Back to Journals » The Application of Clinical Genetics » Volume 8
Sanfilippo syndrome: causes, consequences, and treatments
Authors Fedele A
Received 18 June 2015
Accepted for publication 25 August 2015
Published 25 November 2015 Volume 2015:8 Pages 269—281
DOI https://doi.org/10.2147/TACG.S57672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Anthony O Fedele
Lysosomal Diseases Research Unit, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
Abstract: Sanfilippo syndrome, or mucopolysaccharidosis (MPS) type III, refers to one of five autosomal recessive, neurodegenerative lysosomal storage disorders (MPS IIIA to MPS IIIE) whose symptoms are caused by the deficiency of enzymes involved exclusively in heparan sulfate degradation. The primary characteristic of MPS III is the degeneration of the central nervous system, resulting in mental retardation and hyperactivity, typically commencing during childhood. The significance of the order of events leading from heparan sulfate accumulation through to downstream changes in the levels of biomolecules within the cell and ultimately the (predominantly neuropathological) clinical symptoms is not well understood. The genes whose deficiencies cause the MPS III subtypes have been identified, and their gene products, as well as a selection of disease-causing mutations, have been characterized to varying degrees with respect to both frequency and direct biochemical consequences. A number of genetic and biochemical diagnostic methods have been developed and adopted by diagnostic laboratories. However, there is no effective therapy available for any form of MPS III, with treatment currently limited to clinical management of neurological symptoms. The availability of animal models for all forms of MPS III, whether spontaneous or generated via gene targeting, has contributed to improved understanding of the MPS III subtypes, and has provided and will deliver invaluable tools to appraise emerging therapies. Indeed, clinical trials to evaluate intrathecally-delivered enzyme replacement therapy in MPS IIIA patients, and gene therapy for MPS IIIA and MPS IIIB patients are planned or underway.
Keywords: lysosomal storage disease, Sanfilippo syndrome, mucopolysaccharidosis III
Introduction
Lysosomal storage disorders are a group of more than 50 inherited monogenic disorders. Each is caused by a deficiency of an enzyme responsible for the degradation of a metabolic product, whose accumulation results in lysosomal malfunction and disease.1 Classification is primarily based on the nature of stored material, and secondarily by the enzyme whose activity has been impeded. In Australia, the combined incidence of lysosomal storage disorders is at least 12.9 per 100,000 live births, thus indicating a significant health and economic impact on the family and the community.2
Sanfilippo syndrome, or mucopolysaccharidosis (MPS) type III, refers to one of five autosomal recessive, neurodegenerative lysosomal storage disorders whose symptoms are due to the incomplete lysosomal degradation of heparan sulfate.3 The subtypes are caused by the deficiency of: sulfamidase (MPS IIIA);4 α-N-acetylglucosaminidase (NAGLU, MPS IIIB);5 heparan acetyl CoA: α-glucosaminide N-acetyltransferase (HGSNAT, MPS IIIC);6 N-acetylglucosamine 6-sulfatase (GNS, MPS IIID);7 or N-glucosamine 3-O-sulfatase (arylsulfatase G or ARSG, the currently putative MPS IIIE).8 Phenotypic, enzymatic, and genetic classifications of MPS III subtypes are summarized in Table 1. A listing of reported prevalence is presented in Table 2.
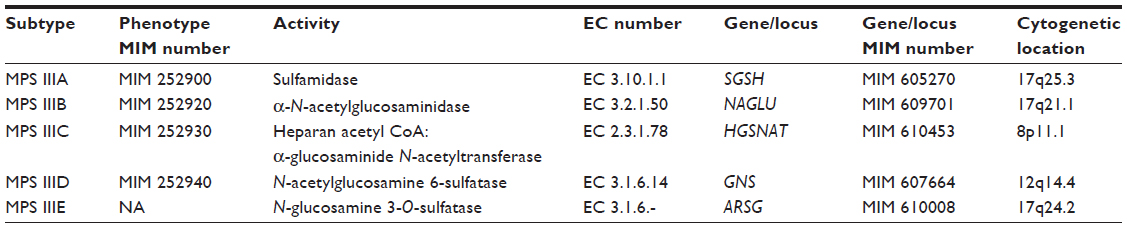 | Table 1 Summary of the phenotypic, enzymatic, and genetic classification of the subtypes of MPS III |
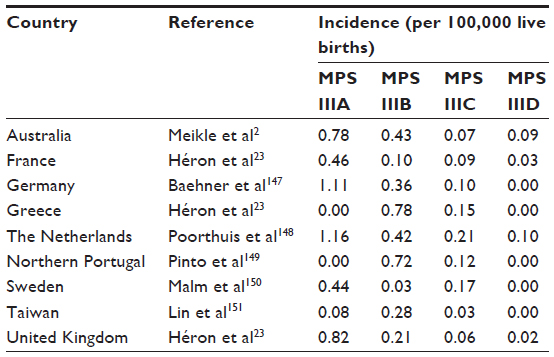 | Table 2 Summary of reported prevalence studies of MPS III |
There is currently no therapy available for MPS III. Animal models exist for all forms, whether spontaneous or generated via gene targeting.8–18 These have contributed to a better understanding of MPS III, and will continue to deliver invaluable tools to evaluate emerging therapies.
Clinical symptoms of MPS III
Somatic symptoms in humans can include coarse facial features with broad eyebrows, dark eyelashes, dry and rough hair, and skeletal pathology that affects growth and causes degenerative joint disease, hepatosplenomegaly, macrocrania, and hearing loss.19–22
The primary characteristic of MPS III is however degeneration of the central nervous system (CNS), resulting in mental retardation and hyperactivity, typically commencing during childhood, although there is some heterogeneity with respect to severity and age of onset.
Pre-natal and early stages of post-natal development are usually normal. The initial stages of disease may begin between the ages of 1 and 3 years, which manifest as delayed cognitive development and/or aggressive behavioral problems, as well as hindered speech development.19,20,23
Behavioral difficulties may become increasingly severe between the ages of 3 and 5 years, commonly manifesting as a combination of hyperactivity, which is often violent and destructive, as well as sleep disturbances.20,24–26
Patients may remain in this state for between 5 and 10 years, after which there is a regression in behavioral disturbances. This is associated with a progressive and severe loss of intellectual processes (such as speech) and motor functions (including walking and swallowing). MPS III patients ultimately regress to a vegetative state until death, which can occur anywhere between the early teens in the most severe scenarios, to as late as the sixth decade in attenuated forms.19,20,27–30
Biochemistry of genes involved in heparan sulfate degradation
Heparan sulfate is a negatively charged polysaccharide or glycosaminoglycan (GAG) covalently bound to a number of proteins at the cell surface and in the extracellular matrix. It in turn consists of repeating disaccharide units of either l-iduronic acid or d-glucuronic acid and α-linked glucosamine.3,8,31,32 The O-sulfation of any subunit can occur, while the α-glucosamine may be acetylated, N-sulfated, or unmodified.
Heparan sulfate GAGs exist in the cell as proteoglycans33 that are catabolized within the lysosome. This step may commence with the action of the endohydrolase heparanase on full-length heparan sulfate GAG chains at the plasma membrane, or early endosomal vesicles. This activity yields heparan oligosaccharide fragments of approximately 10–20 monosaccharide residues, whose subsequent degradation occurs via sequential exolytic processes in the lysosome.34
Depending on the non-reducing end of these heparan oligosaccharides, this pathway may commence with the desulfation of 2-sulfated iduronic acid residues via the action of iduronate 2-sulfatase (Enzyme Commission [EC] 3.1.6.13),35,36 and is followed by the hydrolysis of the subsequent terminal l-iduronic acid by α-l-iduronidase (EC 3.2.1.76).37,38 Both enzymes are also involved in the degradation of dermatan sulfate and their deficiencies result in MPS II (MIM 309900) and subtypes of MPS I (MIM 607014, MIM 607015, and MIM 607016), respectively. At this stage, ARSG desulfates 3-O-sulfated N-sulfoglucosamine.8,39 Sulfamidase then hydrolyses the sulfates bound to the amino group of glucosamine,40,41 and the exposed amino group is in turn acetylated by the activity of HGSNAT.42,43 NAGLU is consequently able to recognize and remove the generated N-acetylglucosaminide from the neighboring uronic acid.44,45 Desulfation of any 2-O-sulfated glucuronic acid residues will then occur with glucoronate 2-sulfatase,46 a deficiency of which is yet to be detected. β-d-glucuronidase (EC 3.2.1.31) then hydrolyzes the terminal glucuronic acid.47 This is also required to degrade dermatan sulfate, and its deficiency causes MPS VII (MIM 253220). Finally, GNS removes the sulfate group from sulfated N-acetlyglucosamine residues.48
The catalytic activation of all eukaryotic sulfatases, including those deficient in MPS IIIA, MPS IIID, and MPS IIIE, undergo post-translational modification in which a conserved cysteine within the active site is catalytically converted in the endoplasmic reticulum to a Cα-formylglycine by the enzyme sulfatase modifying factor 1 (SUMF1).49,50 SUMF1 deficiency thus ablates the function of all sulfatases, which manifests as the symptoms of multiple sulfatase deficiency.51
Pathophysiology of MPS III
The order of events from heparan sulfate accumulation through downstream changes in the levels of other biomolecules within the cell and ultimately the clinical symptoms of MPS III, particularly with respect to CNS degeneration, is probably the least understood aspect.
Monosialic gangliosides GM2 and GM3 accumulate in lysosomes and other organelles (such as mitochondria and Golgi bodies) secondary to heparan sulfate accumulation,18,52–55 either by direct GAG-mediated inhibition of lysosomal enzymes responsible for ganglioside degradation,56 or, alternatively, by deregulated trafficking or synthesis of gangliosides.53
Not only is the reason for GM2 and GM3 accumulation still a matter of debate, but also their contribution to neurodegenerative aspects of MPS III. It is speculated from studies with MPS III animal models that both heparan sulfate and gangliosides initiate an inflammatory response that may exacerbate damage within the brain.12,18,57–59 This may occur as heparan sulfate released from lysosomes (probably via exocytosis) as well as ganglioside-engorged and damaged neurons are phagocytosed by microglia (immunologically active cells involved in CNS defense to damage).
The overall degradative function of lysosomes also relies on the capacity of the lysosomal membranes to associate with target membranes, such as those of autophagic vacuoles.60 Interfering with the composition of lysosomal membranes has been shown to affect trafficking and fusion within the endo-lysosomal network. Using embryonic fibroblasts derived from mouse models of MPS IIIA and multiple sulfatase deficiency, it has been demonstrated that cholesterol abnormally accumulates in the endo-lysosomal membrane as a consequence of disease. This sequesters soluble N-ethylmaleimide-sensitive factor attachment protein receptors, which are in turn required for the function of the fusion machinery in these membranes, thus inhibiting this function.61 This impairment may have wide reaching consequences, such as the pathology of the mitochondrial system observed in the MPS IIIC mouse, caused by the accumulation of dysfunctional mitochondria otherwise eliminated by normal autophagy and lysosomal function.18
Genetics of MPS III
The mutations that have been sequenced in the alleles of the causative genes in patients diagnosed with MPS III are summarized in Table 3. A commentary of those deemed of interest due to frequency and/or phenotypic consequences follows.
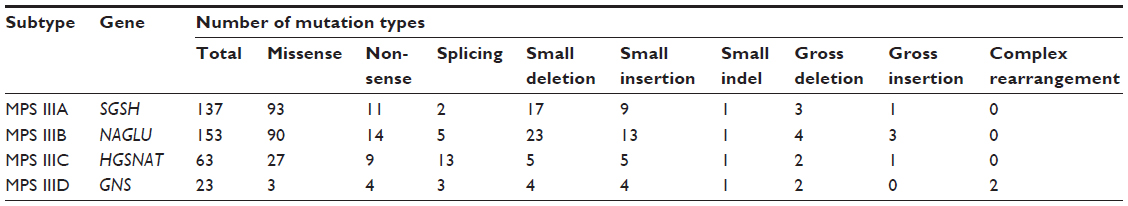 | Table 3 Summary of mutation types found in the alleles of the causative genes of patients diagnosed with MPS III |
MPS IIIA
The first patient known to be diagnosed with symptoms associated with an MPS caused by heparan sulfate accumulation was described by Sanfilippo et al.62
Later, it was observed that medium conditioned by the fibroblasts of a particular MPS III patient was able to reverse mucopolysaccharide accumulation in some but not all of a cohort of other MPS III fibroblasts.63 This cross-correction was repeated with urine from these patients. This suggested that the disease could be caused by the absence of one of two factors, and as such was arbitrarily classified as types A or B, based on which agent was evidently deficient.
Soon after, the increased mucopolysaccharide in MPS IIIA was purified from disease fibroblasts and shown to migrate with the mobility of heparan sulfate. In addition, a compound capable of reducing the accumulation of mucopolysaccharide was purified from the urine of healthy individuals and demonstrated to display N-sulfoglucosamine sulfohydrolase (SGSH, or sulfamidase) activity,64 an absence of which was observed in fibroblasts, leucocytes, and lymphoblasts from MPS IIIA patients.4,65 Eventually, the gene for MPS IIIA was identified.41 The gene for sulfamidase (SGSH) is localized to chromosome 17 q25.3 and contains eight exons.41,66 The cDNA codes for a product of approximately 55 kDa (excluding the signal peptide sequence). Within this, there are up to five asparagine-linked glycosylation sites.41
Specific sulfamidase mutations have been found in high frequencies in distinct geographical locations. The missense mutation p.R74C, which replaces a large, positively charged R group in the active site with one that is small and non-polar, occurs at a frequency of 56% of the Polish population.67 The p.R245H amino acid change is found in 31% of mutant alleles in Australia, 35% of those in Germany, and 58% of those in the Netherlands.67,68 This residue is not conserved among human sulfatases, and has little consequence on specific activity, but it does however affect the stability of the protein.69,70 The p.S66W missense mutation is found in 29% of alleles in a cohort of Italian patients;71 this is not conserved among sulfatases, but is close to the conserved cysteine within the active site that is catalytically converted to a Cα-formylglycine by SUMF1.49,50 A substitution of the small R group of serine with the aromatic R group of tryptophan would thus be predicted to interfere with the active site, and studies of this variant demonstrated a compromise between stability and activity.69,70
MPS IIIB
Soon after the genetically heterogeneous nature of MPS III63 and the factor deficient in MPS IIIA were discovered,64 it was shown that MPS IIIB was caused by a critically reduced activity of α-2-acetamido-2-deoxy-d-glucoside acetamidodeoxyglucohydrolase, (α-N-acetylglucosaminidase, or NAGLU).72
The gene for MPS IIIB was cloned around the same time as that for MPS IIIA.45,44 NAGLU is on chromosome 17q21.1 and contains six exons. The cDNA codes for a polypeptide of 743 amino acids, which contains a cleavable signal sequence and six asparagine-linked glycosylation sites. The mature protein’s 720 amino acids yield a molecular mass of approximately 80 kDa.
Unlike MPS IIIA, there are no common mutations in MPS IIIB. Rather, most of the known mutant alleles in MPS IIIB patients occur at low frequencies or not more than once. However, the p.F48L, p.G69S, p.S612G, and p.R643C missense mutations have been associated with a later-onset phenotype.73–75
MPS IIIC
Primarily due to the biochemical properties of the deficient protein, the gene for MPS IIIC remained more elusive. Originally, it was demonstrated that skin fibroblasts from MPS IIIC patients displayed a lack of HGSNAT activity.6 The responsible enzyme was then localized to the lysosomal membrane and shown to catalyze a transmembrane acetylation of the terminal glucosamine residue of intra-lysosomal heparan sulfatase.76–79 Over 20 years later, the gene was identified.42,43 HGSNAT is on chromosome 8p11.1 and consists of 18 exons. The cDNA encodes a polypeptide of 635 amino acids, which contains eleven transmembrane domains and five asparagine-linked glycosylation sites. The amino-terminal 42 amino acids form a signal peptide crucial for integration into the lysosomal membrane, where it is then post-translationally modified into a 27 kDa α chain and a 44 kDa β chain.80,81
There is contrasting but compelling evidence for two different models of the mechanism of HGSNAT activity (Figure 1). One proposes that the enzyme binds acetyl CoA from the cytoplasmic side of the lysosomal membrane, and is itself acetylated at an active site histidine. A conformational change allows for the transfer of the acetyl group into the lysosome. Once heparan sulfate interacts with the active site, the terminal glucosamine acquires the acetyl group, thus forming N-acetylglucosaminide.76–78,80 A second model suggests that catalysis occurs via a random ternary order complex, in one step, and with no direct intermediary acetylation of the enzyme.79,81
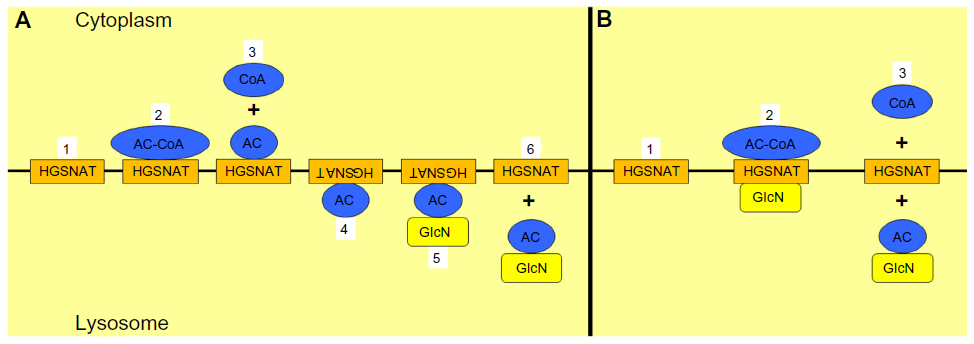 | Figure 1 Proposed models for the mechanism of heparan acetyl CoA: α-glucosaminide N-acetyltransferase (HGSNAT) activity. |
The missense mutations p.R344C and p.S518F account for 22% and 29%, respectively, of mutant alleles in the Netherlands.20 Furthermore, the deleterious nature of nearly all known missense mutations can be established via their occurrence in amino acids present in a transmembrane domain and/or conservation among orthologs.42,43,82 The protein and activity of the majority of the known missense mutations have been exogenously expressed in cell culture.83,84 Transfection of 18 of these resulted in negligible HGSNAT activity, while the remaining four displayed protein and function comparable to those of wild type, and so are therefore currently considered clinically insignificant polymorphisms.
Interestingly, mutations in HGSNAT have been found in six patients with the non-syndromic form of retinitis pigmentosa, each of whom was diagnosed in the third to fifth decade of life.85 These mutations result in reduced HGSNAT activities compared with healthy controls but sufficiently high to avoid neurodegeneration and other symptoms of MPS IIIC.85 Retinitis pigmentosa was also reported in the two patients with late-onset MPS IIIC.20,86 Their genotypes were identified as compound heterozygous for two missense mutations (p.G262R and p.S539C), both of which occur in amino acids predicted to be in the membrane,42,43,82 and whose separate overexpression in cell culture resulted in activity that was essentially undetectable.83,84
MPS IIID
Discounting MPS IIIE, which has not yet been described in humans, MPS IIID was the last to be classified, but the first to have its causative gene discovered. In 1980, skin fibroblasts from two patients with the clinical symptoms of MPS III and high urinary concentrations of heparan sulfate were demonstrated to lack the enzymatic activity required to release sulfate from N-acetylglucosamine 6-sulfate linkages. This was thus designated Sanfilippo type D (MPS IIID).7
GNS activity was subsequently purified and characterized48,87 and Edman sequencing of the isolated form permitted cloning of the coding gene.88,89 GNS is found on chromosome 12q14 and contains a total of 14 exons. The cDNA encodes for a polypeptide of 552 amino acids, and a protein of 78 kDa. In addition to a cleavable amino-terminal signal peptide of 36 amino acids, post-translational modification also results in cleavage by internal peptidases into a 32 kDa amino-terminal and 48 kDa carboxy-terminal species. It also contains 13 potential asparagine-linked glycosylation sites.
Similar to MPS IIIB, there are no common mutations in MPS IIID. It is noteworthy that there are relatively few missense mutations compared with the other MPS III subtypes (13%, see Table 3), and a dominance of deletions, insertions, and rearrangements.
MPS IIIE
The gene for MPS IIIE, N-glucosamine 3-O-sulfatase (ARSG), is localized to chromosome 17q24.2 and contains eleven exons.8,90,91 The cDNA encodes a protein of 525 amino acids, of which four are asparagine-linked glycosylation sites.90,91
Although ARSG has been molecularly characterized, and dog17 and mouse8 models of MPS IIIE exist, ARSG deficiency in humans has yet to be uncovered. These model organisms present with ataxia,17,92 which is only additionally observed in a New Zealand Huntaway dog model of MPS IIIA.13
Transcriptional regulation of the causative genes for MPS III
The expression of most lysosomal genes is mediated by the transcription factor EB (TFEB), a member of the microphthalmia-associated transcription factor E subfamily of basic helix-loop-helix transcription factors.93 This functions by binding to the Coordinated Lysosomal Expression and Regulation (CLEAR) element (a DNA motif consisting of the palindromic consensus sequence GTCACGTGAC) of associated genes, and increasing their transcription.93 Stresses such as starvation and lysosomal storage induce TFEB translocation to the nucleus, thus facilitating transcriptional activity, and contributing to an increase in CLEAR-containing lysosomal genes and lysosomal biogenesis in general.93–96
This phenomenon has been demonstrated to involve the regulatory multi-subunit containing mammalian target of rapamycin complex 1 (mTORC1). In normal conditions (for instance, in the presence of full nutrients and basal lysosomal function), a complex including vacuolar H+ (V)-ATPase, the Ragulator complex, and Rag GTPases is in an active state. In being so, it recruits mTORC1 to the lysosomal surface, where mTORC1 in turn becomes activated and phosphorylates TFEB, thus maintaining TFEB in the cytoplasm in an inactive form. However, in conditions of cellular stress, such as starvation or increased lysosomal storage, the Rag GTPases are inactivated, mTORC1 is detached from the lysosome, and TFEB is no longer phosphorylated. Consequently, TFEB translocates to the nucleus, thus facilitating its capacity as a transcription factor and increasing the expression of CLEAR element containing genes.97–101
The genes for MPS IIIA, MPS IIIB, MPS IIID, and MPS IIIE each contain at least two CLEAR elements (listed in Table 4) and their mRNA levels are increased in cell culture upon exogenous TFEB expression, and reduced via miRNA-mediated inhibition of TFEB.93
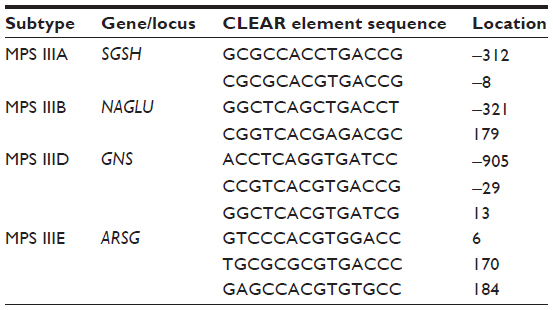 | Table 4 CLEAR element sequences present in the genes for MPS III |
Interestingly, HGSNAT does not contain a CLEAR element, and there is no evidence to suggest that it is directly regulated by TFEB.93,102–104 Furthermore, the mechanisms of HGSNAT transcriptional activation have not been characterized. However, the 5′ untranslated region of HGSNAT has been demonstrated to contain a SINE-VNTR-Alu (SVA) – B element insertion.105 Although its influence on HGSNAT expression requires further investigation, SVA elements are known to be non-autonomous, hominid-specific, non-long terminal repeat (LTR) retrotransposons that possess the capacity to sporadically generate disease-causing insertions.106
Laboratory diagnosis of MPS III
GAG analysis
The detection of heparan sulfate in the urine of suspected MPS III patients, or, more specifically, the GAGs within, is often the first biochemical step in diagnosis.
The traditional method of urinary GAG quantification involves the use of dimethylmethylene blue. Here, dimethylmethylene blue associates with sulfated GAG and the absorbance of the complex at a wavelength of 520 nm can be measured on a spectrophotometer.107,108 An elevation in urinary GAG will suggest a form of MPS.
Subsequent analysis can permit a further characterization of the accumulating product. High-resolution cellulose acetate electrophoresis of urinary samples can separate and identify distinct GAGs, thus providing an indication of MPS III from other forms.109 Additionally, qualitative gradient polyacrylamide gel electrophoretic separation can confirm increased secretion of particular heparan sulfate chains, thus diagnosing the particular MPS III subtype.110
Furthermore, methods comprising tandem mass spectrometry are now being developed. One involves quantification after derivatization of sulfated N-acetylhexosamine and sulfated N-acetylhexosamine-uronic acid.111 This method identifies specific oligosaccharides that accumulate as a result of a specific enzyme deficiency that act at the non-reducing end of heparan sulfate oligosaccharides.34,111,112 This can identify all MPS subtypes, except for MPS IIIB and MPS IIIC. Additionally, extraction and depolymerization of heparan sulfate using methanolysis under acidic conditions, and analysis of the subsequent desulfated disaccharide reaction products by liquid chromatography/tandem mass spectrometry can be used.113,114 This has the disadvantage that it cannot differentiate between MPS III types, but is simple and quick to run and thus a good first screen test. A liquid chromatography/tandem mass spectrometry-based technique that exploits the unique nature of the non-reducing end residue of the accumulated GAG upon the deficiency of a particular enzyme in the pathway has also been developed.115
Activity assay
Two types of artificial substrates that can be catalytically processed by the enzymes in skin fibroblasts and leucocytes otherwise deficient in MPS IIIA to MPS IIID are available: those that are radiolabeled116–119 and those that are fluorogenic.120–123 The basis of radiolabeled substrates is that the relevant enzymatic activity alters the charge of the substrate, thus permitting chromatographic separation. Regarding the latter, a fluorescent 4-methylumbelliferone product is released upon successful completion of the reaction, and can be detected by employing a fluorometric device.
An important fact of the fluorogenic substrate for the MPS IIIC assay is that HGSNAT acetylates it to a non-fluorescing intermediate, which is in turn hydrolyzed to the fluorescing form by β-hexosaminidase; so the presence of the latter in cells that are being evaluated is therefore imperative.122 Also, for MPS IIIA and MPS IIID, regardless of the chosen substrate, activity of a second sulfatase should be tested, in order to categorically exclude multiple sulfatase deficiency.
Genomic DNA sequencing
This is achieved by employing oligonucleotide primers that span the intron/exon junctions of the exons of the gene of interest using the polymerase chain reaction with genomic DNA as the template. Products can be sequenced and cross-referenced to the standard wild-type sequence and known disease-causing mutations and polymorphisms.
This is the definitive method for diagnosing not only disease but also carrier status (the latter is particularly difficult to detect via GAG analysis and activity assays as the levels between carriers and unaffected controls are likely to be similar). However, this is assuming the mutation occurs within one of the boundaries that are being sequenced. Any changes beyond the scope of the amplifying oligonucleotides in concept cannot be screened. Also, if an entire exon is absent in heterozygosity, this cannot be detected via standard qualitative genomic DNA sequencing (which will still have the alternative allele as a template). This would justify the development and application of quantitative techniques for mutation screening, particularly for MPS IIID, where there is a proportionally higher occurrence of large rearrangements.30
Future diagnostic methods
Two motivations for the creation of new screening techniques are decreasing the age at which it can be applied, and increasing output and/or decreasing cost, either by a simpler and more rapid method to detect a particular disease, or by an increased scope in the number of diseases that can be diagnosed using the same test.
A chorionic villus sample taken at 9–10 weeks gestation or cultured cells from 14–16 weeks amniocentesis can be tested for MPS III-causing mutations via genomic DNA sequencing or enzymatic activity assays.124 Increased heparan sulfate, although less reliably, has also been detected using electrophoresis in amniotic fluid taken at 16 weeks gestation from a pregnancy involving an MPS IIIA fetus.125 In each case, the main disadvantage is the intrusiveness of the method of sample collection.
Therefore, steps are being taken to establish multi-tasked newborn screening strategies for MPS III (and other lysosomal storage disorders), whether via detection of protein, activity, or GAG levels. Recent developments have included: measuring the protein amounts of multiple specific enzymes in dried blood spots using a multiplex immune-quantification assay, including sulfamidase and NAGLU;126,127 the synthesis of substrates for the enzymes deficient in MPS IIIA to MPS IIID that measure the reaction products in an in vitro activity assay via tandem mass spectrometry;128 and the use of tandem mass spectrometry to measure and characterize the accumulated GAGs in dried blood spots after digestion into disaccharides.129
Another advance is whole genome or exome sequencing, which would, in theory, permit screening of the entire genome or protein-coding region. These may possess similar caveats as genomic DNA sequencing of a selected gene but could also provide a standard method to test any subject for all sequencing abnormalities.
Treatments in development for MPS III
Due to the lack of a therapy to rescue the primary insult and/or pathophysiology of MPS III, present efforts are generally directed toward regulating behavior and sleep disturbances. However, a number of therapies for MPS III are being developed and evaluated.
Enzyme replacement therapy
The ability for sulfamidase, NAGLU, and GNS to be secreted from and taken up by the lysosomes within cells has provided the basis for the foundation of therapies for MPS III.
The major obstacle thus far in the establishment of enzyme replacement therapy (ERT) is the inability of these same proteins to cross the blood brain barrier (BBB). Injection of human recombinant sulfamidase directly into the brain130 or into cerebrospinal fluid via the cerebellomedullary cistern131 of MPS IIIA mice has been successful in alleviating a number of symptoms but has also emphasized the importance of early detection on account of the irreversibility of certain (particularly neurodegenerative) aspects of the pathophysiology.
The problem of potential invasiveness of targeting the brain has been grappled by engineering forms of sulfamidase to cross the BBB. For instance, a mixture of increasing the dose of recombinant sulfamidase and modifying the chemical structure to remove mannose-6-phosphate glycans has been attempted.132 Despite significantly increasing the effectiveness upon systemic delivery into the MPS IIIA mouse outside the CNS, it was less effective in the brain.
Gene therapy
The use of gene therapy is an extension of ERT insofar as it attempts to introduce the coding sequence of the protein of interest into the cells of the patient via the use of a viral vector, rather than synthesize it in and purify it from another host. Manipulated cells will not only themselves possess enzymatic activity, but will also secrete it into circulation to be taken up by un-altered cells.
Co-delivery of sulfamidase and SUMF1 via intraventricular injection of a recombinant adeno-associated virus (AAV) that drives the expression of both coding sequences resulted in increased sulfamidase activity in the mouse brain; a decrease in lysosomal storage and microglial activation and an enhancement of motor and cognitive capabilities were also observed.133 This strategy is now at the human clinical trial stage.134 Moreover, a therapy using a similar approach and with comparable results for MPS IIIB is being developed based upon successful and efficacious AAV-mediated human NAGLU delivery in MPS IIIB mice and Schipperke dog models.135,136
A recent advance has attempted to address a number of issues (repeated administration, invasiveness of delivery methods, and inability of the normal enzyme to cross the BBB) with one solution.137 Sulfamidase engineered to be fused to both the signal peptide of the highly secreted iduronate-2-sulfatase protein and the BBB binding domain of apolipoprotein B was included in an AAV-based vector designed to target the liver, thus allowing it to serve as a “factory” for perpetual systemic delivery of the modified enzyme. Adult MPS IIIA mice then intravascularly received this once. Not only did the signal peptide enhance secretion, but also the BBB binding domain permitted rescue of sulfamidase activity in the brain, reduction of neuropathology, and a restoration of behavior.137
Genistein
Genistein is an isoflavone purified from soy that exhibits an ability to reduce GAG levels. Although the overall mechanism by which this occurs has not been entirely clarified, its properties as a kinase inhibitor serve to induce TFEB transcriptional activity.103,104 For instance, it would seem that inhibition of the mTORC1-mediated phosphorylation of TFEB by genistein results in the translocation of TFEB to the nucleus from the cytoplasm, an increase in the expression of CLEAR-containing lysosomal genes, and an improvement in the removal of lysosomal storage products.103,104
Supplementation of growth medium with genistein resulted in reduction in GAG levels in skin fibroblasts derived from MPS IIIA and MPS IIIB patients.138 Moreover, treatment of an MPS IIIB mouse model showed an initial reduction in GAG storage139 and an improved clinical outcome when higher doses were provided.140 Most importantly, a pilot study involving five MPS IIIA and five MPS IIIB patients were administered with a genistein-enriched soy isoflavone extract for 12 months, which reduced urinary GAG levels, enhanced hair morphology, and improved behavior.141
Substrate deprivation therapy
In the context of MPS III, the purpose of this strategy is to reduce the levels of heparan sulfate containing GAGs by decreasing the amounts that are initially generated. Treatment of MPS IIIA mice with the non-specific inhibitor of GAG synthesis Rhodamine B resulted in reduced urinary GAG, as well as levels in the liver and the brain, and improved learning and memory as determined by water maze, with no long-term adverse effects.142–144
Development of a therapy for MPS IIIC
The rescue of HGSNAT activity has its own challenges. As with all disorders caused by faulty multi-pass transmembrane protein expression, approaches that rescue deficiencies of soluble proteins that can be secreted and taken up (such as ERT) are not applicable to HGSNAT deficiency. Some success has been achieved with molecular chaperones to rescue HGSNAT activity.83,145 These are typically reversible competitive inhibitors of the enzyme in question, which are administered at sub-inhibiting concentrations. The basis of this is that the inhibitor is capable of associating with its interacting site and aiding the structural restoration of the misfolded protein. In doing so, it restores the biochemical deficiency, which ultimately manifests as the disease in question.146 Modified U1 spliceosomal RNAs that recognize mutated donor sites of selected splice-site mutants and restore RNA splicing of HGSNAT have also been evaluated.145 Yet, such therapies will even in theory be successful in restoring only a subset of mutants (in these cases, missense and donor splice site, respectively). This presents a conundrum not just for MPS IIIC, but all disorders arising from multi-pass transmembrane protein expression deficiencies.
Conclusion
All the genes whose deficiencies cause MPS III have been discovered, and their gene products have been biochemically characterized to varying degrees. Furthermore, a number of genetic and biochemical diagnostic methods have been developed and adopted by diagnostic laboratories. Although MPS III disorders are rare, they are sufficiently debilitating to patients and challenging to parents and carers to warrant attention and research. Despite the current lack of a completely effective therapy for MPS III, there is much promise in therapy development, to the point that clinical trials to evaluate intrathecally-delivered ERT in MPS IIIA and gene therapy for MPS IIIA and MPS IIIB patients are in either clinical trial or being planned.
Acknowledgments
The author is grateful to Sophie Lazenkas and Prof John Hopwood (Lysosomal Diseases Research Unit, South Australian Health and Medical Research Institute, Adelaide, Australia) for critical reading of this manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486. | |
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. | |
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3421–3452. | |
Kresse H. Mucopolysaccharidosis 3 A (Sanfilippo A disease): deficiency of a heparin sulfamidase in skin fibroblasts and leucocytes. Biochem Biophys Res Commun. 1973;54:1111–1118. | |
von Figura K. Human α-N-acetylglucosaminidase. 1. Purification and properties. Eur J Biochem. 1977;80:523–533. | |
Klein U, Kresse H, von Figura K. Sanfilippo syndrome type C: deficiency of acetyl-CoA: α-glucosaminide N-acetyltransferase in skin fibroblasts. Proc Natl Acad Sci U S A. 1978;75:5185–5189. | |
Kresse H, Paschke E, von Figura K, Gilberg W, Fuchs W. Sanfilippo disease type D: deficiency of N-acetylglucosamine-6-sulfate sulfatase required for heparan sulfate degradation. Proc Natl Acad Sci U S A. 1980;77:6822–6826. | |
Kowalewski B, Lamanna WC, Lawrence R, et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci U S A. 2012;109:10310–10315. | |
Thompson JN, Jones MZ, Dawson G, Huffman PS. N-acetylglucosamine 6-sulphatase deficiency in a Nubian goat: a model of Sanfilippo syndrome type D (mucopolysaccharidosis IIID). J Inherit Metab Dis. 1992;15:760–768. | |
Fischer A, Carmichael KP, Munnell JF, et al. Sulfamidase deficiency in a family of Dachshunds: a canine model of mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr Res. 1998;44:74–82. | |
Li HH, Yu WH, Rozengurt N, et al. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding α-N-acetylglucosaminidase. Proc Natl Acad Sci U S A. 1999;96:14505–14510. | |
Bhaumik M, Muller VJ, Rozaklis T, et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology. 1999;9:1389–1396. | |
Jolly RD, Allan FJ, Collett MG, Rozaklis T, Muller VJ, Hopwood JJ. Mucopolysaccharidosis IIIA (Sanfilippo syndrome) in a New Zealand Huntaway dog with ataxia. NZ Vet J. 2000;48:144–148. | |
Aronovich EL, Johnston JM, Wang P, Giger U, Whitley CB. Molecular basis of mucopolysaccharidosis type IIIB in emu (Dromaius novaehollandiae): an avian model of Sanfilippo syndrome type B. Genomics. 2001;74:299–305. | |
Ellinwood NM, Wang P, Skeen T, et al. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N-acetyl-β-D-glucosaminidase deficiency in Schipperke dogs. J Inherit Metab Dis. 2003;26:489–504. | |
Crawley AC, Gliddon BL, Auclair D, et al. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006;1104:1–17. | |
Abitbol M, Thibaud JL, Olby NJ, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci U S A. 2010;107:14775–14780. | |
Martins C, Hulková H, Dridi L, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain. 2015;138:336–355. | |
van de Kamp JJ, Niermeijer MF, von Figura K, Giesberts MA. Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types A, B, and C). Clin Genet. 1981;20:152–160. | |
Ruijter GJ, Valstar MJ, van de Kamp JM, et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab. 2008;93:104–111. | |
de Ruijter J, Maas M, Janssen A, Wijburg FA. High prevalence of femoral head necrosis in Mucopolysaccharidosis type III (Sanfilippo disease): a national, observational, cross-sectional study. Mol Genet Metab. 2013;109:49–53. | |
Buhrman D, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type A. J Inherit Metab Dis. 2014;37:431–437. | |
Héron B, Mikaeloff Y, Froissart R, et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011;155:58–68. | |
Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995;73:77–81. | |
Fraser J, Wraith JE, Delatycki MB. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): a survey of managing clinicians. Clin Genet. 2002;62:418–421. | |
Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J Rare Dis. 2011;6:43. | |
Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: an epidemiological study. J Ment Defic Res. 1983;7:185–203. | |
Valstar MJ, Neijs S, Bruggenwirth HT, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68:876–887. | |
Valstar MJ, Bruggenwirth HT, Olmer R, et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis. 2010;33:759–767. | |
Valstar MJ, Bertoli-Avella AM, Wessels MW, et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010;31:E1348–E1360. | |
Kjellén L, Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. 1991;60:443–475. | |
Lawrence R, Brown JR, Lorey F, Dickson PI, Crawford BE, Esko JD. Glycan-based biomarkers for mucopolysaccharidoses. Mol Genet Metab. 2014;111:73–83. | |
Xu D, Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 2014;83:129–157. | |
Mason KE, Meikle PJ, Hopwood JJ, Fuller M. Characterization of sulphated oligosaccharides in mucopolysaccharidosis type IIIA by electrospray ionization mass spectrometry. Anal Chem. 2006;78:4534–4542. | |
Bielicki J, Freeman C, Clements PR, Hopwood JJ. Human liver iduronate-2-sulphatase: purification, characterization and catalytic properties. Biochem J. 1990;271:75–86. | |
Wilson PJ, Morris CP, Anson DS, et al. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A. 1990;87:8531–8535. | |
Clements PR, Brooks DA, Saccone GT, Hopwood JJ. Human α-l-iduronidase. 1. Purification, monoclonal antibody production, native and subunit molecular mass. Eur J Biochem. 1985;152:21–28. | |
Scott HS, Anson DS, Orsborn AM, et al. Human α-l-iduronidase: cDNA isolation and expression. Proc Natl Acad Sci U S A. 1991;88:9695–9699. | |
Kowalewski B, Lübke T, Kollmann K, et al. Molecular characterization of arylsulfatase G: expression, processing, glycosylation, transport, and activity. J Biol Chem. 2014;289:27992–28005. | |
Freeman C, Hopwood JJ. Human liver sulphamate sulphohydrolase. Determinations of native protein and subunit Mr values and influence of substrate agylcone structure on catalytic properties. Biochem J. 1986;234:83–92. | |
Scott HS, Blanch L, Guo XH, et al. Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Nat Genet. 1995;11:465–467. | |
Fan X, Zhang H, Zhang S, et al. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am J Hum Genet. 2006;79:738–744. | |
Hrebícek M, Mrázová L, Seyrantepe V, et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am J Hum Genet. 2006;79:807–819. | |
Weber B, Blanch L, Clements PR, Scott HS, Hopwood JJ. Cloning and expression of the gene involved in Sanfilippo B syndrome (mucopolysaccharidosis III B). Hum Mol Genet. 1996;5:771–777. | |
Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF. The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci U S A. 1996;93:6101–6105. | |
Freeman C, Hopwood JJ. Human liver glucuronate 2-sulphatase. Purification, characterization and catalytic properties. Biochem J. 1989;259:209–216. | |
Oshima A, Kyle JW, Miller RD, et al. Cloning, sequencing, and expression of cDNA for human β-glucuronidase. Proc Natl Acad Sci U S A. 1987;84:685–689. | |
Freeman C, Clements PR, Hopwood JJ. Human liver N-acetylglucosamine-6-sulfate sulphatase: purification and characterization. Biochem J. 1987;246:347–354. | |
Cosma MP, Pepe S, Annunziata I, et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. | |
Dierks T, Schmidt B, Borissenko LV, et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Cα-formylglycine generating enzyme. Cell. 2003;113:435–444. | |
Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. | |
Jones MZ, Alroy J, Rutledge JC, et al. Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J Neuropathol Exp Neurol. 1997;56:1158–1167. | |
McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol. 2004;480:415–426. | |
Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev Biol. 2004;15:433–444. | |
Dawson G, Fuller M, Helmsley KM, Hopwood JJ. Abnormal gangliosides are localized in lipid rafts in Sanfilippo (MPS3a) mouse brain. Neurochem Res. 2012;37:1372–1380. | |
Avila JL, Convit J. Inhibition of leucocytic lysosomal enzymes by glycosaminoglycans in vitro. Biochem J. 1975;152:57–64. | |
Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. | |
Wilkinson FL, Holley RJ, Langford-Smith KJ, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7:e35787. | |
King B, Marshall N, Beard H, et al. Evaluation of enzyme dose and dose-frequency in ameliorating substrate accumulation in MPS IIIA Huntaway dog brain. J Inherit Metab Dis. 2015;38:341–350. | |
Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. | |
Fraldi A, Annunziata F, Lombardi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607–3620. | |
Sanfilippo SJ, Podosin R, Langer LO, Good RA. Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J Pediatr. 1963;63:837–838. | |
Kresse H, Wiesmann U, Cantz M, Hall CW, Neufeld EF. Biochemical heterogeneity of the Sanfilippo syndrome: preliminary characterization of two deficient factors. Biochem Biophys Res Commun. 1971;42:892–898. | |
Kresse H, Neufeld EF. The Sanfilippo A corrective factor. Purification and mode of action. J Biol Chem. 1972;247:2164–2170. | |
Matalon R, Dorfman A. Sanfilippo A syndrome: sulfamidase deficiency in cultured skin fibroblasts and liver. J Clin Invest. 1974;54:907–912. | |
Karageorgos LE, Guo XH, Blanch L, et al. Structure and sequence of the human sulphamidase gene. DNA Res. 1996;3:269–271. | |
Bunge S, Ince H, Steglich C, et al. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A). Hum Mutat. 1997;10:479–485. | |
Weber B, Guo XH, Wraith JE, et al. Novel mutations in Sanfilippo A syndrome: implications for enzyme function. Hum Mol Genet. 1997;6:1573–1579. | |
Perkins KJ, Byers S, Yogalingam G, Weber B, Hopwood JJ. Expression and characterization of wild type and mutant recombinant human sulfamidase. Implications for Sanfilippo (Mucopolysaccharidosis IIIA) syndrome. J Biol Chem. 1999;274:37193–37199. | |
Perkins KJ, Muller V, Weber B, Hopwood JJ. Prediction of Sanfilippo phenotype severity from immunoquantification of heparan-N-sulfamidase in cultured fibroblasts from mucopolysaccharidosis type IIIA patients. Mol Genet Metab. 2001;73:306–312. | |
Di Natale P, Balzano N, Esposito S, Villani GR. Identification of molecular defects in Italian Sanfilippo A patients including 13 novel mutations. Hum Mutat. 1998;11:313–320. | |
O’Brien JS. Sanfilippo syndrome: profound deficiency of α-acetylglucosaminidase activity in organs and skin fibroblasts from type-B patients. Proc Natl Acad Sci U S A. 1972;69:1720–1722. | |
Zhao HG, Aronovich EL, Whitley CB. Genotype-phenotype correspondence in Sanfilippo syndrome type B. Am J Hum Genet. 1998;62:53–63. | |
Weber B, Guo XH, Kleijer WJ, van de Kamp JJ, Poorthuis BJ, Hopwood JJ. Sanfilippo type B syndrome (mucopolysaccharidosis III B): allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. Eur J Hum Genet. 1999;7:34–44. | |
Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: diagnostic, clinical, and biological implications. Hum Mutat. 2001;18:264–281. | |
Bame KJ, Rome LH. Acetyl coenzyme A: α-glucosaminide N-acetyltransferase. Evidence for a transmembrane acetylation mechanism. J Biol Chem. 1985;260:11293–11299. | |
Bame KJ, Rome LH. Acetyl-coenzyme A: α-glucosaminide N-acetyltransferase. Evidence for an active site histidine residue. J Biol Chem. 1986;261:10127–10132. | |
Bame KJ, Rome LH. Genetic evidence for transmembrane acetylation by lysosomes. Science. 1986;233:1087–1089. | |
Meikle PJ, Whittle AM, Hopwood JJ. Human acetyl-coenzyme A: α-glucosaminide N-acetyltransferase. Kinetic characterization and mechanistic interpretation. Biochem J. 1995;308:327–333. | |
Durand S, Feldhammer M, Bonneil E, Thibault P, Pshezhetsky AV. Analysis of the biogenesis of heparan sulfate acetyl-CoA: α-glucosaminide N-acetyltransferase provides insights into the mechanism underlying its complete deficiency in mucopolysaccharidosis IIIC. J Biol Chem. 2010;285:31233–31242. | |
Fan X, Tkachyova I, Sinha A, Rigat B, Mahuran D. Characterization of the biosynthesis, processing and kinetic mechanism of action of the enzyme deficient in mucopolysaccharidosis IIIC. PLoS One. 2011;6: e24951. | |
Fedele AO, Filocamo M, Di Rocco M, et al. Mutational analysis of the HGSNAT gene in Italian patients with mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Hum Mutat. 2007;28:523. | |
Feldhammer M, Durand S, Pshezhetsky AV. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS One. 2009;4:e7434. | |
Fedele AO, Hopwood JJ. Functional analysis of the HGSNAT gene in patients with mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Hum Mutat. 2010;31:E1574–E1586. | |
Haer-Wigman L, Newman H, Leibu R, et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-α-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet. 2015;24:3742–3751. | |
Berger-Plantinga EG, Vanneste JA, Groener JE, van Schooneveld MJ. Adult-onset dementia and retinitis pigmentosa due to mucopolysaccharidosis III-C in two sisters. J Neurol. 2004;251:479–481. | |
Freeman C, Hopwood JJ. Human liver N-acetylglucosamine-6-sulfate sulphatase. Catalytic properties. Biochem J. 1987;246:355–365. | |
Robertson DA, Freeman C, Nelson PV, Morris CP, Hopwood JJ. Human glucosamine-6-sulfatase cDNA reveals homology with steroid sulfatase. Biochem Biophys Res Commun. 1988;157:218–224. | |
Robertson DA, Callen DF, Baker EG, Morris CP, Hopwood JJ. Chromosomal localization of the gene for human glucosamine-6-sulphatase to 12q14. Hum Genet. 1988;79:175–178. | |
Ferrante P, Messali S, Meroni G, Ballabio A. Molecular and biochemical characterisation of a novel sulphatase gene: arylsulfatase G (ARSG). Eur J Hum Genet. 2002;10:813–818. | |
Frese MA, Schulz S, Dierks T. Arylsulfatase G, a novel lysosomal sulfatase. J Biol Chem. 2008;283:11388–11395. | |
Kowalewski B, Heimann P, Ortkras T, et al. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum Mol Genet. 2015;24:1856–1868. | |
Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. | |
Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–430. | |
Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. | |
Settembre C, Ballabio A. Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harb Perspect Biol. 2014;6: a016907. | |
Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–3258. | |
Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. | |
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. | |
Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. | |
Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200:475–491. | |
Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. | |
Moskot M, Montefusco S, Jakóbkiewicz-Banecka J, et al. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J Biol Chem. 2014;289:17054–17069. | |
Moskot M, Jakóbkiewicz-Banecka J, Kloska A, et al. Modulation of expression of genes involved in glycosaminoglycan metabolism and lysosome biogenesis by flavonoids. Sci Rep. 2015;5:9378. | |
Kwon YJ, Choi Y, Eo J, et al. Structure and expression analyses of SVA elements in relation to functional genes. Genomics Inform. 2013;11:142–148. | |
Raiz J, Damert A, Chira S, et al. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012;40:1666–1683. | |
Farndale RW, Sayers CA, Barrett AJ. A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect Tissue Res. 1982;9:247–248. | |
de Jong JG, Wevers RA, Liebrand-van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin Chem. 1992;38:803–807. | |
Hopwood JJ, Harrison JR. High-resolution electrophoresis of urinary glycosaminoglycans: an improved screening test for the mucopolysaccharidoses. Anal Biochem. 1982;119:120–127. | |
Byers S, Rozaklis T, Brumfield LK, Ranieri E, Hopwood JJ. Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab. 1998;65:282–290. | |
Fuller M, Rozaklis T, Ramsay SL, Hopwood JJ, Meikle PJ. Disease-specific markers for the mucopolysaccharidoses. Pediatr Res. 2004;56:733–738. | |
Mason K, Meikle P, Hopwood J, Fuller M. Distribution of heparan sulfate oligosaccharides in murine mucopolysaccharidosis type IIIA. Metabolites. 2014;4:1088–1100. | |
Auray-Blais C, Bhérer P, Gagnon R, et al. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol Genet Metab. 2011;102:49–56. | |
Trim PJ, Lau AA, Hopwood JJ, Snel MF. A simple method for early age phenotype confirmation using toe tissue from a mouse model of MPS IIIA. Rapid Commun Mass Spectrom. 2014;28:933–938. | |
Lawrence R, Brown JR, Al-Mafraji K, et al. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat Chem Biol. 2012;8:197–204. | |
Hopwood JJ, Elliott H. Sulphamidase activity in leucocytes, cultured skin fibroblasts and amniotic cells: diagnosis of the Sanfilippo A syndrome with the use of radiolabelled disaccharide substrate. Clin Sci. 1981;61:729–735. | |
Hopwood JJ, Elliott H. The diagnosis of the Sanfilippo C syndrome, using monosaccharide and oligosaccharide substrates to assay acetyl-CoA: 2-amino-2-deoxy- α-glucoside N-acetyltransferase activity. Clin Chim Acta. 1981;112:67–75. | |
Hopwood JJ, Elliott H. Detection of the Sanfilippo type B syndrome using radiolabelled oligosaccharides as substrates for the estimation of α-N-acetylglucosaminidase. Clin Chim Acta. 1982;120:77–86. | |
Elliott H, Hopwood JJ. Detection of the Sanfilippo D syndrome by the use of a radiolabeled monosaccharide sulfate as the substrate for the estimation of N-acetylglucosamine-6-sulfate sulfatase. Anal Biochem. 1984;138:205–209. | |
Marsh J, Fensom AH. 4-Methylumbelliferyl α-N-acetylglucosaminidase activity for diagnosis of Sanfilippo B disease. Clin Genet. 1985;27:258–262. | |
He W, Voznyi YaV, Boer AM, Kleijer WJ, van Diggelen OP. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type D (MPS IIID). J Inherit Metab Dis. 1993;16:935–941. | |
Voznyi YV, Karpova EA, Dudukina TV, et al. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease C (MPS III C). J Inherit Metab Dis. 1993;16:465–472. | |
Karpova EA, Voznyi YV, Keulemans JL, et al. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type A (MPS IIIA). J Inherit Metab Dis. 1996;19:278–285. | |
Hopwood JJ. Prenatal diagnosis of Sanfilippo syndrome. Prenat Diagn. 2005;25:148–150. | |
Whiteman P, Henderson H. A method for the determination of amniotic-fluid glycosaminoglycans and its application to the prenatal diagnosis of Hurler and Sanfilippo diseases. Clin Chim Acta. 1977;79:99–105. | |
Meikle PJ, Grasby DJ, Dean CJ, et al. Newborn screening for lysosomal storage disorders. Mol Genet Metab. 2006;88:307–314. | |
Fuller M, Tucker JN, Lang DL, et al. Screening patients referred to a metabolic clinic for lysosomal storage disorders. J Med Genet. 2011;48:422–425. | |
Wolfe BJ, Ghomashchi F, Kim T, et al. New substrates and enzyme assays for the detection of mucopolysaccharidosis III (Sanfilippo Syndrome) types A, B, C, and D by tandem mass spectrometry. Bioconjug Chem. 2012;23:557–564. | |
Tomatsu S, Fujii T, Fukushi M, et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2013;110:42–53. | |
Savas PS, Hemsley KM, Hopwood JJ. Intracerebral injection of sulfamidase delays neuropathology in murine MPS-IIIA. Mol Genet Metab. 2004;82:273–285. | |
Hemsley KM, King B, Hopwood JJ. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab. 2007;90:313–328. | |
Rozaklis T, Beard H, Hassiotis S, et al. Impact of high-dose, chemically modified sulfamidase on pathology in a murine model of MPS IIIA. Exp Neurol. 2011;230:123–130. | |
Fraldi A, Hemsley K, Crawley A, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16:2693–2702. | |
Tardieu M, Zérah M, Husson B, et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther. 2014;25:506–516. | |
Fu H, Kang L, Jennings JS, et al. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther. 2007;14:1065–1077. | |
Ellinwood NM, Ausseil J, Desmaris N, et al. Safe, efficient, and reproducible gene therapy of the brain in the dog models of Sanfilippo and Hurler syndromes. Mol Ther. 2011;19:251–259. | |
Sorrentino NC, D’Orsi L, Sambri I, et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. 2013;5:675–690. | |
Piotrowska E, Jakóbkiewicz-Banecka J, Baranska S, et al. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur J Hum Genet. 2006;14:846–852. | |
Malinowska M, Wilkinson FL, Bennett W, et al. Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide IIIB mice. Mol Genet Metab. 2009;98:235–242. | |
Malinowska M, Wilkinson FL, Langford-Smith KJ, et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS One. 2010;5:e14192. | |
Piotrowska E, Jakóbkiewicz-Banecka J, Tylki-Szymanska A, et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: An open-label, pilot study in 10 pediatric patients. Curr Ther Res Clin Exp. 2008;69:166–179. | |
Roberts AL, Fletcher JM, Moore L, Byers S. Trans-generational exposure to low levels of rhodamine B does not adversely affect litter size or liver function in murine mucopolysaccharidosis type IIIA. Mol Genet Metab. 2010;101:208–213. | |
Roberts AL, Rees MH, Klebe S, Fletcher JM, Byers S. Improvement in behaviour after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol Genet Metab. 2007;92:115–121. | |
Roberts AL, Thomas BJ, Wilkinson AS, Fletcher JM, Byers S. Inhibition of glycosaminoglycan synthesis using rhodamine B in a mouse model of mucopolysaccharidosis type IIIA. Pediatr Res. 2006;60:309–314. | |
Matos L, Canals I, Dridi L, et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J Rare Dis. 2014;9:180. | |
Parenti G, Andria G, Valenzano KJ. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther. 2015; 1138–1148. | |
Baehner F, Schmiedeskamp C, Krummenauer F, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011–1017. | |
Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156. | |
Pinto R, Caseiro C, Lemos M, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12:87–92. | |
Malm G, Lund AM, Månsson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr. 2008;97:1577–1581. | |
Lin HY, Lin SP, Chuang CK, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am J Med Genet A. 2009;149:960–964. | |
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.