrates a 16-compartment model of compound pharmacokinetics and was used to simulate and predict drug exposure of three chemically and biologically distinct drug-like compounds. Machine learning optimization was composed of two components: exhaustive search algorithm (coarse-tuning) and descent (fine-tuning) integrated with the platform used to quantitatively determine parameters influencing pharmacokinetics (eg permeability, kperm) of test compounds.
Results: The model successfully predicted drug exposure (AUC, Cmax and Tmax) following transdermal application of morphine, buprenorphine and nicotine in human subjects, mostly with less than two-fold absolute average fold error (AAFE). The model was further able to successfully characterize the relationship between observed systemic exposure and intended pharmacological effect. The predicted systemic concentration of morphine and plasma levels of endogenous pain biomarkers were used to estimate the effectiveness of a given therapeutic regimen.
Conclusion: BIOiSIM marks a novel approach to in silico prediction that will enable leveraging of machine learning technology in the pharmaceutical space. The approach to model development outlined results in scalable, accurate models and enables the generation of large parameter/coefficient datasets from in vivo clinical data that can be used in future work to train quantitative structure activity relationship (QSAR) models for predicting likelihood of compound utility as a transdermally administered therapeutic.
Keywords: transdermal absorption, computational modeling, machine learning, BIOiSIM
Introduction
Transdermal drug delivery (TDD) has been gaining traction over recent years due to increasing incidences of chronic diseases, technological development in health care and biotechnology, advent of new minimally invasive products, and a rise in regulatory approval of medical devices.1 In 2018, the global TDD system market was estimated at $5.8 billion and is projected to reach about $8 billion by 2025.2 Transdermal application poses a safer, widely accepted alternative to oral and parenteral administration of therapeutics, specifically hypodermic injections, and is also highly relevant to the field of cosmetics.1 After application on skin, the drug permeates through the stratum corneum into the epidermal and dermal layers (see Figure 1), and enters the systemic circulation through the dermal microcirculatory system.3,4 Xenobiotics with poor oral bioavailability can be potentially administered transdermally as the route circumvents first-pass metabolic pathways and provides minimal systemic exposure to avoid undesired side effects.1 Transdermal application can also enable dosing in patients who are unconscious or are susceptible to gastric disorders, and is associated with better patient compliance.1 There has been rising interest in using TDD as an inexpensive and painless method for administering vaccines and other protein/large molecule drugs.
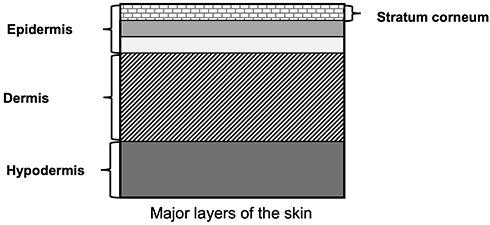 |
Figure 1 Major layers of human skin. Note: The figure was generated in house and data from Tortora and Derrickson.4 |
According to the FDA, transdermal dosage forms can be characterized as semi-solid (cream, ointment, gel, paste, powder), liquid (emulsion, lotion, solution, spray, suspension) and “special formulation” (microemulsion, nanoemulsion, patch).5 Most of these formulations are designed to ensure steady drug delivery to the active site by exerting a local effect following topical application. However, the formulation can be modified by adding excipients that aid deeper drug entry to the deeper skin layers and possibly into systemic circulation. The formulation composition has a significant impact on dermal absorption; one such study conducted by Stahl et al observed that ibuprofen (5%) exhibited highest transdermal absorption when administered as an aqueous solution and gel (containing 2-propanol) followed by the cream (with propylene glycol).5 The transdermal route is mostly suitable for drugs with a certain degree of lipophilicity, limiting its use in administering hydrophilic xenobiotics. However, innovative techniques such as chemical enhancers (propylene glycol, sodium lauryl sulfate, ethanol), biochemical enhancers (magainin), electroporation, iontophoresis, ultrasound, microneedles and thermal ablation have shown promise in improving TDD for drugs that are low or moderately lipophilic by maintaining steady drug concentrations while simultaneously minimizing adverse effects associated with high systemic exposure.3,5 The magnitude of complexity in such transdermal formulations needs to be appropriately captured in order to predict absorption kinetics with reasonable confidence.
Numerous test models are available for predicting dermal absorption. Animal models have been by far the most extensively used for performing risk assessment for drugs with topical formulations. However, more recently this inhumane nature of drug testing has met with strong resistance and criticism from industry, academia and regulatory bodies, especially in the EU.6 In addition to the ethical issues, the clinical translatability of animal models to predict pharmacokinetics (PK) in humans is questionable, especially when using traditional allometric scaling practices. There are significant differences in the composition of dermal layers, density of hair follicles and skin thickness that can inevitably lead to distinct absorption profiles in animal species compared to humans.7
Computational modeling has been widely used to predict and simulate systemic exposure following topical administration of a compound.8–12 However, the existing paradigm results in models that are extremely drug-specific and with physiological considerations relevant only for the given drug, limiting their scope of application (as evidenced by the plethora of PBPK publications with distinct methodologies).8–11 Most current transdermal models are complex, modeling the skin as a series of interconnected compartments with parameters that can vary significantly between species and subjects; while useful in specific applications where these parameters have been characterized to a high degree of accuracy with in vitro or in vivo studies, unfortunately the requirement to capture excessively drug-specific parameters results in models that are highly data-intensive and unscalable. These inputs, which can span physics associated with drug diffusion, binding, and permeation through the dermal layers, further introduce variability in the prediction and make the models more error-prone. Effectively, the outputs obtained from such a complex model are difficult to interpret because they can be frequently overfitted and thus the underlying mechanistic factors contributing to the observed results cannot be correctly identified. This tends to constrain the scope of model application to only a single drug under very specific experimental conditions.
VeriSIM Life (VSL) has developed BIOiSIM, a dynamic, biology-driven platform which provides a scalable computational solution through the use of machine learning (ML) integrated physiological modeling to make accurate and faster predictions that can be applied to larger compound datasets. Integration of ML with mechanistic modeling allows BIOiSIM to fill in missing data gaps commonly found in biological datasets. The methodology is built on creating mechanistic computational models which incorporate only the key parameters that influence drug disposition, thus broadening their domain of applicability. The aim of this study was to develop and validate a simple and physiologically relevant transdermal model built using BIOiSIM. The model was further applied to describe the relationship between systemic exposure and intended therapeutic effect. A dataset comprising three biologically and chemically distinct drug-like compounds with prevalent transdermal application was chosen to validate the model; namely nicotine, morphine, and buprenorphine.
Methods
Overview of BIOiSIM Platform
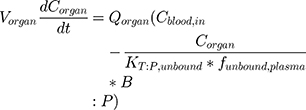
BIOiSIM is a dynamic drug PK prediction platform that learns, adapts and refines its predictions with each iteration of training. The core of the platform is a 16-compartment physiological, mechanistic model with compartments corresponding to vital organs and fluid reservoirs in the body (Figure 2), and ordinary differential equations used to define the standard mass balance relationships across the compartments (as a function of organ-specific fluid dynamics, physiological parameters, and partitioning), an approach described previously.13 Derived from mass transport, these equations follow the general form:
(1)
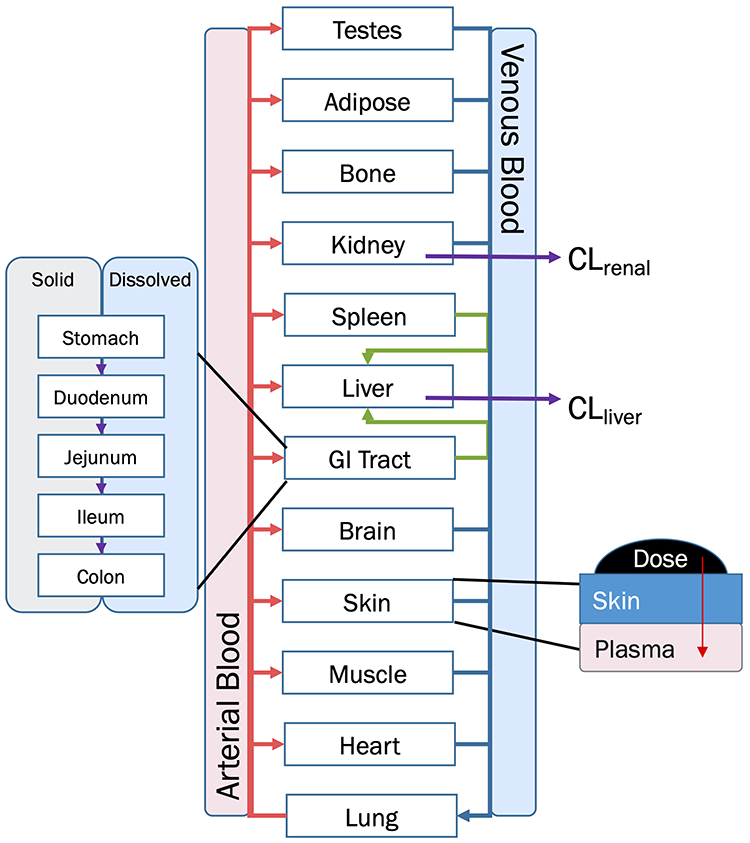 |
Figure 2 Overview diagram of BIOiSIM Mechanistic Model. Note: Some compartments are omitted for clarity. Abbreviations: CLliver, hepatic clearance; CLrenal, renal clearance. |
for distribution compartments, and for metabolizing and clearance compartments (such as liver and kidney):
(2)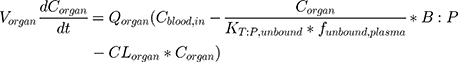
where organ-specific parameters include V (volume), Q (flow rate), CL (organ-level clearance), K (organ-plasma unbound partition coefficient); B:P is blood:plasma binding, and C is the concentration in a specific compartment. The main interactions characterized with the model are passive diffusion into and out of organs driven by concentration gradient of the compound species, fluid dynamics and mass balances across organ compartments, and clearance in hepatic and renal compartments.
The model has been validated using an internal test set of structurally diverse drug-like compounds. In keeping with the principles of good modeling (“as simple as possible, but no simpler”), the BIOiSIM model includes additional complexity (eg enzyme-specific metabolism, transporter dynamics) only for cases where those model inputs are reliable and the presence of these mechanisms is associated explicitly with the PK of a specific compound class. Model development is guided relative to its performance across the training set, thus ensuring that it is complex enough to capture drug-specific disposition in a mechanistic/semi-mechanistic manner and to use ML techniques to refine and extend the model beyond one specific drug or species. The system-dependent parameters capture physiological characteristics (perfusion rate, organ composition, organ volume, etc) specific for a given species—human or animal. Further characterization in the physiological parameters was achieved by performing simulations using values specific for the species, ethnicity, and gender used in the observed clinical study to make translatable predictions across different populations. The physiological parameters were derived from previously published reputable sources.14–18 A set of well-defined and experimentally determined drug-dependent parameters (clearances, unbound plasma fractions, bioavailability, log P, pKa, solubility, etc) incorporate relevant information about the drug’s physicochemical and PK properties to enable prediction of their absorption, distribution, metabolism, and excretion (ADME) mechanisms.19–31 Another set of important drug-dependent parameters are tissue-to-plasma partition coefficients (Kp) for the organs included in the mechanistic model. Kp values characterize the pattern of drug distribution throughout the body and degree of accumulation at specific anatomical locations. The platform also stores study design related information such as dosing regimen, patient population, and formulation characteristics to simulate the observed study as closely as possible. BIOiSIM allows incorporation of variability in drug and system dependent parameters to predict exposure in target populations in lieu of a “one-size-fits-all” approach. In this study, the variability and sensitivity evaluation was focused on only the unknown drug-specific parameters and demonstrated using convergence plots of error associated with outputs AUC, Cmax, and tmax. Biological datasets with missing drug parameters are simultaneously optimized using ML models trained on pharmacological outputs. BIOiSIM is cloud-based, and translates to faster computation and a linearly scalable overall high throughput.
Transdermal Modeling
Key assumptions were made when modeling transdermal drug permeation regarding both the skin physiology and physics. For the purpose of this study, skin was treated as a uniform barrier—this assumption is predicated on the assumption that the cornified layer (stratum corneum) of the skin is the main barrier to compound diffusion for intact skin, and that in de-epithelialized skin the dermis is the rate-limiting barrier. The skin was treated as an isotropic and static barrier at a constant hydration level. Passive, one-dimensional Fickian diffusion was assumed to be the dominant transport mechanism, with compounds undergoing no chemical/enzymatic reactions, and species in the formulation were assumed to be mostly nonionized.32 The plasma compartment was acting as an infinite sink when describing the concentration gradient attained during topical exposure to a therapeutic—ie, the concentration of drug at the surface of the skin was significantly higher than the concentration of drug in the plasma compartment. It was assumed that there was a single rate-limiting barrier to diffusion: the stratum corneum. The compound vehicle was assumed to be well-mixed—concentration of compound uniform at any point in the vehicle—nonevaporating, and not containing any penetrants, moisturizers or other agents that alter skin/stratum corneum properties, moisture content, or clearance.
The amount of compound that is available for transdermal absorption was defined by two variables: the dose (a concentration of compound in formulation) and the surface area of application. Nicotine and buprenorphine were administered as a patch and had no information regarding the thickness of the patch applied or the concentration of compound within it; given that the main force behind passive diffusion is the concentration gradient across a barrier, the patch thickness for both buprenorphine and nicotine was assumed to be 100 μm and an effective administration volume was calculated from the surface area of application and thickness to enable concentration calculation.
There are two compound-barrier specific parameters that describe the time-dependent kinetics and extent of transdermal absorption of a compound: the skin barrier permeability (kperm, cm/h) and compound dermal bioavailability (Fderm). Most changes in transdermal permeation of compounds can be ascribed to different factors influencing these parameters. This includes environmental factors (eg humidity) and formulation (eg moisturizers).33 The relation can be summarized as:
(3)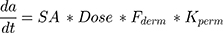
where dA/dt represents the rate of mass transport, and SA is the surface area of formulation application. Optimization for missing parameters in the system was done with proprietary ML algorithms (based on Monte Carlo and derivative optimization) that converged on optimal parameter values consistent across the different routes and doses for a compound.
Test Dataset
A chemically diverse dataset with distinct pharmacological indications was chosen to validate the model. Tables 1 and 2 summarize information on some basic physicochemical properties of the test drugs essential to their PK simulation. These compounds were selected based on the availability of PK parameters (protein binding, clearance, dissociation), their chemical diversity, and the robustness of in vivo human datasets.
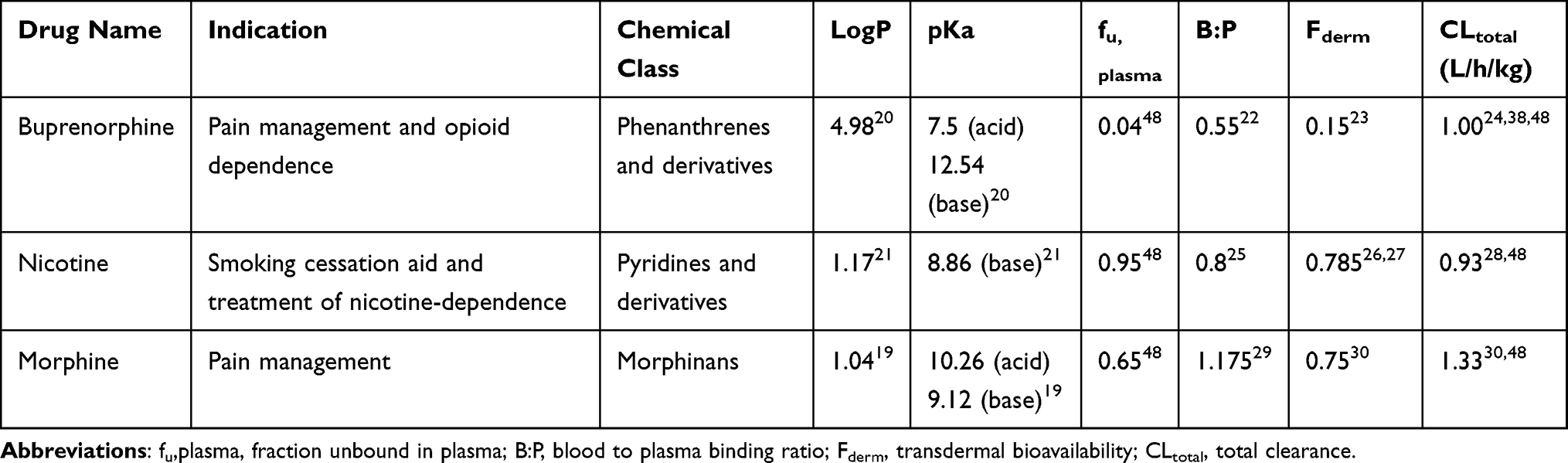 |
Table 1 Physicochemical and Physiological Properties of Compounds |
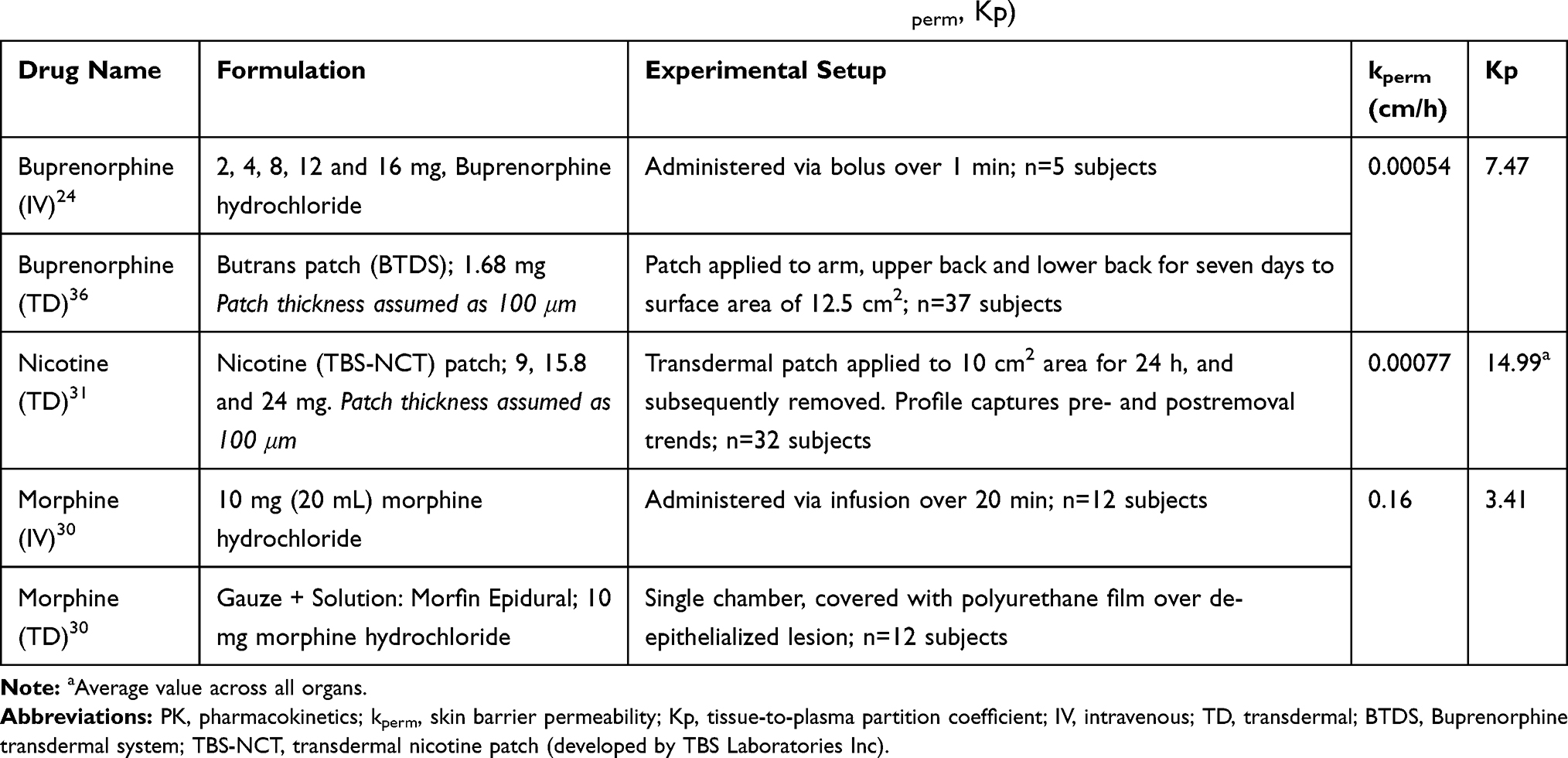 |
Table 2 Experimental Setup and BIOiSIM-optimized PK Parameter Values (kperm, Kp) |
Subjects
The species-specific parameters (eg organ flow, volume, composition) for the different physiological compartments in the BIOiSIM model were adapted from reputed literature sources.14–18
Statistics and Tools
Datasets (including error bars) were manually digitized from source publications using “WebPlotDigitizer” version 4.2.34 All model development and validation was done in Python; libraries used included matplotlib (v2.0.2) and Numpy (v1.14.2). Microsoft Excel (2016) and GraphPad Prism version 8.4.1 (GraphPad Software, San Diego, CA, USA) were utilized for calculations and statistical analysis. Sensitivity of the model to optimized parameters was evaluated using convergence plots generated during optimization of the parameters. Model validation and analysis of model goodness-of-fit/accuracy was conducted using three quantitative metrics: absolute average fold error (AAFE), average fold-error (AFE), and chi-squared statistic (χ2) and associated p-value (null hypothesis defined as no difference in predicted vs experimental measurements). AAFE and AFE were utilized to evaluate the accuracy of the PK outputs AUC, Cmax, and tmax using the general equations:
(4) (5)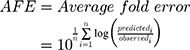
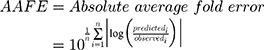
where n is the total number of compounds used in the analysis, and Predicted_i/Observed_i correspond to predicted and observed values of PK parameters, respectively. Chi-squared was calculated using the relationship:
(6)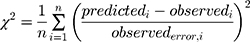
where observederror is defined as standard error in the measurements in the individual experimental data timepoints, obtained by digitizing the error bars from the respective publications. Optimization convergence was driven and measured by AAFE of AUC, Cmax, and tmax.
In addition to quantitative metrics, visual comparison of the ascending and terminal portions of the PK curves as well as tests of significance comparison of the curve plots against the standard error of the experimental measurements was conducted.
Results
Simulation Accuracy
To characterize the performance of the base model simulation across different compounds, comparison between experimental data (with standard error as extracted from the references) and the simulation profiles was conducted using AAFE, χ2, p-value and visual analysis of the goodness-of-fit (summarized in Table 3). Buprenorphine profiles were predicted accurately across different doses for IV administration, and translated well between transdermal and IV routes, with all outputs predicted within 1.5 average absolute fold error (AAFE, see Table 3). Visualization of the plots indicates that the BIOiSIM platform captured both the early-phase and terminal profiles well (Figure 3A) and within the confidence interval, as defined by the standard error bars.
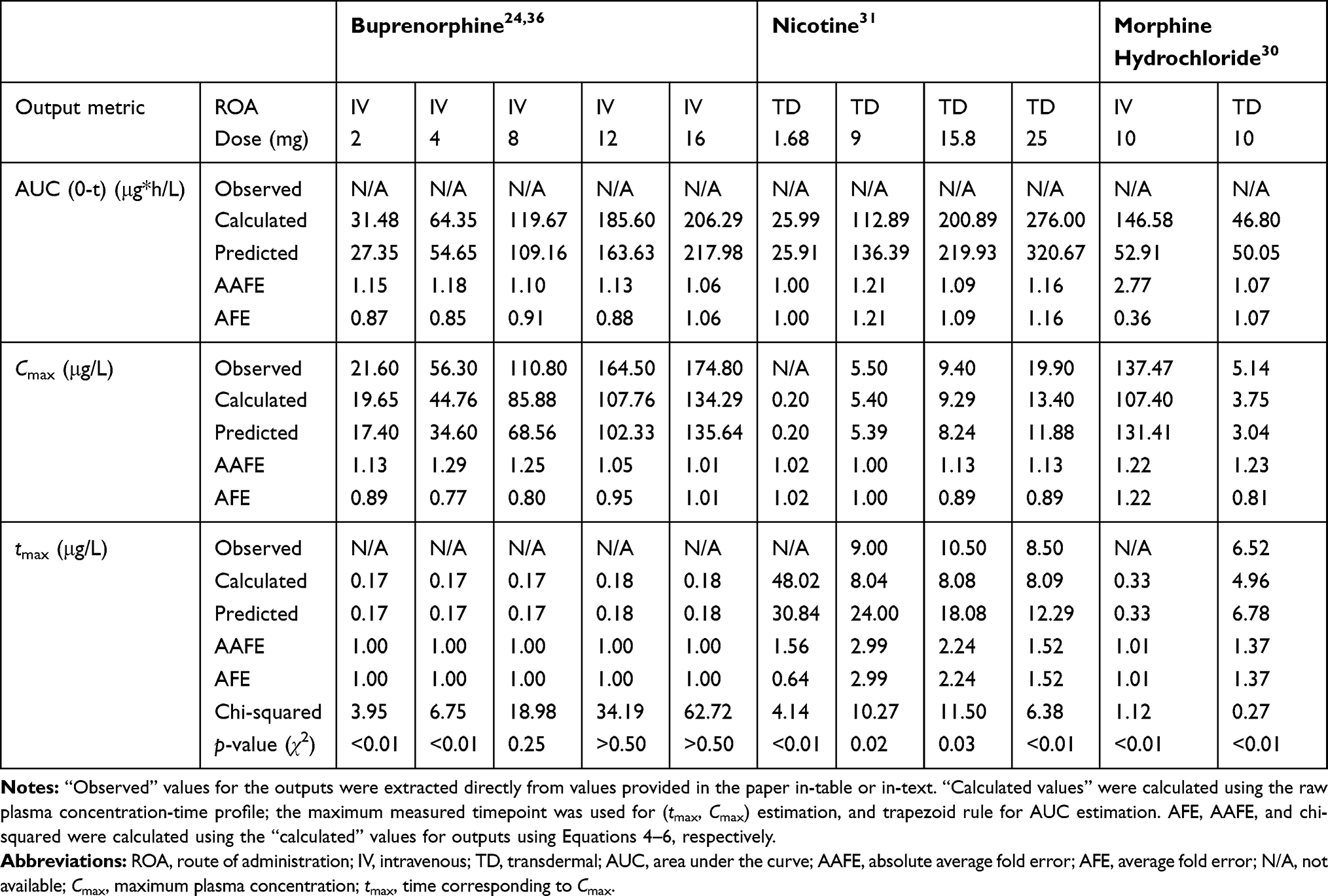 |
Table 3 Comparison of Model-predicted Outputs to Experimentally Derived Outputs |
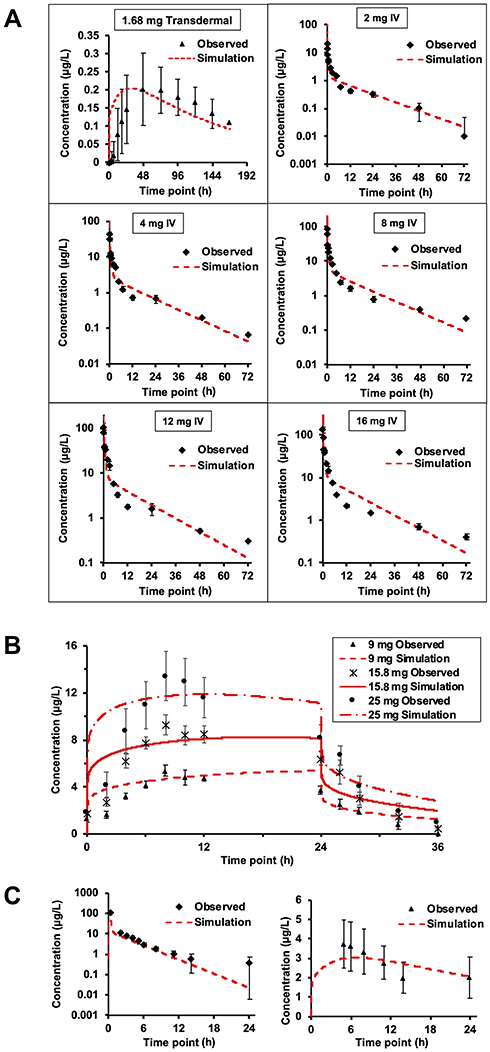 |
Figure 3 Dose and route-of-administration dependent prediction of compound plasma concentration for (A) Buprenorphine, transdermal administration (top left) and IV administration (different doses). (B) Nicotine transdermal administration for three doses; (C) Morphine, IV administration (left) transdermal administration (right). Red lines correspond to BIOiSIM simulation outputs. Error bars and individual data points were digitized from the original publications (citations in output Table 3), and correspond to standard error/standard deviation, as presented in the original work. Abbreviation: IV, intravenous. |
For nicotine, the transdermal profiles across the different doses are of similar accuracy (average AAFE: AUC = 1.15 ± 0.05, Cmax = 1.09 ± 0.06, tmax = 2.25 ± 0.60) indicating that the mechanistic model captured the appropriate mechanisms involved in drug disposition (Figure 3B). Prediction of morphine disposition across the two routes of administration was consistent, and evident after inspection of the log-linear plots (Figure 3C). The AAFE for AUC0-t post-IV administration was higher; although methodology of the paper indicated that the concentration the researchers obtained post administration was higher, that section of the profile was not clearly depicted in the publication and therefore excluded during parameter optimization.
Predicted Morphine Dose
The topical dose of morphine used in this study showed a maximum predicted Cmax plasma concentration of 3.04 μg/L. Morphine is reported to have a minimum effective concentration of 10 μg/L to treat pain.35 BIOiSIM prediction dictates that a dose of approximately 33 mg would be required to maintain the analgesic effect of topical morphine. It should be noted that this dose suggestion assumes that the volume of the formulation remains constant; given that passive diffusion is driven entirely by the concentration gradient across some barrier, it is critical that the driving force have this scalar increase in magnitude to maintain similar PK.
Sensitivity and Convergence Testing
To evaluate how well the optimization algorithms were able to converge on a confident set of parameter values, convergence plots were generated to compare absolute minimum with regard to cost. For each compound, a discrete parameter combination was converged upon during coarse optimization (Figure 4). For morphine, these values were: [kperm = 0.1, Kp = 4]; for buprenorphine: [kperm = 0.001, Kp = 10]; and for nicotine: [kperm = 0.001, Kp = 20]. For buprenorphine, variance around the converged parameter kperm = 0.001 cm/h combinations was highest amongst the compounds (39%), indicating that population variability in organ composition (lipids, water) could have a more significant effect on the disposition of the compound in vivo. For nicotine, variance around kperm = 0.001 cm/h was 7.3% across different coefficients. For morphine, at the fixed permeability of 0.1 cm/h the sensitivity to changes in Kp were relatively low; from the set of Kp tested {0.4, 1, 4, 10, 40} the variance in the cost was 19%, minimal given the significant deviations in partition coefficients tested. This gives higher confidence in the utility of model predictions, as small variations in populations are unlikely to have an impact.
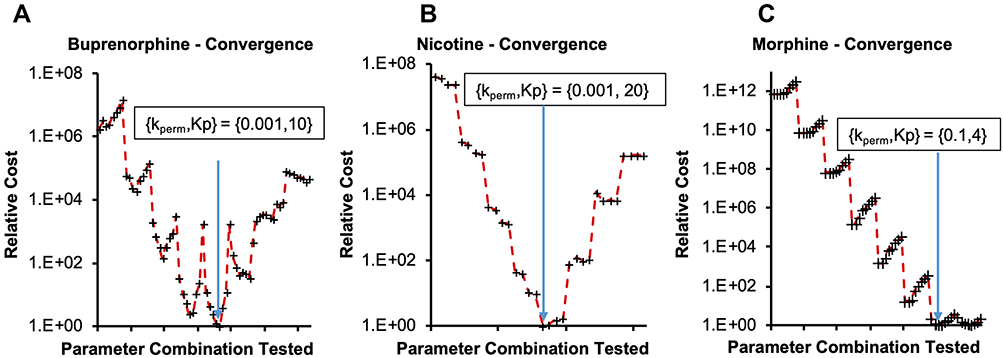 |
Figure 4 Convergence onto discrete parameter combinations during coarse optimization for (A) buprenorphine, (B) nicotine and (C) morphine. Relative cost on the y-axis is calculated as: Costrelative = AAFECmax*AAFEtmax*AAFEAUC, normalized to the lowest Cost calculation for a parameter combination. Abbreviations: kperm, skin barrier permeability; Kp, tissue-to-plasma partition coefficient; AAFECmax, AAFEtmax; AAFEAUC, absolute average fold error; Cmax, maximum plasma concentration; tmax, time corresponding to Cmax; AUC, area under the curve predictions. |
Discussion
In recent years, transdermal drug delivery has been gaining popularity as a painless and noninvasive route of delivery with ease of access due to the large surface area of the skin. Computational modeling has been extensively used for developing transdermal therapeutics; however, several of these published approaches have a restrictive scope of application. Owing to their excessively specific nature, these models are useful only under limited experimental conditions and often only to a small set of compounds. Hence, the aim of this study was to develop a model that can be globally employed to predict transdermal disposition of a wide variety of compounds. As a proof of concept, the disposition of three physicochemically distinct and popular transdermal drugs, ie buprenorphine, morphine, and nicotine was successfully predicted using a novel ML-integrated modeling platform, BIOiSIM. Unlike current modeling approaches, BIOiSIM’s proprietary ML algorithms can help fill in knowledge gaps by optimizing missing experimental information within the bounds of physiology.
The model successfully predicted the disposition of buprenorphine post intravenous and transdermal application. The transdermal validation data from the published study was significantly variable (CV=0.403).36 However, in spite of this experimental variability, the predicted results match the profile closely and even the tmax AAFE (1.56) is similar. While the lower doses of the compound were accurately simulated (p-values <0.05), doses greater than 4 mg had a higher p-value, indicating that the predicted values were not as similar. Given the goodness-of-fit seen in visual analysis, it is possible that small differences in the sampling timepoints (not accounted for in the probability analysis) can lead to the differential chi-squared calculation at the individual points being higher than expected. Additionally, the variability in experimental physiological parameters is not surprising, given the difference in skin composition/thickness between different application sites (the study involved applying patches to the arm, upper/lower back) and the variability in clearance between different patients; interestingly, however, the simulation results using the same parameters closely matched even though the data for buprenorphine comes from two distinct studies (and effectively two distinct healthy patient populations), indicating that there may be a normal distribution for parameters across these patient populations.37–40
Similarly to buprenorphine, the PK of nicotine was well-predicted by the BIOiSIM model. However, the ascending phase of the curve was captured better for high doses, indicating that there may be some change in accuracy as a result of the lower limit of detection (the higher error at higher doses is indicative of this as well). The terminal phase was modeled with the assumption that at t=24 h (upon the removal of the patch), the topical dose disappears instantaneously; it is possible that there is a temporary reservoir remaining after patch removal in the in vivo studies simply because of compound partitioning into the skin local to the site of administration. The terminal phase simulation differs slightly from the experimental data, indicating that either the average experimental clearance value used for prediction or the population used in the study deviated from a normally distributed subset. Overall, translation between different doses (9, 15.8, and 24 mg) and accurate fit of the profiles (AAFE <1.3 for AUC, Cmax; AAFE <3 for tmax; p-values <0.05 for all doses) is evidence of the model’s predictive capabilities.
Morphine was chosen to demonstrate the utility of extrapolating the PK disposition to predict effectiveness of observed pharmacodynamic effects. We selected morphine because it has proven to be an effective analgesic agent and researchers are increasingly studying its topical applications to treat inflammation and pain in palliative care settings.41,42 Morphine administration was modeled accurately by the base model across both the IV and transdermal route of administration, as measured by AAFE <3.0 for AUC0-t and AAFE <1.5 for Cmax, tmax across both experimental profiles. The higher optimized permeability value (0.16 cm/h) is expected given that the skin at the application site was de-epithelialized prior to compound administration.
This is evidenced additionally by a visual interrogation of the plasma concentration-time profile, where the predicted datapoints fall within the confidence interval for the experimental measurements, especially in the terminal phase. The p-value based on the reduced chi-squared metric is <0.01 for both doses, indicating that there significant similarity between the predicted and observed profiles.
The premise that topical opioids can prove effective in treating pain is based on data that indicates the presence of a similar population of opioid receptors on peripheral nerve terminals, as its central nervous system counterpart.35 For successful pain management, it is essential to strike a balance between achieving pain relief without experiencing excessive undesirable effects. Higher systemic exposure to opiates like morphine is associated with adverse effects such as nausea, constipation, respiratory depression, cognitive impairment, etc.43 Transdermally dosed morphine was successful in managing chronic pain in patients suffering from arthritis pain. In this study, the authors detected the presence of morphine (31–191 ng/mL) in the urine of three arthritis patients over time, suggesting possible contribution of systemically absorbed morphine to the observed analgesic effect.44 More recently, Ciałkowska-Rysz et al found self-administered topical morphine to be a highly effective and safe medication for treating mucosal and skin lesions in palliative cancer patients, providing sustained pain relief for over 28 days.41 The studies discussed above also suggest that the transdermal route is associated with a higher patient compliance as it offers evasive dosing, the possibility of self-administration, and flexibility in adjusting dosing regimen to experience rapid and long-lasting pain relief, hence the interest in accurate modeling and simulation.
Interestingly, while the type of formulation for administration varied between the different compounds—buprenorphine and nicotine were applied via patches, while morphine was applied as a solution to de-epithelialized skin—the prediction accuracy was still high (AAFE=1.38 across outputs) for all of the compounds, indicating that the assumption of a single rate-limiting barrier holds valid across different experimental setups. The validated model can be utilized to optimize therapeutic regimens by incorporating subject-specific parameters (eg polymorphic metabolism, disease conditions, gender, age, etc) that capture population variability and can cater to personalized medical needs. With this validation, predictions from BIOiSIM can be further correlated to the biomarker levels observed in healthy and diseased patients to obtain valuable pharmacodynamic insights. Following the morphine example above, the biopathway of pain and nociceptor response can be characterized by the presence of several endogenous biomarkers such as cystatin C, substance P, nociception, tau proteins and others.45–47 A study by Albo et al found that there is a marked elevation in the tryptase and substance P levels in patients with chronic pain.46 The levels of such endogenous mediators can be linked to the expected drug concentrations in plasma or active site to gain a deeper understanding of the interaction of drug with the active site components and their downstream pharmacological effects—integration of these biomarkers with VSL’s BIOiSIM platform can extend the prediction of safety and efficacy to drug therapies in healthy and diseased populations.
Conclusion
A powerful outcome that BIOiSIM enables from this approach is the generation of datasets that can be utilized for ML training and prediction. Work from Lombardo et al, has focused on accumulating datasets from existing literature to aid these types of efforts, and the parameters optimized utilizing the framework applied in this work augments the existing efforts.48 BIOiSIM presents researchers with the opportunity to extend this type of approach to internal datasets and to continually re-analyze databases to extract parameters that were not explicitly measured or calculated previously. This is evidenced by the extraction of partition coefficient values and, more specifically, kperm from datasets where these parameters were not of explicit interest to researchers at the time. The accumulated dataset can afterwards be used for training QSAR models to predict the likelihood of specific characteristics of xenobiotics (eg solubility, ionization state, molecule size) contributing to successful transdermal drug delivery. Vehicle characteristics (eg lotions, patches) can be evaluated as well, and cross-validation between different therapeutics and formulations will enable researchers to make prediction of transdermally administered compound success either at an earlier stage in drug development or for drug repurposing without the need for testing in preclinical in vivo subjects.
Acknowledgments
Neha Maharao and Victor Antontsev have contributed equally and are co-first authors on this manuscript.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
All authors are affiliated with VeriSIM Life Inc. Dr Jyotika Varshney reports a patent WO/2019/104101 pending. The authors report no other conflict of interest in this work.
References
1. Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008;26(11):1261–1268. doi:10.1038/nbt.1504
2. Zion Market Research. Global transdermal drug delivery system market will reach USD 8,127 Million By 2025: zion Market Research. Available from: https://www.globenewswire.com/news-release/2019/09/05/1911711/0/en/Global-Transdermal-Drug-Delivery-System-Market-Will-Reach-USD-8-127-Million-By-2025-Zion-Market-Research.html.
3. Brown MB, Martin GP, Jones SA, Akomeah FK. Dermal and transdermal drug delivery systems: current and future prospects. Drug Deliv. 2006;13(3):175–187. doi:10.1080/10717540500455975
4. Tortora GJ, Derrickson BH. Principles of Anatomy and Physiology.
5. Stahl J. Dermal and Transdermal Formulations: How They Can Affect the Active Compound. Springer-Verlag Berlin Heidelberg;2015.
6. Fischer K. animal testing and marketing bans of the EU cosmetics legislation. Eur J Risk Regul. 2015;6(4):613–621. doi:10.1017/S1867299X00005158
7. Todo H. transdermal permeation of drugs in various animal species. Pharmaceutics. 2017;9:3. doi:10.3390/pharmaceutics9030033
8. Polak S, Ghobadi C, Mishra H, et al. Prediction of concentration-time profile and its inter-individual variability following the dermal drug absorption. J Pharm Sci. 2012;101(7):2584–2595. doi:10.1002/jps.23155
9. Zhang F, Bartels M, Clark A, et al. Performance evaluation of the GastroPlus(TM) software tool for prediction of the toxicokinetic parameters of chemicals. SAR QSAR Environ Res. 2018;29(11):875–893. doi:10.1080/1062936X.2018.1518928
10. Yamashita F, Hashida M. Mechanistic and empirical modeling of skin permeation of drugs. Adv Drug Deliv Rev. 2003;55(9):1185–1199. doi:10.1016/S0169-409X(03)00118-2
11. Chen T, Lian G, Kattou P. In silico modelling of transdermal and systemic kinetics of topically applied solutes: model development and initial validation for transdermal nicotine. Pharm Res. 2016;33(7):1602–1614. doi:10.1007/s11095-016-1900-x
12. Maharao N, Antontsev V, Wright M, Varshney J. Entering the era of computationally driven drug development. Drug Metab Rev. 2020;1–16. doi:10.1080/03602532.2020.1726944
13. Kuepfer L, Niederalt C, Wendl T, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol. 2016;5(10):516–531. doi:10.1002/psp4.12134
14. Integrated Physiology Database. PK-Sim®, Bayer Technology Services GmbH, Leverkusen, Germany. Available from: http://www.systems-biology.com/products/pk-sim.html.
15. US Environmental Protection Agency Office of Health and Environmental Assessment. Physiological Parameter Values for PBPK Models: A Report Prepared by the International Life Sciences Institute and Risk Sciences Institute; 1994.
16. ICRP. Basic anatomical and physiological data for use in radiological protection reference values. Ann ICRP. 2002;32:3–4.
17. Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–484. doi:10.1177/074823379701300401
18. Peters S. Physiologically‐Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry. John Wiley & Sons, Inc.; 2012.
19. Drug bank – Morphine. Available from: https://www.drugbank.ca/drugs/DB00295.
20. Drug bank – Buprenorphine. Available from: https://www.drugbank.ca/drugs/DB00921.
21. Drug bank - Nicotine Available from: https://www.drugbank.ca/drugs/DB00184.
22. Kalluri HV, Zhang H, Caritis SN, Venkataramanan R. A physiologically based pharmacokinetic modelling approach to predict buprenorphine pharmacokinetics following intravenous and sublingual administration. Br J Clin Pharmacol. 2017;83(11):2458–2473. doi:10.1111/bcp.13368
23. Food and Drug Administration (FDA). Prescribing information - BUTRANS (June 2014).
24. Huestis MA, Cone EJ, Pirnay SO, Umbricht A, Preston KL. Intravenous buprenorphine and norbuprenorphine pharmacokinetics in humans. Drug Alcohol Depend. 2013;131(3):258–262. doi:10.1016/j.drugalcdep.2012.11.014
25. Robinson DE, Balter NJ, Schwartz SL. A physiologically based pharmacokinetic model for nicotine and cotinine in man. J Pharmacokinet Biopharm. 1992;20(6):591–609. doi:10.1007/BF01064421
26. Benowitz NL, Chan K, Denaro CP, Jacob P
27. Benowitz NL, Hukkanen J, Jacob P
28. Feyerabend C, Ings RM, Russel MA. Nicotine pharmacokinetics and its application to intake from smoking. Br J Clin Pharmacol. 1985;19(2):239–247. doi:10.1111/j.1365-2125.1985.tb02637.x
29. Skopp G, Potsch L, Ganssmann B, Aderjan R, Mattern R. A preliminary study on the distribution of morphine and its glucuronides in the subcompartments of blood. J Anal Toxicol. 1998;22(4):261–264. doi:10.1093/jat/22.4.261
30. Westerling D, Hoglund P, Lundin S, Svedman P. Transdermal administration of morphine to healthy subjects. Br J Clin Pharmacol. 1994;37(6):571–576. doi:10.1111/j.1365-2125.1994.tb04306.x
31. Lane J, Westman E, Gail R, Wu J, Chiang C, Rose J. Pharmacokinetics of a transdermal nicotine patch compared to nicotine gum. Drug Dev Ind Pharm. 1993;19(16):1999–2010. doi:10.3109/03639049309069337
32. Hadgraft J. Transdermal Drug Delivery Systems: Revised and Expanded. CRC Press; 2002.
33. Iikura H, Uchida K, Ogawa-Fuse C, et al. Effects of temperature and humidity on the skin permeation of hydrophilic and hydrophobic drugs. AAPS PharmSciTech. 2019;20(7):264. doi:10.1208/s12249-019-1481-1
34. Rohatgi A. WebPlotDigitizer; 2019. Available from: https://automeris.io/WebPlotDigitizer.
35. Paice JA, Von Roenn JH, Hudgins JC, Luong L, Krejcie TC, Avram MJ. Morphine bioavailability from a topical gel formulation in volunteers. J Pain Symptom Manage. 2008;35(3):314–320. doi:10.1016/j.jpainsymman.2007.04.016
36. Kapil RP, Cipriano A, Friedman K, et al. Once-weekly transdermal buprenorphine application results in sustained and consistent steady-state plasma levels. J Pain Symptom Manage. 2013;46(1):65–75. doi:10.1016/j.jpainsymman.2012.06.014
37. Elkader A, Sproule B. Buprenorphine: clinical pharmacokinetics in the treatment of opioid dependence. Clin Pharmacokinet. 2005;44(7):661–680. doi:10.2165/00003088-200544070-00001
38. Kuhlman JJ
39. Maharao NV, Joshi AA, Gerk PM. Inhibition of glucuronidation and oxidative metabolism of buprenorphine using GRAS compounds or dietary constituents/supplements: in vitro proof of concept. Biopharm Drug Dispos. 2017;38(2):139–154. doi:10.1002/bdd.2050
40. Maharao N, Venitz J, Gerk PM. Use of generally recognized as safe or dietary compounds to inhibit buprenorphine metabolism: potential to improve buprenorphine oral bioavailability. Biopharm Drug Dispos. 2019;40(1):18–31. doi:10.1002/bdd.2166
41. Ciałkowska-Rysz A, Dzierzanowski T. Topical morphine for treatment of cancer-related painful mucosal and cutaneous lesions: a double-blind, placebo-controlled cross-over clinical trial. Arch Med Sci. 2019;15(1):146–151. doi:10.5114/aoms.2018.72566
42. LeBon B, Zeppetella G, Higginson IJ. Effectiveness of topical administration of opioids in palliative care: a systematic review. J Pain Symptom Manage. 2009;37(5):913–917. doi:10.1016/j.jpainsymman.2008.06.007
43. Cherny N, Ripamonti C, Pereira J, et al. Strategies to manage the adverse effects of oral morphine: an evidence-based report. J Clin Oncol. 2001;19(9):2542–2554. doi:10.1200/JCO.2001.19.9.2542
44. Wilken M, Ineck JR, Rule AM. Chronic arthritis pain management with topical morphine: case series. J Pain Palliat Care Pharmacother. 2005;19(4):39–44.
45. Marchi A, Vellucci R, Mameli S, Rita Piredda A, Finco G. Pain biomarkers. Clin Drug Investig. 2009;29 Suppl 1:41–46. doi:10.2165/0044011-200929001-00006
46. Albo C, Kumar S, Pope M, et al. Characteristics and potential biomarkers of adult sickle cell patients with chronic pain. Blood. 2018;132:857. doi:10.1182/blood-2018-99-115084
47. Cao S, Fisher DW, Yu T, Dong H. The link between chronic pain and Alzheimer’s disease. J Neuroinflammation. 2019;16(1):204. doi:10.1186/s12974-019-1608-z
48. Lombardo F, Berellini G, Obach RS. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 1352 drug compounds. Drug Metab Dispos. 2018;46(11):1466–1477. doi:10.1124/dmd.118.082966
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.