Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Sanfilippo Syndrome: Optimizing Care with a Multidisciplinary Approach
Authors Cyske Z, Anikiej-Wiczenbach P, Wisniewska K, Gaffke L, Pierzynowska K, Mański A ![]() , Wegrzyn G
, Wegrzyn G
Received 7 August 2022
Accepted for publication 6 September 2022
Published 19 September 2022 Volume 2022:15 Pages 2097—2110
DOI https://doi.org/10.2147/JMDH.S362994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Zuzanna Cyske,1 Paulina Anikiej-Wiczenbach,2 Karolina Wisniewska,1 Lidia Gaffke,1 Karolina Pierzynowska,1 Arkadiusz Mański,2 Grzegorz Wegrzyn1
1Department of Molecular Biology, Faculty of Biology, University of Gdansk, Gdansk, 80-308, Poland; 2Psychological Counselling Centre of Rare Genetic Diseases, University of Gdansk, Gdansk, 80-309, Poland
Correspondence: Grzegorz Wegrzyn, Department of Molecular Biology, Faculty of Biology, University of Gdańsk, Wita Stwosza 59, Gdańsk, 80-308, Poland, Tel +48 58 523 6024, Fax +48 58 523 5501, Email [email protected]
Abstract: Sanfilippo syndrome, or mucopolysaccharidosis type III (MPS III), is a disease grouping five genetic disorders, four of them occurring in humans and one known to date only in a mouse model. In every subtype of MPS III (designed A, B, C, D or E), a lack or drastically decreased activity of an enzyme involved in the degradation of heparan sulfate (HS) (a compound from the group of glycosaminoglycans (GAGs)) arises from a genetic defect. This leads to primary accumulation of HS, and secondary storage of other compounds, combined with changes in expressions of hundreds of genes and many defects in organelles and various biochemical processes in the cell. As a result, dysfunctions of tissues and organs occur, leading to severe symptoms in patients. Although changes in somatic organs are considerable, the central nervous system is especially severely affected, and neurological, cognitive and behavioral disorders are the most significant changes, making the disease enormously burdensome for patients and their families. In the light of the current lack of any registered therapy for Sanfilippo syndrome (despite various attempts of many research groups to develop effective treatment, still no specific drug or procedure is available for MPS III), optimizing care with a multidisciplinary approach is crucial for managing this disease and making quality of patients’ life passable. This includes efforts to make/organize (i) accurate diagnosis as early as possible (which is not easy due to various possible misdiagnosis events caused by similarity of MPS III symptoms to those of other diseases and variability of patients), (ii) optimized symptomatic treatment (which is challenging because of complexity of symptoms and often untypical responses of MPS III patients to various drugs), and (iii) psychological care (for both patients and family members and/or caregivers). In this review article, we focus on these approaches, summarizing and discussing them.
Keywords: mucopolysaccharidosis type III, accurate diagnosis, symptomatic treatment, psychological care
Introduction – Brief Overview of Sanfilippo Syndrome
Sanfilippo syndrome (other names: Sanfilippo disease, mucopolysaccharidosis type III, MPS III) is a rare, inherited metabolic disease from the group of mucopolysaccharidoses (MPS), belonging to lysosomal storage diseases (LSD). The primary biochemical defect of MPS III is lysosomal accumulation of partially degraded molecules of heparan sulfate (HS) (a compound belonging to glycosaminoglycans (GAGs)). The HS storage is caused by mutations in genes coding for enzymes involved in the decay of this complex carbohydrate.1–4
There are five subtypes of MPS III (A, B, C, D, and E), classified on the basis of the kind of mutated gene, and deficiency of specific enzyme (Table 1).5–7 Four of these subtypes (A, B, C, and D) have been found in humans, while subtype E is currently known only from studies on an animal model, a mutant mice, constructed in the laboratory, with dysfunctional ARSG gene.8–10 Therefore, although incompletely degraded HS is stored in each Sanfilippo disease subtype, every individual subtype is characterized by inhibition of decay of this GAG at specific stage of the process, being formally a separate metabolic disorder. All these diseases are inherited in an autosomal recessive manner, as residual activity of any enzyme involved in HS degradation at the level of 10–20% of the normal activity may ensure efficient decay process.1–7
 |
Table 1 Subtypes of Sanfilippo Syndrome (Mucopolysaccharidosis Type III; MPS III) |
Because HS is present in virtually all tissues and organs, its storage affects majority of organs. Nevertheless, in Sanfilippo syndrome, symptoms occurring in visceral organs are milder than those in other types of MPS.11,12 They include hepatomegaly, frequent infections of the respiratory system, face dysmorphology, carpal tunnel syndrome, hip dysplasia, general skeletal problems like scoliosis, kyphosis and lumbar lordosis, hirsutism, changes in hair morphology, and others.2 On the other hand, dysfunctions of the central nervous system (CNS) occurring in MPS III are among the most severe ones not only in the group of LSD but also within a large group of neurodegenerative disorders. All MPS III patients, irrespective of the subtype, develop similar CNS-related symptoms, though their intensity may vary considerably between subtypes (with MPS IIIA and IIIB being considered the most severe, and MPS IIIC and IIID somewhat less severe) and between individuals. The most characteristic symptoms of Sanfilippo syndrome include severe developmental delay, cognitive decline, severe delay in speech or a lack of speech, hyperactivity, aggression-like behavior, sleep disorders, and seizures. Importantly, MPS III patients are born without any disease symptoms which develop usually at the age of several months or a few years.1–4 The disease is progressive, and symptoms become more and more severe in time, which leads to a significantly shortened life span, estimated for 2–3 decades on average.2
Unfortunately, despite many attempts and intensive work of researches on various potential therapies, no specific and effective treatment of Sanfilippo syndrome is currently available. Proposed therapeutic approaches include enzyme replacement therapy, substrate reduction therapy, gene therapy, and others, however, none of them revealed sufficient efficacy in clinical trials, and no drug for MPS III has been registered. These problems have been reviewed recently in details by various authors1–6,11,13–17; thus, we will not focus on them in this review article. However, due to severity of the disease and a lack of specific treatment, there are serious problems with managing this condition. Since the state of patients worsens continuously, and the neurological and psychiatric manifestations predominate, affected people require intensive care, round the clock, from the age of a few years to the end of life. Such a disease course is debilitating and stressful not only for patients but also for their families and/or caregivers. Thus, optimizing care with a multidisciplinary approach is crucial for managing this disease and for ensuring quality of life of patients and their families passable. In this paper, we will concentrate on the difficulties of Sanfilippo disease and indicate crucial points and key aspects of MPS III patient care.
Complexity of Sanfilippo Disease Pathomechanisms
Although all subtypes of Sanfilippo syndrome are monogenic diseases, the pathomechanism of this disease is not simple. Despite the fact that the primary biochemical defect is HS storage, this is only the initial point of severe changes that occur in cells of patients, and then in tissues, organs and the whole organisms. Definitely, HS accumulation in lysosomes is not the only, or even the major problem causing dysfunctions of cells, tissues and organs. Obviously, the storage is the onset of the disease, but it triggers a series of other events which eventually lead to dramatic consequences for the physiology of cells and the organism. We suppose that this might be one of reasons for relatively low clinical efficacy of potential therapies, tested to date for MPS III, which despite reducing GAG levels could not remove all symptoms. This concerns various types of therapies, including enzyme replacement therapy (provided intrathecally), gene therapy, and substrate reduction therapy.18–21
The physical storage of HS inhibits lysosomal functions; however, it is hardly possible to explain all cellular and organismal defects observed in Sanfilippo disease solely by accumulation of this GAG.2 Nevertheless, if huge amounts of HS cannot be accommodated inside lysosomes, the undegraded molecules can remain in either the cytoplasm, due to a block in further transport to the target organelle, or outside the cell, where GAGs play their major physiological roles.22 Moreover, overloaded lysosomes might be broken, liberating partially degraded HS molecules into the cytoplasm. Theoretical analyses of chemical properties of incomplete HS decay products led to the proposal that chemical moieties present at the ends of such molecules can be highly reactive and might be involved in biochemical reactions interfering with functions of cells, especially neurons.23 In fact, exposure of cells to exogenous HS fragments resulted in focal adhesion stimulation, followed by pathological modulations of both interactions of cells with the extracellular matrix and their migration orientation.24 Moreover, the presence of high levels of HS was postulated to result in elevated amounts of oxidative stress markers.25 As discussed thoroughly in a recent article,26 oxidative imbalance causes appearance of reactive oxygen species which interact with various biomolecules, causing their damage in MPS cells; if this occurs in neurons, microglial activation and subsequent neurodegeneration are likely.
On the other hand, HS storage can lead to a battery of secondary changes which are considerably more destructive than the primary agglomeration of this GAG. Blockage of lysosomal functions results in severely impaired activities of other acid hydrolases (apart from the mutated enzyme involved in HS decay), like cathepsins,27 which leads to inefficient degradation of various macromolecules, including dermatan sulfate,28 gangliosides,29 and other compounds, like ceramides, galactosylceramides and sphingomyelin.30 All of them can affect cells as severely as HS derivatives, discussed above.
The secondary changes in cell physiology apparently induce stress responses and defensive reactions which alter expressions of many genes. Although such modulations of genes’ activities and resultant modifications of cellular processes were rather underestimated previously,31 recent studies indicated that they can significantly contribute to the pathomechanisms of all MPS types. Sanfilippo syndrome revealed the highest dysregulation of gene expression among MPS diseases, with numbers of down- and up-regulated transcripts (relative to healthy cells) exceeding 700 in every MPS III subtype.32 Such global changes in levels of transcripts and proteins encoded by them result in dysmorphology and dysfunctions of different cellular organelles (like nucleus, mitochondria, endoplasmic reticulum, Golgi apparatus),33 as well as abnormalities in various cellular processes.34 Among them, apoptosis,35 cell activation,36 proteasomal degradation,37 homeostasis of different ions (especially Ca2+, Fe2+ and Zn2+),38 signal transduction,39 and cell cycle,40 are especially strongly affected. It was suggested that even behavioral disorders appearing in Sanfilippo disease and some other MPS types might be partially caused by disturbed regulation of expression of specific genes.41 Interestingly, it appears likely that such extensive changes in expressions of hundreds of genes in MPS cells arise from dysregulation of a group of transcription factors and other agents involved in the control of gene expression.42 Furthermore, some of these changes in gene expression regulation could not be reversed after effective decreasing levels of stored GAGs, suggesting that these dysfunctions are either irreversible or require additional manipulations (along with abolition of the storage) to be corrected.42 Indeed, it was demonstrated experimentally that some cellular abnormalities could not be corrected by reduction of GAG storage; these included changes in organelles,33 and proteasomal functions.37 Very recent investigations demonstrated that some proteins involved in the signal transduction-mediated control of gene expression, like GPER1 and OXTR receptors, form aggregates which arise as effects of their interactions with undegraded GAGs.39 Therefore, we suggest that the cascade of cellular changes in MPS cells may include interactions of specific control agents (like factors involved in gene expression regulation) with GAGs, their inactivation due to formation of complexes and aggregates, subsequent dysregulation of a battery of genes, and resultant abnormalities in structures and functions of cellular organelles as well as in the courses of cellular processes. If interactions of the above mentioned agents with GAGs are sufficiently strong, or if further changes in cellular structures and functions involve formations of specific mechanical defects, these disturbances might be impossible to reverse solely by abolition of lysosomal GAG storage.
The above hypothesis can be supported by previous findings. GAGs are known to bind to various growth factors, exemplified by fibroblast growth factors (FGFs). In fact, proliferation of astrocytes and their differentiation are dependent on interactions between HS and FGF2.43 It was demonstrated that intermediates of HS decay might interfere with proper signaling by FGF2, leading to dysregulation of expression on many genes and disturbed cell differentiation.44 Furthermore, changes in levels of FGF1 and FGF2 were observed in the brains of mice used as an animal model of Sanfilippo disease subtype B which correlated with overactivation of astrocytes.45 These types of changes cause disruptions of cellular homeostatic networks, and provoke stress responses which, however, cannot resolve the disorders due to their complexity and severity. Moreover, the secondary biochemical changes and global dysregulation of expression of genes result is disturbances and inefficiencies of as important cellular responses as autophagy and ferroptosis.46,47
The enormous dysregulations of cellular processes occurring in MPS III result in severe changes in tissues and organs, especially in the brain. They lead to neurodegenerative processes which have been summarized and deeply discussed recently,48,49 thus, they will not be presented in detail here. Nevertheless, it is necessary to mention that above mentioned disturbances in cell signaling, dysfunctions of mitochondria and enhanced oxidative stress, and impaired autophagy and ferroptosis, lead to further pathological processes, like increased neuroinflammation and defective neurotransmission. Finally, all these disorders contribute to neuronal cell death.
Since the neurodegenerative process is not sudden but proceeds for many years, many symptoms appear in patients. The huge complexity of changes is responsible for significant differences in the intensity of many symptoms occurring in various patients as different pathological processes may proceed with different intensities depending on a large number of factors. In fact, the presence of various mutations in Sanfilippo disease patients result in significant differences in severity and the course of this condition.50–55 These can be enhanced by the occurrence of untypical mutations which make phenotypes of patients especially unusual,56,57 or even by combination of MPS III with another ultra-rare genetic condition.58 Moreover, processes of gene expression regulation which operate with different intensities in every person, like activities of promoters, actions of micro-RNA molecules, alternative mRNA splicing, and posttranslational modifications of proteins might considerably modulate the MPS disease phenotypes, as discussed recently.59 One specific process which was demonstrated experimentally to affect the course of Sanfilippo disease is GAG production, which depends on many biochemical and genetic transactions and proceeds with different intensities in various persons, irrespectively of MPS. Synthesis of GAGs contributes to final levels of these compounds and in combination with the presence or absence of residual activities of enzymes involved in their degradation can meaningly modulate the time of appearance of the first symptoms as well as their severity.60,61
All the above described complex pathomechanisms of Sanfilippo syndrome result is variability of phenotypes of patients, differential course of the disease, and different severities of specific symptoms. In combination with a small number of patients, they cause significant problems with proper diagnosis which can significantly influence the quality of life of patients and their families. These problems are discussed in the next section.
Importance of Accurate Diagnosis and Diagnostic Problems in Sanfilippo Syndrome
Because of the lack of specific and efficient therapy, and due to severity of the disease,1–5 it is crucial to make accurate diagnosis of Sanfilippo syndrome as early as possible to start to manage numerous problems relative early in patient’s life. Although the biochemical and genetic diagnosis can be unequivocal by determining urinary/plasma HS levels, measuring activity of specific lysosomal enzyme (see Table 1) in leukocytes or fibroblasts, and identifying specific mutations,2,7,15,62,63 the rarity of MPS III and variability of early symptoms make the proper diagnosis challenging. In fact, it is common that it takes a few or even several years to diagnose Sanfilippo disease.63–65 Therefore, not the diagnostic procedures themselves but rather difficulty in coming up with an idea to test a patient for Sanfilippo disease prolongs a search for proper conclusion regarding MPS III. This is not only very frustrating for families and/or caregivers but also (or even predominantly) delays the onset of the best possible management, and exposes patients to unnecessary, ineffective, and often burdensome medical procedures.
To understand diagnostic challenges in MPS III, it is crucial to remind the clinical course of patients. As mentioned in the preceding section, patients are born without any specific symptoms. They appear between the age of a dozen of months and 3 years.2,64 First, some signs of developmental slowdown can be detected, sometimes accompanied with mild-to-moderate facial dysmorphology. Frequent cases of diarrhea and respiratory tract infection can also occur.64 However, due to nonspecific symptoms, suspicion of Sanfilippo disease is rare at this stage, and other possible disorders are usually considered. Then, more worrisome problems arise, including deterioration of cognitive processes, disturbance of sleep, impulsive reactions, aggressive-like behavior, and anxiety disorders. These problems augment in time. Then, at the age of several years, some problems with bones and joints may develop, together with deterioration of motor functions. Finally, dementia and loss of any communication and cognitive skills progress continuously and considerably, together with losing the ability to move and even to consume food independently. The expected life span is around 2 decades.2,64 However, it is necessary to stress again that disease severity and progress may differ significantly from patient to patient, and some symptoms may be either extremely highly pronounced or not occurring at all.
Such a clinical picture of MPS III patients may cause misdiagnosis, especially at the early or middle stages of the disease.7,64 The most often false suspicion is autism spectrum disorders (ASD), predominantly due to similarities of some symptoms of both diseases.64,66 Another quite common diagnostic error is prediction of attention deficit/hyperactivity disorder (ADHD).64,67,68 Misdiagnosis is often disturbing, as it results in the abandonment of further diagnostic procedures and can lead to the wrong treatment. Therefore, proper diagnosis is delayed while the unmanaged disease progresses significantly. These problems were recognized as crucial in the management of Sanfilippo syndrome, and a special need for improving the diagnostic procedures, and for developing a clinical algorithm for the early diagnosis of MPS III, has been postulated.69 Importantly, progressive character of symptoms of patients which were initially diagnosed for ASD or ADHS may indicate a possible misdiagnosis; this should stimulate next diagnostic approaches. On the other hand, it is not an easy task as Sanfilippo disease symptoms may resemble not only ASD or ADHS, but also other diseases. For example, joint contractures accompanied with mild neurological problems occur both at early stages of MPS III and in juvenile idiopathic arthritis.70 Furthermore, cognitive impairment, speech difficulties, disturbed social interactions, and aggressive-like behavior might be similar to symptoms of the Landau–Kleffner syndrome.71,72 Misdiagnosis of the Rett syndrome, another neurogenetic disorder characterized by communication regression and deterioration of motor skills, has also been reported in a patient who actually suffered from Sanfilippo disease.73 These examples strengthen the need for development of precise diagnostic algorithm that should be employed when MPS III is suspected or even plausible.69 In fact, a diagram for the diagnostic procedures towards MPS has been proposed recently.7 However, Sanfilippo syndrome is a somewhat quaint type of this disease due to the extremely severe neurological component and relative mild somatic defects; thus, special care must be taken when suggesting a specific neurodegenerative disease in a child. Therefore, perhaps more precise recommendations might be suggested for diagnosis of MPS III, like testing urinary GAG levels (which is a quick and cheap assay) if any cognitive problems are observed in a child. It is also tempting to propose that whole exome sequencing (WES) should be considered when neuronopathy is observed in a pediatric patient, as many neurodegenerative diseases are genetic disorders. Indeed, there are examples published in the literature that WES analyses allowed final diagnosis of MPS III patients which were difficult to classify clinically.58,73 Summary of recommended diagnostic procedures to identify Sanfilippo disease is presented in Figure 1.
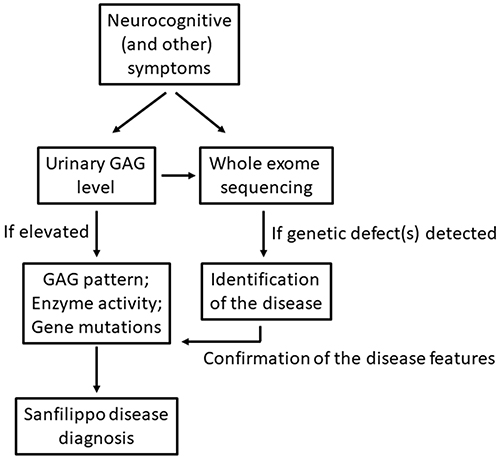 |
Figure 1 Summary of recommended diagnostic procedures to identify Sanfilippo disease. |
Optimized Symptomatic Treatment of Sanfilippo Disease
In the light of the absence of available therapy for Sanfilippo syndrome, the only way to manage this disease is to keep patients in as good condition as possible for a relatively long time (taking into consideration the expected life span). There are two possible methods to achieve this, (i) optimized symptomatic treatment and (ii) psychological care. These approaches are discussed in more detail in this and the next sections, respectively.
Definitely, the major clinical problems in MPS III are neurological, cognitive and behavioral disorders, as summarized recently in an article presenting comprehensive analyses of natural histories of neuronopathic MPS types.74 Unfortunately, symptomatic treatment of such patients is difficult, and pharmacological methods are often ineffective as children with Sanfilippo disease may respond atypically to psychotropic drugs. Ensuring physically safe home environment which allows to avoid accidental injuries is often helpful to minimize the risk of dangerous incidents.72 Risperidone treatment was suggested as a possible management of hyperactivity with some efficacy.76 Sleep disorders are frequent in this disease, and the use of melatonin might allow patients to improve sleep deficits.77 It is important to test if sleep apnea occurs, to support breathing mechanically if necessary.76,77
Bone, joint and muscle disorders occur commonly in Sanfilippo disease, but they are usually not as severe as in other MPS types. Nevertheless, symptomatic treatment of these disorders may improve the quality of life of patients. Vitamin D supplementation can be considered as a pharmacological treatment.78 Surgery might be helpful; however, such intervention should always be considered carefully, as MPS III patients reveal an increased risk during anesthesia.79,80 Moreover, the procedures should not be highly invasive due to very restricted contact with patients and their difficult convalescence after surgery.75
Otorhinolaryngological manifestations are often among MPS III patients, and the most frequent complications include chronic or recurrent rhinosinusitis, upper airway obstruction, hearing loss, and acute otitis media.81 Standard pharmacological treatment, specific for these disorders, can be used, with early antibiotic application being especially effective in the case of infections. However, it is also recommended to consider more invasive laryngological interventions if necessary, especially in the case of potentially life-threatening complications. Adenoidectomy, tympanostomy, and tracheostomy were reported in MPS III patients.81
Cardiovascular anomalies are less frequent in Sanfilippo syndrome than in other MPS types.82 Nevertheless, valvular heart disease, aortic valve abnormalities and valvular stenosis were reported in MPS III patients,83 as well as in cellular and animal models of the disease.84,85 Due to the milder character of these disorders relative to the rest of MPS patients and difficulties with convalescence of children suffering from Sanfilippo disease, invasive therapeutic methods should be considered only if evidently necessary.
Gastrointestinal manifestations are common in MPS III patients, though they are usually underestimated relative to the severe neurological symptoms. Nevertheless, there are cases of deaths of such patients caused by gastrointestinal problems.86 The most severe gastrointestinal complications in MPS III, reported to date, include bleeding from the digestive tract, hemorrhagic pancreatitis, perforation of the tract due to gastrostomies, paralytic ileus, and emaciation.86 On the other hand, infections of the digestive tract may be exhausting for patients and such disorders can significantly decrease the quality of life. Gut infections might be caused by untypical bacterial or viral pathogens when occurring in MPS III; however, it is important to note that in many cases they can be effectively treated with antimicrobial agents and/or the use of probiotics, as reported for another MPS.87 Intolerance of specific food products occurs relatively often, and a special diet is helpful to relieve symptoms in such cases, despite the fact that dietary treatment is believed to be ineffective in slowing down the diseases course.
In summary, because of many different complications in the course of Sanfilippo syndrome progression, the symptomatic treatment is important, irrespective of the fact that it cannot reverse the primary cause of the disease. Keeping patients in relatively good condition can significantly improve the quality of life and prolong the life span. Optimal management of musculoskeletal, otorhinolaryngological, cardiovascular, and gastrointestinal manifestations can considerably decrease the risk of death of MPS III patients, especially in the first decade of life, and may allow to reduce their suffering significantly. Such a care is important to both patients and their family members and/or caregivers. The common possibilities of symptomatic treatment of patients suffering from Sanfilippo disease are summarized in Figure 2.
 |
Figure 2 Possibilities of symptomatic treatments of patients suffering from Sanfilippo disease. |
Psychological Approach in Sanfilippo Syndrome
As indicated in previous sections, Sanfilippo disease is a neurodegenerative disorder characterized by progressive intellectual decline, finally resulting in severe dementia.74 In psychological approach, it is crucial to focus on neurocognitive development of patients; thus, before presenting specific recommendations, we will summarize characteristic features of this aspect of MPS III in more detail here.
According to the literature, the majority of patients with the severe, or so-called “classical”, phenotype of Sanfilippo syndrome have normal development by the age of 2 years.1–4 However, on the basis of the data obtained from interviews with parents (during the authors’ practical work) it can be concluded that developmental delays occur even earlier, but the focus is paid to other symptoms and medical conditions. Then, the development slows down of up to full stagnation at around the age of 3–4 years. At this time, regression of cognitive capacities occurs. Regarding the high variability of the progression of the disease, especially from 36 to 78 months, the prediction of cognitive functioning for a single patient is very difficult. However, the group of MPS III patients as a whole is characterized by a steady loss of skills after 54 months, and a stable low level of functioning at around 6 years of age.74 During their teenage years, the patients become fully dependent on others’ care.1 However, it should be underlined that there are also reported cases with mild phenotypes and delayed disease progression.56
The deterioration of the cognitive functioning during a child’s life results not only in intelligence quotient (IQ) loss and skill regression but also intense behavioral abnormalities.74 This affects the patients’ and their families’ life profoundly. Patients with the classical severe phenotype of MPS III generally reach a maximal developmental age of approximately 3–4 years.88 Moreover, it should be considered that more than 20% of the patients show a disharmonic developmental pattern of functioning.88 This results in some difficulties in assessing the overall development quotient (DQ) or IQ.
In the light of the severity and progressive character of neurocognitive manifestations, early access to clinical and psychological services is crucial for the families with children suffering from Sanfilippo disease. These services should be prepared for the support in this challenging, rare disease. The health care institutions should provide some assistance, especially in managing difficult behaviors.89 For the planning and evaluation of early interventions for children with Sanfilippo disease, the assessment of behavioral, cognitive, and emotional functioning should be done. Moreover, assessment of cognitive functioning is important to quantify the decline of intellectual abilities and to develop the patterns of the natural history of this disease.74 What is more, the assessment will be essential in order to evaluate treatment efficacy and for the sake of improving the quality of life.1–4 However, the diagnosis of cognitive functioning is much more difficult in this group of children, especially due to behavioral problems. These include restlessness and hyperactivity, temper tantrums, aggression, unusual affect (ie, screaming, crying, laughing), and hyperorality.89 In some cases, neurocognitive assessment should be conducted in familiar places.67,88 Moreover, noisy behavior, unwillingness, and throwing or biting of test materials usually occur.88 Attention deficits are also frequent in these patients, and are manifested in several problems with the continuation of cognitive testing, distraction during longer tasks, or with following instructions (especially those more detailed).88 Therefore, the elasticity of the diagnostician, some breaks, positive reinforcements, and the thoughtful placement of test materials are needed.67
During testing, MPS III patients are more often interested in other people than in the materials used. This should be taken into consideration by a diagnostician who can make the testing procedure more manageable due to showing his/her interest on it. Another symptom hindering the diagnostic process is aggressive behavior. Therefore, parental assistance could be helpful in this case, so the child does not hurt himself/herself and others. Other hindering factors are stereotypic behavior or language delay and aphasia, which may complicate the assessment in this field. However, the authors’ recommendation is to try to assess the abilities in this field to estimate the developmental age of the use of language by the patient, and to follow the changes in this regard over the coming years. If the child has aphasia, the better solution is the use of only nonverbal scales. The diagnostician should be also aware that many patients tend to perseverate; thus, he/she should be ready to help the child to stop the perseverative or stereotypical behaviors, for example, by drawing the child’s attention to other aspects of the test.67 Finally, testing the deterioration of physical function could be another obstacle to perform the diagnosis. Indeed, in some patients, the problems in the field of fine and gross motor skills generate a lot of frustration and negative emotions during the testing. Unfortunately, no large-scale studies on cognitive levels in Sanfilippo disease, using formal psychometric tests allowing the comparison of normatively developing children, have been reported.
Since Sanfilippo disease is inherited in an autosomal recessive manner, parents do not observe any signs or symptoms of the condition in their bodies, and the disease is typically not seen in many generations in the family. Parents do not suspect any possibility that their expected child could be sick. They often desperately need answers to questions about health problems of their child, especially during a long diagnostic process which proceeds sometimes several years. This process consists of many visits to the specialists (sometimes also extra paid), conduction of invasive testing (that could be lengthy and futile), and answering many questions, which is accompanied with a lack of knowledge and hope.90 This process undoubtedly carries significant personal costs. The situation of having a child with a rare genetic disease may frustrate the needs of its members and limit their individual development.91 In fact, parents of children with disabilities often experience extreme stress.92 In the case of Sanfilippo disease, characterized by the deterioration of functioning, the fulfilment of social requirements of parental roles is even more difficult. Additionally, this could be complicated by other roles that they have to attend (like medical guardian, teacher, and others), and finally because of the lack of institutional support.
The consequences of neurodegeneration and cognitive impairment are manifested in challenges with the daily functioning of patients and their families. Children with MPS III usually experience severe problems with communication and with the deterioration of their cognitive and motor functioning. Except for the symptoms described earlier, several behavioral disturbances, particularly impacting the safety of patients and other people, may occur.22,87 Somatic disturbances and pain were also reported. Among them, there were headaches, pain in the joints, gastrointestinal pain, and episodes of distress.94 Moreover, children meet problems to adapt to changes and new situations.23,93 Another challenging feature is expressed as sleeping problems, including restlessness and waking at night.95 As the disease progresses, aspiration problems and dysphagia are also present which force the parent to take a number of specialized nursing activities.96 Negative changes in their functioning are progressive and dynamic. Almost always, parents are left emotionally unprepared for such a difficult process. Indeed, parents often have to struggle with the powerlessness, caused by the inability to treat the disease, as well as the symptoms and challenges that the disease brings. Results of psychological analyses indicated that parents of children with MPS III are less future-oriented and goal-directed than parents of children with other intellectual disabilities.97 Considering the prognosis of this disease, parents usually try not to look ahead, not to make plans, and the future fills them with fear. Mothers of children with Sanfilippo disease met the criteria for clinically relevant anxiety and depression more frequently compared to mothers of children with other intellectual disabilities. Regarding depression, the same occurrence was noted in fathers. They more often met the criteria for clinically relevant depression compared to fathers of children with other intellectual disabilities. Furthermore, clinically relevant distress was highly prevalent in mothers and fathers of MPS III patients compared to reference parents.98 The daily functioning of these families, often filled with negative emotions and enormous fear, is undoubtedly a large field of challenges for psychologists and medical workers.
In summary, the neurocognitive assessment of patients with Sanfilippo disease as well as testing and interviewing their families are crucial for the improvement of the quality of life of the whole family. First, they are important for collecting data for studies and to provide information about the natural history of this disease, thus, contributing to the search for therapy. Second, they provide appropriate, individually dedicated, and interdisciplinary interventions for both child and his/her family who experienced the hardships of caring for a child with Sanfilippo disease. Unfortunately, the parents of these children are often left without support which has a direct impact on their daily functioning, and further on their relationship with the child. As shown in the previous paragraphs, parents often experience a lower quality of life and suffer from depressed mood and even depression. The psychological care (including regular individual or group therapy), support groups, psychoeducation, and provision of the reliable knowledge by medical workers are necessary. Parents of children with Sanfilippo disease may also benefit from education about sleep hygiene early in their child’s life, in order to reduce the impact of sleep disturbance, and hence, the well-being of all family. Moreover, it should be underlined that the mission of people who take care of patients with this rare disease should be the dissemination of knowledge. Thanks to this, it will be possible to recognize the disease faster at the early stages of the child’s life, an appropriate diagnostic process (including both medical and psychological diagnosis), faster and more appropriate implementation of dedicated interventions, and also – if it is possible – implementation of experimental therapies. Summary of the psychological approach in Sanfilippo syndrome is depicted schematically in Figure 3.
 |
Figure 3 Summary of the psychological approach in Sanfilippo disease. |
Recommendations Regarding Special Care and Schedules of Assessment
Despite the common primary cause of the Sanfilippo syndrome, a genetic defect resulting in impaired degradation of HS, the course of the disease may vary considerably from patient to patient.1–7 Therefore any recommendations for management of the disease in every MPS III patient are extremely difficult, if not impossible. Nevertheless, as discussed in previous chapters, some general advices can be proposed which should be helpful in providing the most effective care to the patients and their families.
The first point is to make a proper diagnosis as soon as possible. Therefore, as depicted in Figure 1, any neurocognitive symptoms in young children, especially those who were born without such problems and developed normally for the first several months, should signal a possibility of one of LSD. Determination of GAG levels in urine, which can be done using simple tests, can be recommended as one of early diagnostic steps in such cases, since detection of increased amounts of these compounds may shorten the diagnostic procedure significantly. If HS storage is confirmed, further biochemical and genetic tests are mandatory to confirm the diagnosis. Ideally, newborn screening for elevated urinary GAG would be the optimal procedure. However, it is not available now (mainly due to economical reasons), thus, because of unspecific symptoms appearing at the beginning of the course of MPS III, and in the light of the existence of many diseases characterized by similar symptoms at early stages,7 we strongly recommend to perform the whole genome sequencing (WES) analyses whenever possible. The use of this method can shorten the way to obtain accurate diagnosis significantly which is of great benefit to patients, their families, and physicians.
When the final diagnosis of Sanfilippo syndrome is obtained, it is crucial to provide a complex care to the patient. As depicted in Figure 2, there are many potential manifestations which may or may not appear in different patients; the only exception is occurrence of neurocognitive disorders in virtually all children suffering from MPS III.1–7 Nevertheless, this means that it is not sufficient to ensure the care of a geneticist, neurologist and specialist in the field of metabolic diseases. Regular visits to orthopedic, ENT (ear, nose and throat), pulmonary, cardiology, and gastroenterology clinics are also highly recommended. Early interventions of such specialists may allow to avoid severe complications of relative mild disorders which otherwise might progress to serious negative effects, high morbidity and significant worsening of the general condition of the patient. Optimally, visits in all clinics mentioned above should be planned every year, and as quickly as possible after appearing respective problems.
Early and constant access to psychological services can be as important as that to clinical specialists. As indicated in Figure 3, psychological care is crucial for both patients and their families. This can make family members mentally stronger and more motivated to organize the best possible care to their children. Moreover, specific psychological advices, based on proper assessment of mental and cognitive abilities of patients, facilitate choosing the best possible environment for functioning of MPS III children. Visits in a psychological center should be planned as often as recommended by the psychologist, with regular assessment of both cognitive development of the patient and mental state of parents.
Organization of relatively frequent visits in clinics is not an easy task for families of affected children. Therefore, it appears that development of clinical centers focused on rare/genetic/metabolic diseases is perhaps the best way to provide such complex clinical and psychological services that are invaluable for MPS III patients and their families.
Concluding Remarks
Sanfilippo syndrome is a severe, life-threatening, inherited metabolic disease which significantly affects not only the patients but also the life of whole families and/or caregivers. In the light of the lack of specific and effective therapy, it is crucial to provide complex and optimized care with a multidisciplinary approach. Three major fields were recognized in such an approach. They include (i) accurate diagnosis as early as possible, (ii) optimized symptomatic treatment, and (iii) psychological care for both patients and family members and/or caregivers. These are, however, not easy tasks, as (i) various misdiagnosis events are possible due to similarity of MPS III symptoms to those of other diseases and variability of phenotypes of patients is significant; (ii) clinical manifestations are complex and symptoms are often severe, while untypical responses of MPS III patients to various drugs occur frequently and invasive medical interventions are usually risky; and (iii) behavior of patients and their psychological features are significantly changed, and extreme stress of parents/caregivers has a direct impact on their daily functioning, respectively. Nevertheless, in this article, we propose specific schemes of actions and provide detailed recommendations for managing the care of patients with Sanfilippo disease and their families.
Abbreviations
ADHD, attention deficit/hyperactivity disorder’ASD, autism spectrum disorders; CNS, central nervous system; DQ, development quotient; FGF, fibroblast growth factor; GAG, glycosaminoglycan; HS, heparan sulfate; IQ, intelligence quotient; LSD, lysosomal storage disease(s); MPS, mucopolysaccharidosis; WES, whole exome sequencing.
Acknowledgments
This work was supported by Fundacja Sanfilippo, Poland.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Benetó N, Vilageliu L, Grinberg D, Canals I. Sanfilippo syndrome: molecular basis, disease models and therapeutic approaches. Int J Mol Sci. 2020;21(21):7819. doi:10.3390/ijms21217819
2. Pierzynowska K, Rintz E, Gaffke L, et al. Mucopolysaccharidosis type III (Sanfilippo disease) subtypes A, B, C, D: molecular mechanism and therapeutic effect. In: Surendran S, editor. Neurochemistry of Metabolic Diseases - Lysosomal Storage Diseases, Phenylketonuria and Canavan Disease. Hauppauge NY, USA: Nova Science Publishers, Inc; 2020:51–101.
3. Spahiu L, Behluli E, Peterlin B, et al. Mucopolysaccharidosis III: molecular basis and treatment. Pediatr Endocrinol Diabetes Metab. 2021;27:201–208. doi:10.5114/pedm.2021.109270
4. Kaczor-Kamińska M, Kamiński K, Wróbel M. Heparan sulfate, Mucopolysaccharidosis IIIB and sulfur metabolism disorders. Antioxidants. 2022;11:678. doi:10.3390/antiox11040678
5. Fedele AO. Sanfilippo syndrome: causes, consequences, and treatments. Appl Clin Genet. 2015;8:269–281. doi:10.2147/TACG.S57672
6. Węgrzyn G, Pierzynowska K, Pavone LM. Editorial: molecular aspects of Mucopolysaccharidoses. Front Mol Biosci. 2022;9:874267. doi:10.3389/fmolb.2022.874267
7. Wiśniewska K, Wolski J, Gaffke L, Cyske Z, Pierzynowska K, Węgrzyn G. Misdiagnosis in mucopolysaccharidoses. J Appl Genet. 2022;63:475–495. doi:10.1007/s13353-022-00703-1
8. Kowalewski B, Lamanna WC, Lawrence R, et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci U S A. 2012;109:10310–10315. doi:10.1073/pnas.1202071109
9. Kowalewski B, Heimann P, Ortkras T, et al. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum Mol Genet. 2015;24:1856–1868. doi:10.1093/hmg/ddu603
10. Kruszewski K, Lüllmann-Rauch R, Dierks T, Bartsch U, Damme M. Degeneration of photoreceptor cells in arylsulfatase G-deficient mice. Invest Ophthalmol Vis Sci. 2016;57:1120–1131. doi:10.1167/iovs.15-17645
11. McBride KL, Flanigan KM. Update in the Mucopolysaccharidoses. Semin Pediatr Neurol. 2021;37:100874. doi:10.1016/j.spen.2021.100874
12. Nagpal R, Goyal RB, Priyadarshini K, et al. Mucopolysaccharidosis: a broad review. Indian J Ophthalmol. 2022;70:2249–2261. doi:10.4103/ijo.IJO_425_22
13. Kong W, Yao Y, Zhang J, Lu C, Ding Y, Meng Y. Update of treatment for mucopolysaccharidosis type III (Sanfilippo syndrome). Eur J Pharmacol. 2020;888:173562. doi:10.1016/j.ejphar.2020.173562
14. Seker Yilmaz B, Davison J, Jones SA, Baruteau J. Novel therapies for mucopolysaccharidosis type III. J Inherit Metab Dis. 2021;44(1):129–147. doi:10.1002/jimd.12316
15. Zhou J, Lin J, Leung WT, Wang L. A basic understanding of mucopolysaccharidosis: incidence, clinical features, diagnosis, and management. Intractable Rare Dis Res. 2020;9:1–9. doi:10.5582/irdr.2020.01011
16. Gaffke L, Pierzynowska K, Piotrowska E, Węgrzyn G. How close are we to therapies for Sanfilippo disease? Metab Brain Dis. 2018;33:1–10. doi:10.1007/s11011-017-0111-4
17. Rajan DS, Escolar ML. Evolving therapies in neuronopathic LSDs: opportunities and challenges. Metab Brain Dis. 2022. doi:10.1007/s11011-022-00939-0
18. Wijburg FA, Whitley CB, Muenzer J, et al. A multicenter open-label extension study of intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A. Mol Genet Metab. 2021;134:175–181. doi:10.1016/j.ymgme.2021.07.001
19. Wijburg FA, Heap F, Rust S, et al. Long-term safety and clinical outcomes of intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A. Mol Genet Metab. 2021;134:317–322. doi:10.1016/j.ymgme.2021.09.003
20. Deiva K, Ausseil J, de Bournonville S, et al. Intracerebral gene therapy in four children with Sanfilippo B syndrome: 5.5-year follow-up results. Hum Gene Ther. 2021;32:1251–1259. doi:10.1089/hum.2021.135
21. Ghosh A, Rust S, Langford-Smith K, et al. High dose genistein in Sanfilippo syndrome: a randomised controlled trial. J Inherit Metab Dis. 2021;44:1248–1262. doi:10.1002/jimd.12407
22. De Pasquale V, Pavone LM. Heparan sulfate proteoglycans: the sweet side of development turns sour in mucopolysaccharidoses. Biochim Biophys Acta Mol Basis Dis. 2019;1865:165539. doi:10.1016/j.bbadis.2019.165539
23. Węgrzyn G, Jakóbkiewicz-Banecka J, Narajczyk M, et al. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Med Hypotheses. 2010;75:605–609. doi:10.1016/j.mehy.2010.07.044
24. Bruyère J, Roy E, Ausseil J, et al. Heparan sulfate saccharides modify focal adhesions: implication in mucopolysaccharidosis neuropathophysiology. J Mol Biol. 2015;427:775–791. doi:10.1016/j.jmb.2014.09.012
25. Trudel S, Trécherel E, Gomila C, et al. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B. J Neurosci Res. 2015;93:424–432. doi:10.1002/jnr.23497
26. Pierzynowska K, Gaffke L, Cyske Z, et al. Oxidative Stress in Mucopolysaccharidoses: pharmacological Implications. Molecules. 2021;26:5616. doi:10.3390/molecules26185616
27. De Pasquale V, Moles A, Pavone LM. Cathepsins in the pathophysiology of mucopolysaccharidoses: new perspectives for therapy. Cells. 2020;9:979. doi:10.3390/cells9040979
28. Lamanna WC, Lawrence R, Sarrazin S, Esko JD. Secondary storage of dermatan sulfate in Sanfilippo disease. J Biol Chem. 2011;286:6955–6962. doi:10.1074/jbc.M110.192062
29. Dawson G, Fuller M, Helmsley KM, Hopwood JJ. Abnormal gangliosides are localized in lipid rafts in Sanfilippo (MPS3a) mouse brain. Neurochem Res. 2012;37:1372–1380. doi:10.1007/s11064-012-0761-x
30. Baydakova G, Ilyushkina A, Gaffke L, et al. Elevated LysoGb3 Concentration in the Neuronopathic Forms of Mucopolysaccharidoses. Diagnostics. 2020;10:155. doi:10.3390/diagnostics10030155
31. Gaffke L, Pierzynowska K, Podlacha M, Brokowska J, Węgrzyn G. Changes in cellular processes occurring in mucopolysaccharidoses as underestimated pathomechanisms of these diseases. Cell Biol Int. 2021;45:498–506. doi:10.1002/cbin.11275
32. Gaffke L, Pierzynowska K, Podlacha M, et al. Underestimated Aspect of Mucopolysaccharidosis Pathogenesis: global Changes in Cellular Processes Revealed by Transcriptomic Studies. Int J Mol Sci. 2020;21:1204. doi:10.3390/ijms21041204
33. Gaffke L, Pierzynowska K, Rintz E, Cyske Z, Giecewicz I, Węgrzyn G. Gene Expression-Related Changes in Morphologies of Organelles and Cellular Component Organization in Mucopolysaccharidoses. Int J Mol Sci. 2021;22:2766. doi:10.3390/ijms22052766
34. Gaffke L, Pierzynowska K, Krzelowska K, Piotrowska E, Węgrzyn G. Changes in expressions of genes involved in the regulation of cellular processes in mucopolysaccharidoses as assessed by fibroblast culture-based transcriptomic analyses. Metab Brain Dis. 2020;35:1353–1360. doi:10.1007/s11011-020-00614-2
35. Brokowska J, Pierzynowska K, Gaffke L, Rintz E, Węgrzyn G. Expression of genes involved in apoptosis is dysregulated in mucopolysaccharidoses as revealed by pilot transcriptomic analyses. Cell Biol Int. 2021;45:549–557. doi:10.1002/cbin.11332
36. Rintz E, Gaffke L, Podlacha M, et al. Transcriptomic changes related to cellular processes with particular emphasis on cell activation in lysosomal storage diseases from the group of Mucopolysaccharidoses. Int J Mol Sci. 2020;21:3194. doi:10.3390/ijms21093194
37. Pierzynowska K, Gaffke L, Jankowska E, et al. Proteasome composition and activity changes in cultured fibroblasts derived from Mucopolysaccharidoses patients and their modulation by genistein. Front Cell Dev Biol. 2020;8:540726. doi:10.3389/fcell.2020.540726
38. Gaffke L, Szczudło Z, Podlacha M, et al. Impaired ion homeostasis as a possible associate factor in mucopolysaccharidosis pathogenesis: transcriptomic, cellular and animal studies. Metab Brain Dis. 2022;37:299–310. doi:10.1007/s11011-021-00892-4
39. Pierzynowska K, Żabińska M, Gaffke L, Cyske Z, Węgrzyn G. Changes in expression of signal transduction-related genes, and formation of aggregates of GPER1 and OXTR receptors in mucopolysaccharidosis cells. Eur J Cell Biol. 2022;101:151232. doi:10.1016/j.ejcb.2022.151232
40. Brokowska J, Gaffke L, Pierzynowska K, Cyske Z, Węgrzyn G. Cell cycle disturbances in mucopolysaccharidoses: transcriptomic and experimental studies on cellular models. Exp Biol Med. 2022. doi:10.1177/15353702221114872
41. Pierzynowska K, Gaffke L, Podlacha M, Węgrzyn G. Genetic base of behavioral disorders in Mucopolysaccharidoses: transcriptomic studies. Int J Mol Sci. 2020;21:1156. doi:10.3390/ijms21031156
42. Cyske Z, Gaffke L, Pierzynowska K, Węgrzyn G. Complex changes in the efficiency of the expression of many genes in monogenic diseases, Mucopolysaccharidoses, may arise from significant disturbances in the levels of factors involved in the gene expression regulation processes. Genes. 2022;13:593. doi:10.3390/genes13040593
43. Gómez-Pinilla F, Vu L, Cotman CW. Regulation of astrocyte proliferation by FGF-2 and heparan sulfate in vivo. J Neurosci. 1995;15:2021–2029. doi:10.1523/JNEUROSCI.15-03-02021.1995
44. Lemonnier T, Blanchard S, Toli D, et al. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum Mol Genet. 2011;20:3653–3666. doi:10.1093/hmg/ddr285
45. Li HH, Zhao HZ, Neufeld EF, Cai Y, Gomez-Pinilla F. Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J Neurosci Res. 2002;69:30–38. doi:10.1002/jnr.10278
46. Pierzynowska K, Gaffke L, Podlacha M, Brokowska J, Węgrzyn G. Mucopolysaccharidosis and autophagy: controversies on the contribution of the process to the pathogenesis and possible therapeutic applications. Neuromolecular Med. 2020;22:25–30. doi:10.1007/s12017-019-08559-1
47. Pierzynowska K, Rintz E, Gaffke L, Węgrzyn G. Ferroptosis and its modulation by autophagy in light of the pathogenesis of lysosomal storage diseases. Cells. 2021;10:365. doi:10.3390/cells10020365
48. Bigger BW, Begley DJ, Virgintino D, Pshezhetsky AV. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab. 2018;125:322–331. doi:10.1016/j.ymgme.2018.08.003
49. Heon-Roberts R, Nguyen ALA, Pshezhetsky AV. Molecular bases of neurodegeneration and cognitive decline, the major burden of Sanfilippo disease. J Clin Med. 2020;9:344. doi:10.3390/jcm9020344
50. Zhao HG, Aronovich EL, Whitley CB. Genotype-phenotype correspondence in Sanfilippo syndrome type B. Am J Hum Genet. 1998;62:53–63. doi:10.1086/301682
51. Di Natale P, Villani GR, Di Domenico C, Daniele A, Dionisi Vici C, Bartuli A. Analysis of Sanfilippo A gene mutations in a large pedigree. Clin Genet. 2003;63:314–318. doi:10.1034/j.1399-0004.2003.00053.x
52. Jansen AC, Cao H, Kaplan P, et al. Sanfilippo syndrome type D: natural history and identification of 3 novel mutations in the GNS Gene. Arch Neurol. 2007;64:1629–1634. doi:10.1001/archneur.64.11.1629
53. Valstar MJ, Neijs S, Bruggenwirth HT, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68:876–887. doi:10.1002/ana.22092
54. Héron B, Mikaeloff Y, Froissart R, et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011;155A:58–68. doi:10.1002/ajmg.a.33779
55. Delgadillo V, O’Callaghan Mdel M, Gort L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orphanet J Rare Dis. 2013;8:189. doi:10.1186/1750-1172-8-189
56. Pierzynowska K, Mański A, Limanówka M, et al. Untypically mild phenotype of a patient suffering from Sanfilippo syndrome B with the c.638C>T/c.889C>T (p.Pro213Leu/p.Arg297Ter) mutations in the NAGLU gene. Mol Genet Genomic Med. 2020;8(9):e1356. doi:10.1002/mgg3.1356
57. Lorenz D, Musacchio T, Kunstmann E, et al. A case report of Sanfilippo syndrome - the long way to diagnosis. BMC Neurol. 2022;22:93. doi:10.1186/s12883-022-02611-7
58. Anikiej-Wiczenbach P, Mański A, Milska-Musa K, et al. Highly diverse phenotypes of mucopolysaccharidosis type IIIB sibling patients: effects of an additional mutation in the AUTS2 gene. J Appl Genet. 2022. doi:10.1007/s13353-022-00702-2
59. Kong W, Lu C, Ding Y, Meng Y. Molecular environment and atypical function: what do we know about enzymes associated with Mucopolysaccharidoses? Orphanet J Rare Dis. 2022;17:112. doi:10.1186/s13023-022-02211-1
60. Perkins KJ, Muller V, Weber B, Hopwood JJ. Prediction of Sanfilippo phenotype severity from immunoquantification of heparan-N-sulfamidase in cultured fibroblasts from mucopolysaccharidosis type IIIA patients. Mol Genet Metab. 2001;73:306–312. doi:10.1006/mgme.2001.3190
61. Piotrowska E, Jakóbkiewicz-Banecka J, Tylki-Szymańska A, Czartoryska B, Wegrzyn A, Wegrzyn G. Correlation between severity of mucopolysaccharidoses and combination of the residual enzyme activity and efficiency of glycosaminoglycan synthesis. Acta Paediatr. 2009;98:743–749. doi:10.1111/j.1651-2227.2008.01153.x
62. Jakobkiewicz-Banecka J, Gabig-Ciminska M, Kloska A, et al. Glycosaminoglycans and mucopolysaccharidosis type III. Front Biosci. 2016;21:1393–1409. doi:10.2741/4463
63. Bodamer OA, Giugliani R, Wood T. The laboratory diagnosis of mucopolysaccharidosis III (Sanfilippo syndrome): a changing landscape. Mol Genet Metab. 2014;113:34–41. doi:10.1016/j.ymgme.2014.07.013
64. Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013;102:462–470. doi:10.1111/apa.12169
65. Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML. Sanfilippo syndrome: overall review. Pediatr Int. 2015;57:331–338. doi:10.1111/ped.12636
66. Wolfenden C, Wittkowski A, Hare DJ. Symptoms of autism spectrum disorder (ASD) in individuals with Mucopolysaccharide disease type III (Sanfilippo Syndrome): a systematic review. J Autism Dev Disord. 2017;47:3620–3633. doi:10.1007/s10803-017-3262-6
67. Anikiej-Wiczenbach P, Rudnik A, Limanówka M, Wierzba J, Mański A. Diagnosis of children with Sanfilippo disease—Psychological, social and motor assessment. Acta Neuropsychol. 2020;18:525–535. doi:10.5604/01.3001.0014.5316
68. Urgancı N, Kalyoncu D, Gümüştekin R. Is mucopolysaccharidosis a cause of sleep and speech disorders? Report of four cases. J Acad Res Med. 2020;10:204–207. doi:10.4274/jarem.galenos.2020.3255
69. Escolar M, Bradshaw J, Tharp Byers V, et al. Development of a clinical algorithm for the early diagnosis of Mucopolysaccharidosis III. J Inborn Errors Metabol Screening. 2020;8:2. doi:10.1590/2326-4594-JIEMS-2020-0002
70. Cimaz R, Coppa GV, Koné-Paut I, et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatr Rheumatol Online J. 2009;7:18. doi:10.1186/1546-0096-7-18
71. Ahmed M, Saleem A, Nasir S, Ariff M, Iftikhar P. Landau-Kleffner syndrome: a diagnostic challenge. Cureus. 2020;12:e7182. doi:10.7759/cureus.7182
72. Rezayi A, Feshangchi-Bonab M, Taherian R. An uncommon presentation of Mucopolysaccharidosis type IIIb similar to the Landau-Kleffner syndrome. Iran J Child Neurol. 2019;13:105–111. doi:10.22037/ijcn.v13i3.16536
73. Zeng Q, Fan Y, Wang L, Huang Z, Gu X, Yu Y. Molecular defects identified by whole exome sequencing in a child with atypical mucopolysaccharidosis IIIB. J Pediatr Endocrinol Metab. 2017;30:463–469. doi:10.1515/jpem-2016-0333
74. Shapiro EG, Eisengart JB. The natural history of neurocognition in MPS disorders: a review. Mol Genet Metab. 2021;133:8–34. doi:10.1016/j.ymgme.2021.03.002
75. Wagner VF, Northrup H, et al. Mucopolysaccharidosis Type III. In: Adam MP, Mirzaa GM, Pagon RA, editors. GeneReviews®. Seattle: University of Washington, Seattle; 2019.
76. Kalkan Ucar S, Ozbaran B, Demiral N, Yuncu Z, Erermis S, Coker M. Clinical overview of children with mucopolysaccharidosis type IIIA and effect of Risperidone treatment on children and their mothers psychological status. Brain Dev. 2010;32:156–161.
77. Mahon LV, Lomax M, Grant S, et al. Assessment of sleep in children with mucopolysaccharidosis type III. PLoS One. 2014;9:e84128. doi:10.1371/journal.pone.0084128
78. Nur BG, Nur H, Mihci E. Bone mineral density in patients with mucopolysaccharidosis type III. J Bone Miner Metab. 2017;35:338–343. doi:10.1007/s00774-016-0762-y
79. Cohen MA, Stuart GM. Delivery of anesthesia for children with Mucopolysaccharidosis Type III (Sanfilippo syndrome): a review of 86 anesthetics. Paediatr Anaesth. 2017;27:363–369. doi:10.1111/pan.13075
80. Clark BM, Sprung J, Weingarten TN, Warner ME. Anesthesia for patients with mucopolysaccharidoses: comprehensive review of the literature with emphasis on airway management. Bosn J Basic Med Sci. 2018;18:1–7. doi:10.17305/bjbms.2017.2201
81. Murgasova L, Jurovcik M, Jesina P, et al. Otorhinolaryngological manifestations in 61 patients with mucopolysaccharidosis. Int J Pediatr Otorhinolaryngol. 2020;135:110137. doi:10.1016/j.ijporl.2020.110137
82. Nijmeijer SCM, de Bruin-Bon RHACM, Wijburg FA, Kuipers IM. Cardiac disease in mucopolysaccharidosis type III. J Inherit Metab Dis. 2019;42:276–285. doi:10.1002/jimd.12015
83. Lin HY, Chen MR, Lin SM, et al. Cardiac characteristics and natural progression in Taiwanese patients with mucopolysaccharidosis III. Orphanet J Rare Dis. 2019;14:140. doi:10.1186/s13023-019-1112-7
84. De Pasquale V, Pezone A, Sarogni P, et al. EGFR activation triggers cellular hypertrophy and lysosomal disease in NAGLU-depleted cardiomyoblasts, mimicking the hallmarks of mucopolysaccharidosis IIIB. Cell Death Dis. 2018;9:40. doi:10.1038/s41419-017-0187-0
85. Schiattarella GG, Cerulo G, De Pasquale V, et al. The murine model of mucopolysaccharidosis IIIB develops cardiopathies over time leading to heart failure. PLoS One. 2015;10:e0131662. doi:10.1371/journal.pone.0131662
86. Thomas S, Ramaswami U, Cleary M, Yaqub M, Raebel EM. Gastrointestinal manifestations in Mucopolysaccharidosis Type III: review of death certificates and the literature. J Clin Med. 2021;10:4445. doi:10.3390/jcm10194445
87. Wegrzyn G, Kurlenda J, Liberek A, et al. Atypical microbial infections of digestive tract may contribute to diarrhea in mucopolysaccharidosis patients: a MPS I case study. BMC Pediatr. 2005;5:9. doi:10.1186/1471-2431-5-9
88. Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J Rare Dis. 2011;6:43. doi:10.1186/1750-1172-6-43
89. Cross EM, Hare DJ. Behavioural phenotypes of the mucopolysaccharide disorders: a systematic literature review of cognitive, motor, social, linguistic and behavioural presentation in the MPS disorders. J Inherit Metab Dis. 2013;36:189–200. doi:10.1007/s10545-012-9572-0
90. Hartley T, Lemire G, Kernohan KD, Howley HE, Adams DR, Boycott KM. New diagnostic approaches for undiagnosed rare genetic diseases. Annu Rev Genomics Hum Genet. 2020;21:351–372. doi:10.1146/annurev-genom-083118-015345
91. Cuzzocrea F, Larcan R, Westh F. Family and parental functioning in parents of disabled children. Nordic Psychology. 2013;65:271–287. doi:10.1080/19012276.2013.824201
92. Walker A, Alfonso ML, Colquitt G, Weeks K, Telfair J. “When everything changes:” Parent perspectives on the challenges of accessing care for a child with a disability. Disabil Health J. 2016;9:157–161. doi:10.1016/j.dhjo.2015.06.002
93. Cross EM, Grant S, Jones S, et al. An investigation of the middle and late behavioural phenotypes of Mucopolysaccharidosis Type-III. J Neurodev Disord. 2014;6:46. doi:10.1186/1866-1955-6-46
94. Congedi S, Orzalesi M, Di Pede C, Benini F. Pain in Mucopolysaccharidoses: analysis of the Problem and Possible Treatments. Int J Mol Sci. 2018;19:3063. doi:10.3390/ijms19103063
95. Porter KA, O’Neill C, Drake E, et al. Caregivers’ assessment of meaningful and relevant clinical outcome assessments for Sanfilippo syndrome. J Patient Rep Outcomes. 2022;6:40. doi:10.1186/s41687-022-00447-w
96. Shapiro E, Ahmed A, Whitley C, Delaney K. Observing the advanced disease course in mucopolysaccharidosis, type IIIA; a case series. Mol Genet Metab. 2018;123:123–126. doi:10.1016/j.ymgme.2017.11.014
97. Grant S, Cross E, Wraith JE, et al. Parental social support, coping strategies, resilience factors, stress, anxiety and depression levels in parents of children with MPS III (Sanfilippo syndrome) or children with intellectual disabilities (ID). J Inherit Metab Dis. 2013;36:281–291. doi:10.1007/s10545-012-9558-y
98. Conijn T, Nijmeijer SCM, van Oers HA, Wijburg FA, Haverman L. Psychosocial functioning in parents of MPS III patients. JIMD Rep. 2019;44:33–41. doi:10.1007/8904_2018_119
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.