Back to Journals » International Journal of Nanomedicine » Volume 15
Salinomycin-Loaded Small-Molecule Nanoprodrugs Enhance Anticancer Activity in Hepatocellular Carcinoma
Authors Wang J, Zhuo J, Tao Y, Xu S, Chen Z, Yang F, Ke Q, Xie H, Zheng S, Wang H ![]() , Xu X
, Xu X
Received 3 November 2019
Accepted for publication 10 August 2020
Published 15 September 2020 Volume 2020:15 Pages 6839—6854
DOI https://doi.org/10.2147/IJN.S236928
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Mian Wang
Jianguo Wang,1,* Jianyong Zhuo,2,* Yaoye Tao,2 Shengjun Xu,2 Zun Chen,2 Fan Yang,2 Qinghong Ke,2,3 Haiyang Xie,2,3 Shusen Zheng,2– 4 Hangxiang Wang,2,3 Xiao Xu1
1Department of Hepatobiliary and Pancreatic Surgery, Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou 310006, People’s Republic of China; 2NHC Key Laboratory of Combined Multi-Organ Transplantation, Hangzhou 310003, People’s Republic of China; 3Department of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, People’s Republic of China; 4Department of Hepatobiliary and Pancreatic Surgery, Shulan (Hangzhou) Hospital, Hangzhou 310003, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiao Xu
Department of Hepatobiliary and Pancreatic Surgery, Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou 310006, People’s Republic of China
Tel/Fax +86-571-87236567
Email [email protected]
Background: There is currently no effective treatment for advanced hepatocellular carcinoma (HCC), and chemotherapy has little effect on long-term survival of HCC patients, largely due to the cancer stem cell (CSC) chemoresistance of HCC.
Methods: We constructed a small-molecule nanometer-sized prodrug (nanoprodrug) loaded with salinomycin (SAL) for the treatment of HCC. SAL was encapsulated by the prodrug LA-SN38 (linoleic acid modified 7-ethyl-10-hydroxycamptothecin) to construct a self-assembled nanoprodrug further PEGylated with DSPE-PEG2000. We characterized this codelivered nanoprodrug and its antitumor activity both in vitro in human HCC cell lines and in vivo in mice.
Results: Delivery of the SAL- and LA-SN38-based nanoprodrugs effectively promoted apoptosis of HCC cells, exerted inhibition of HCC tumor-sphere formation as well as HCC cell motility and invasion, and reduced the proportion of CD133+ HCC-CSC cells. In nude mice, the nanoprodrug suppressed growth of tumor xenografts derived from human cell lines and patient.
Conclusion: Our results show that SAL-based nanoprodrugs are a promising platform for treating patients with HCC and a novel strategy for combination therapy of cancers.
Keywords: small-molecule prodrugs, salinomycin, self-assemble, cancer stem cells, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is an extremely lethal malignancy with a high rate of recurrence and limited treatment options. For patients with early HCC, surgical resection is the most favorable treatment available; however, an effective treatment for advanced HCC is lacking and chemotherapy has little effect on the long-term survival of HCC patients, largely due to HCC chemoresistance.1 Cancer stem cells (CSCs) have been found to contribute to HCC chemoresistance.2 CSCs can initiate new tumors and self-renew, and evidence suggests that CSCs are critical for tumor regeneration, metastasis, and recurrence. Hence, CSCs have become key targets for cancer-prevention strategies.
Several studies have shown that CSCs can be eliminated by specific drugs.3 For example, EpCAM+ CSCs resistant to 5-FU become sensitive to chemotherapy when combined with a Wnt-signaling inhibitor.4 In HCC cells with activated CDK1/PDK1 and β-Catenin signaling, CD133+ and CD90+ CSC subpopulations are significantly decreased by the combinatorial of a CDK1 inhibitor and the chemotherapeutic sorafenib.5 These studies suggest that targeting CSCs in combination with chemotherapy is a promising treatment strategy to overcome HCC-CSC resistance and viability.
However, in recent years, combination therapy for cancer treatment has provoked widespread concern because the administration of drugs in combination is accompanied by challenges, such as plasma instability, low bioavailability, and systemic toxicity.6,7 Therefore, the codelivery of two or multiple drugs with nanoparticles has been proposed to achieve targeted tumor delivery, extend circulation time, and reduce side effects.
Liposomes have been used clinically as vehicles for drug release and are therapeutically benign. Thus, a promising method for overcoming the disadvantages of previous drug carrier efforts is to use amphiphilic-synthesis to self-assemble pure prodrugs. Thus, we previously modified SN-38, the active ingredient of CPT-11—a clinical anticancer drug—with the polyunsaturated fatty-acid linoleic acid (LA), a hydrophobic moiety. Then, the modified SN38 were amphiphilic and could induce self-assembly in aqueous solutions. In our previous study, the prepared supramolecular nanometer-sized prodrug (nanoprodrug) showed excellent anti-tumor effects in vivo and in vitro.8 However, whether LA-SN38 can be used as a pharmaceutical carrier loaded with compounds that possess CSC-specific toxicity to achieve combinational therapeutic effects remains to be investigated.
Salinomycin (SAL) is an antibacterial therapeutic drug extracted from Streptomyces albus that has the capacity to suppress breast cancer CSCs.9,10 In recent years, an increasing number of studies have suggested that SAL can eliminate CSCs in various cancers including HCC.11,12 However, the poor aqueous solubility of SAL as well as its neural and muscular toxicity hinders its clinical application. There have been efforts to develop nanodrug delivery systems with reduced side effects by increasing the water solubility of SAL. Nanoparticles are one solution for this delivery problem, as nanoparticles loaded with SAL that exhibits excellent anti-tumor effects against CSCs have previously been prepared.13,14 Nanoparticle codelivery systems have yet to be successfully used with SAL to treat HCC-CSCs.
Here, we constructed a SAL-loaded self-assembled LA-SN38 prodrug nanoparticle PEGylated with DSPE-PEG2000 and evaluated its inhibitory effects on HCC cells in vitro and in vivo. We found that the nanoprodrug codelivered system effectively improved therapeutic efficacy in HCC and suppressed tumor growth in a nude-mouse-transplanted tumor model.
Materials and Methods
Materials
Salinomycin (SAL) and SN38 (7-ethyl-10-hydroxycamptothecin) were purchased from MedChemExpress (New Jersey, USA). LA-SN38 was synthesized in our lab. DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000]) was purchased from A.V.T. (Shanghai, China).
Patients and Specimens
Fresh tumor tissues were collected from HCC patients. The specimen used in this study isolated from a patient with tumor recurrence and not received chemotherapy before surgery. Signed informed consent was obtained from each patient before sample acquisition. This study was approved by the ethics committee of the First Affiliated Hospital of Zhejiang University.
Cell Cultures
The human HCC cell lines, HCC-LM3, Hep3B, and PLC/PRF/5, were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai Institute of Cell Biology). HCC-LM3 cells were cultured in DMEM (Gibco, USA) supplemented with 10% FBS (Gibco, USA). Hep3B and PLC/PRF/5 lines were cultured in MEM (Gibco, USA) supplemented with 10% FBS (Gibco, USA).
Preparation of Nanoparticles
First, SAL, LA-SN38, and DSPE-PEG2000 (100 mg/mL) were dissolved in dimethyl sulfoxide (DMSO). For LA-SN38 nanoparticles, the prodrugs were premixed with DSPE-PEG2000 and rapidly injected into water. The co-loaded nanoparticles were prepared by mixing SAL, LA-SN38, and DSPE-PEG2000 solutions and then injecting the solutions into water. The prepared nanoparticles were designated as follows: (1) LA-SN38 prodrug nanoparticles PEGylated by DSPE-PEG2000 (LDN), and (2) SAL and LA-SN38 coloaded prodrug nanoparticles PEGylated by DSPE-PEG2000 (CDN).
Particle morphology and size were analyzed by dynamic light scattering (DLS), transmission electron microscopy (TEM), and a Malvern Nano-ZS 90 laser particle-size analyzer. TEM samples were prepared with 1% uranyl acetate and were observed using a TECNAL 10 transmission electron microscope (Philips) at an acceleration voltage of 80 kV.
Stability of Prodrug-Assembled LDN and CDN
Prepared LDN and CDN were suspended in PBS (containing 10% FBS) at 37°C for 2 days. At predetermined time intervals (2, 4, 8, 24, and 48 h), particle sizes were measured on a Malvern Nano-ZS 90 laser particle-size analyzer.
In vitro Drug Release of LDN and CDN
The drug release from LDN and CDN was investigated using a dialysis diffusion method. Briefly, 5 mL of prepared LDN and CDN (0.5 mg/mL) were placed into the dialysis bag (molecular weight cutoff: 3000 Da) and suspended in 40 mL PBS (containing 0.1% Tween 80). The sealed tube was incubated at 37°C with stirring (100 rpm). At predetermined time intervals (0.5, 1, 2, 4, 8, 24, 48, 72, and 96 h), the release media (1 mL) were collected and fresh media (1 mL) were supplemented. The content of the released drug was determined by ultraviolet spectrophotometer.
Determination of the Encapsulation Efficacy (EE) and Drug Loading (DL)
After careful preparation of LDN and CDN, solution was centrifuged by centrifugal filter devices (Amicon Ultra, MWCO 10 kDa, Millipore) to remove the drugs that are not entrapped inside NPs. The filtrate was collected, and the amounts of free SN38 were determined at 368 nm using ultraviolet spectrophotometer (UV2700, SHIMADZU, Japan). The amounts of free SAL were determined as described previously.15 Briefly, the filtrate derivatized with vanillin (Aladdin, China) in an acidic medium at 72°C for 40 min. The derivatization mixture was determined at 527 nm by ultraviolet spectrophotometer. The EE of SN38 or SAL was calculated from the following formula: (We – Wf)/We × 100%. The DL of SN38 or SAL was calculated from the following formula: (We – Wf)/Wt × 100%. We, Wf, and Wt were defined as the quality of totally added SN38 or SAL, the quality of free LA-SN38 or SAL, and the quality of NPs, respectively.
In vitro Cellular Uptake
Confocal microscopic studies were performed to observe the in vitro cellular uptake of LDN and CDN in HCC-LM3 cells. Cy5.5-labeled DSPE-PEG2000 was used to prepare the NPs. HCC-LM3 cells were initially seeded on 35 mm glass-bottom cell-culture dish (Thermo Scientific Nunc, USA) at a density of 2 × 105 cells dish−1 and incubated for 24 h. Later, NPs were added to the cells, which were then incubated for 2 hours. After incubation, the cells were washed with PBS and immediately visualized under confocal laser scanning microscopy (Olympus Fluoview FV-3000, Japan) excitation at 640 nm.
Cytotoxicity and Combined Effect Evaluation
To measure the cytotoxicity of these nanoparticles and the anti-tumor synergistic effect of SAL and LA-SN38, HCC cells were seeded in 96-well plates at a density of 5000 cells/well. The cells were incubated overnight and treated with two doses of SAL (0.25 and 0.5 μM) were used in combination with different concentrations of SN38 (0, 0.1, 0.5, 1, 2, 5 and 10 μM), respectively. These three groups were defined as LDN, LDN+ SAL 0.25 μM and LDN+ SAL 0.5μM. After 48h incubation, the CCK-8 assay was used to examine cell viability. All experiments were performed in triplicate.
The coefficient of drug interaction (CDI) was used to analyze the synergistic effect between LDN and SAL.16 CDI was calculated as follows: CDI=AB/(A×B). AB represents the ratio of the CDN group to the control group in OD490, and A or B means the ratio of LDN/SAL group to the control group in OD490. CDI<1 indicates synergism, CDI<0.7 indicates a significant synergistic effect, CDI=1 indicates additivity and CDI>1 indicates antagonism.
Colony Formation Assay
Cells (1×103cells/plate) were seeded in 6-well plates and incubated with free SAL, LDN, or CDN for 2 weeks. Then, the cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed twice with PBS and stained with Wright‐Giemsa (NJJCBIO, China).
EdU (5-Ethynyl-2ʹ-Deoxyuridine) Proliferation Assay
To evaluate cell-proliferation, HCC cells were cultured with free SAL, LDN, or CDN. Subsequently, the cells were incubated with 50 mM EdU for 6 h according to the manufacturer’s protocol. The cell nuclei were stained with DAPI (Sigma) at a concentration of 1 mg/mL for 20 min. The results were determined by fluorescence microscopy (Nikon, 80i, Japan).
Apoptosis Analysis
HCC-LM3 and PLC/PRF/5 cells were treated with DMSO, free SAL (5 μM), LDN (5 μM), or CDN (5 μM) for 48 h. The cells were then harvested and washed twice with cold PBS. Subsequently, the cells were resuspended in 100 μL of binding buffer and 5 μL of FITC AnnexinV and 5 μL of propidium iodide were added. Then, the cells were incubated in the dark for 15 min. The apoptosis rate of the HCC cells was quantified with a flow cytometer (BD Biosciences, San Jose, CA, USA).
Transwell Invasion and Wound-Healing Assays
HCC cell invasion was evaluated using the transwell assay. Briefly, transwell cell-culture chambers were pre-coated with 50 μL of Matrigel (BD) in the upper chamber. After 30 min of incubation at 37°C, 5 × 104 cells in 200 μL of DMEM medium (without FBS) were added to the upper chamber. Then, 600 μL of DMEM medium supplemented with 10% FBS was added to the lower chambers. The cells were treated with DMSO, free SAL, LDN, or CDN and were incubated at 37°C for 24 h. A cotton swab was used to remove the non-invasive cells. The chambers were fixed in methanol for 10 min and stained with 0.5% crystal violet for 15 min. Six random microscopic fields (magnification × 100) were counted per well. All experiments were repeated three times.
Next, 5 × 105 cells were seeded into six-well plates. After incubation for 24 h, a 200 μL pipette tip was used to scratch the cells. Subsequently, the cells were treated with medium containing DMSO, free SAL, LDN, or CDN. The cells were photographed at 0 and 48 h in the same position. Wound closure was calculated as follows: (original wound area – actual wound area)/area of the original wound × 100.
Stem Cell Sorting and Analysis
HCC cells were stained with anti-CD133-PE (AC133, Miltenyi Biotec; Auburn, CA, USA). The cell samples were sorted on a FACS Calibur apparatus, and the percentage of CD133+ cells was analyzed using Cell Quest software (BD Biosciences).
Tumor-Spheroid Formation
Spheroid formation was calculated to determine the effect of SAL-induced stem-cell inhibition on HCC cells. Cells were treated with DMSO, free SAL, LDN, or CDN and seeded in six-well ultra-low-attachment plates (Corning) at a density of 500 cells per well. Spheres were incubated in serum-free DMEM/F12 supplemented with 2% B27 (Invitrogen), 20 ng/mL human recombinant fibroblast growth factor 2 (FGF-2), 20 ng/mL epidermal growth factor (EGF), 0.4% BSA, and 5 μg/mL insulin (Sigma). Seven days after plating, the spheres were photographed and counted under a microscope.
Animal Experiments
For the cell-derived tumor-xenograft (CDX) model, 5 × 106 HCC-LM3 cells were subcutaneously injected into male Balb/C nude mice. For the patient-derived tumor-xenograft (PDX) model, fresh patient HCC tissues were grafted into male NOD/SCID-IL-2R-γc-KO (NSG) mice by subcutaneous injections. Once the tumor volume reached 200 mm3, the tumors were cut into pieces and were subsequently implanted into Balb/C nude mice for subsequent experiments. Once tumors were established to be 100 mm3, mice were randomized and treated with sterile saline, free SAL (4 mg/kg), LDN (10 mg/kg), and CDN (10 mg/kg + 4 mg/kg) for two weeks (every two days by intravenous injection). Tumor volume and body weight were estimated every three days using electronic Vernier calipers. The tumor-volume-calculation formula used was as follows: (length × width2)/2. All animal experiments were approved by the ethics committee of the First Affiliated Hospital of Zhejiang University. All experiments met the standards of the institutional animal Care and Use committee of the Zhejiang University School of Medicine.
Statistical Analysis
Statistical analyses were performed using SPSS 11.0 software. All quantitative data are expressed as the mean ± SD. The statistical significance among the treatment groups were assessed using two-tailed unpaired Student’s t-test. P-values <0.05 (*), <0.01 (**), or <0.001 (***) were considered to indicate statistical significance.
Results
Preparation and Characterization of Nanoparticles
As a proof of concept, we prepared nanoformulations by quickly injecting the compounds into water. DSPE-PEG2000, a widely used amphiphilic copolymer with the advantages of reduced clearance by the reticuloendothelial system and prolonged circulation time in vivo for drug delivery, was adopted for PEGylation of self-assembled prodrug nanoparticles.17 Briefly, LA-SN38 and DSPE-PEG2000 were premixed in DMSO at a 1:5 (w/w) ratio and then rapidly injected into water to obtain LDN. To prepare codelivered nanoparticles, SAL, LA-SN38, and DSPE-PEG2000 were mixed at a 2:5:35 (w/w/w) ratio and then injected into water to obtain CDN (Figure 1). DLS was used to measure the hydrodynamic diameters of LDN and CDN, which were approximately 70 ± 0.6 nm and 61.7 ± 1.6 nm (Figure 2A), respectively. The results indicated that the hydrodynamic diameter of CDN is smaller than the diameter of LDN. It is well known that the enhanced permeability and retention (EPR) effect is the basis for nanoparticle accumulation and penetration in tumor tissues and that particle size is considered one of the main factors of the EPR effect.18,19 Increasing evidence suggests that reducing particle size will improve tumor penetration and distribution.20–22 Therefore, compared with the accumulation of LDNs, the accumulation of CDN may be enhanced in tumor tissues, potentially enabling therapeutic effects. TEM was used to observe the morphology of LDN and CDN, and showed that LDN and CDN were dispersed as individual particles with well-defined spherical structures (Figure 2B).
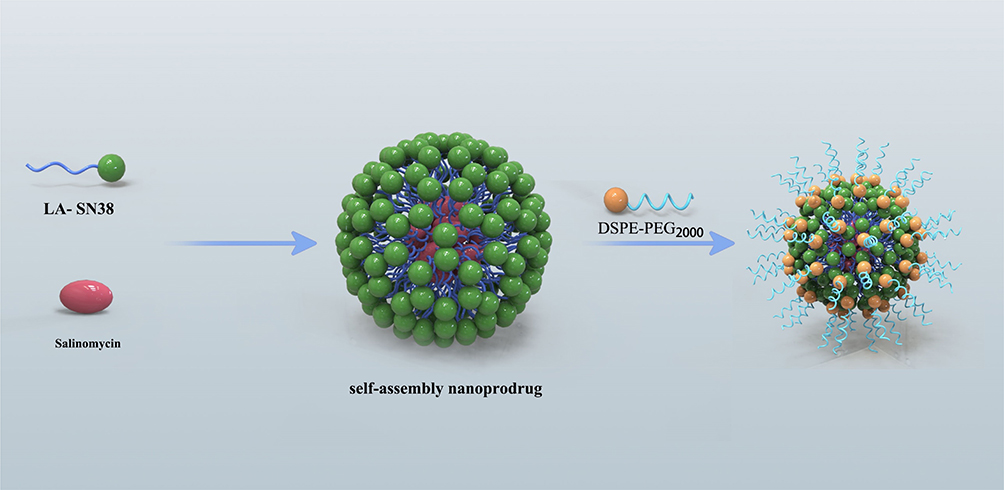 |
Figure 1 Schematic illustration of the amphiphilic self-assembly of small-molecule prodrugs as drug-carrier platforms for systemic drug codelivery. |
 |
Figure 2 Characterization of the nanoparticles. (A) LDN and CDN size distribution and (B) NP morphology, as observed by TEM. Scale bar: 100 nm. (C) Hydrodynamic size of LDN and CDN in the presence of 10% serum at 37 °C. (D) Release profiles of SN38/SAL from NPs. |
The stability of LDN and CDN was studied by measuring the hydrodynamic size of LDN and CDN in the PBS (containing 10% FBS) at 37°C for 48h. The results showed that the structural stability of CDN was maintained for 48 h of incubation in serum-conditioned media, while the size of LDN fluctuated between 50 nm and 100 nm after 24h of incubation (Figure 2C). The drug release profile is an important evaluation index of drug delivery system. Therefore, the SN38 and SAL release profile from LDN and CDN was evaluated at pH 7.4. About 70% of SN38 loaded in the LDN was released within 4h; however, SN38 and SAL loaded in the CDN were released slowly in 96h (Figure 2D). As shown in Table 1, the drug EE of LDN or CDN was both >95%. The SN38 and SAL drug loading of CDN was about 33.64% and 32.71%, respectively, indicating a high drug-loading capacity of CDN.
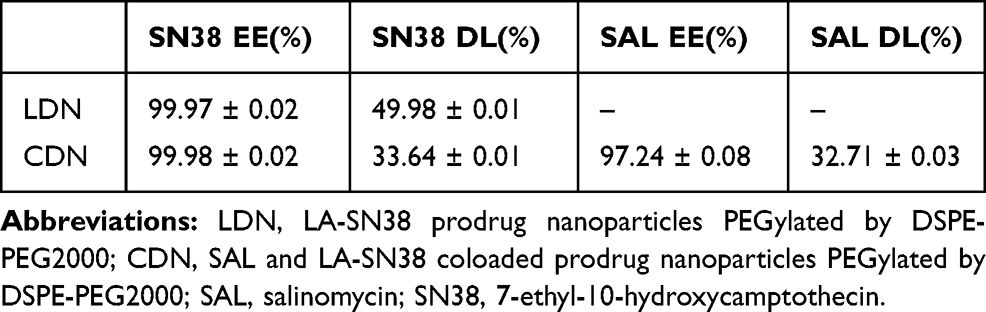 |
Table 1 Encapsulation Efficiency (EE) and Drug Loading (DL) of LDN and CDN |
In vitro Cellular Uptake of LDN and CDN
To calculate the in vitro cellular uptake of LDN and CDN, Cy5.5-labeled DSPE-PEG2000 was used to prepare the NPs. The prepared LDN and CDN were added to HCC-LM3 culture medium and incubated for 2 hours. Confocal microscopy was used to evaluate the cellular uptake. The images indicated that LDN and CDN were effectively internalized in HCC-LM3 cells (Figure 3).
 |
Figure 3 Cellular uptake of LDN and CDN. HCC-LM3 cells were treated with LDN and CDN and incubated for 2h. The blue fluorescence of Cy5.5 was analyzed by a confocal microscope. After 2h of incubation, blue fluorescence signal was observed in HCC-LM3 cells indicating that LDN and CDN had been internalized by tumor cells. Scale bar: 100 μm. |
In vitro Cytotoxicity
We next evaluated the anti-tumor effects of these formulations. In vitro cytotoxicity of the HCC cell lines HCC-LM3, Hep3B, and PLC/PRF/5 was evaluated by the CCK-8 assay in. The tested cells showed dose-dependent viability inhibition after treated with LDN, LDN+ SAL 0.25 μM, and LDN+ SAL 0.5μM for 48 h at 37°C (Figure 4A). The IC50 values are exhibited in Table 2. As expected, compared to monotherapy with LDN, the combination therapy significantly enhanced the anti-proliferative effects of cancer cells. These results suggest that SAL decreases the IC50 values of LA-SN38.
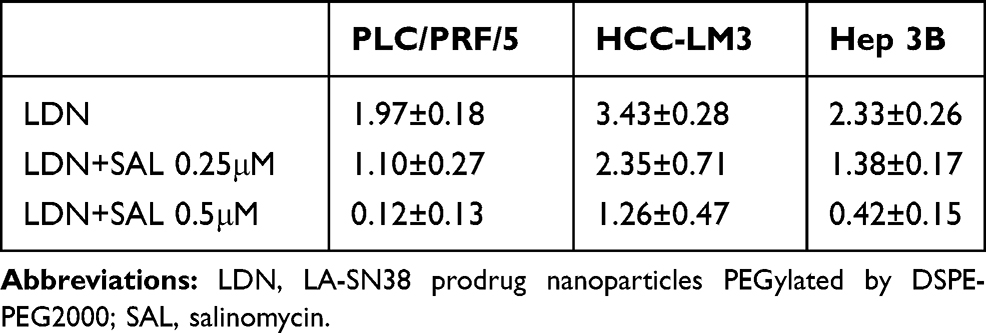 |
Table 2 Results of the Cell-Viability Assay (IC50 values, μM) Following Treatment with LDN and LDN Combination with SAL in HCC-LM3, Hep3B, and PLC/PRF/5 Cell Lines |
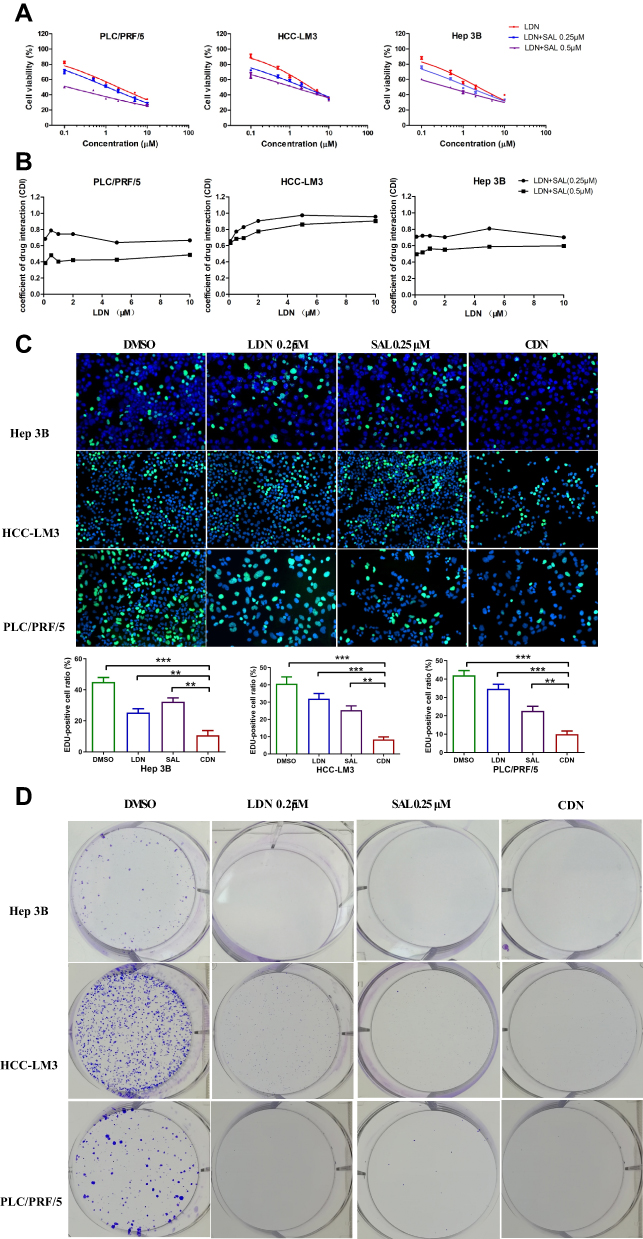 |
Figure 4 Salinomycin enhances the effects of LA-SN38 treatment in HCC cells. (A) The CCK-8 assay was used to detect the viability of PLC/PRF/5, HCC-LM3, and Hep 3B cells following SAL, LDN and LDN combination with SAL treatment. The combination therapy significantly inhibited the proliferative effects of cancer cells. (B) The CDI of LDN combined with SAL on PLC/PRF/5, HCC-LM3, and Hep 3B cells. The CDI was <1 in all cell lines, which represents a synergistic effect between LDN and SAL. (C) Cell proliferation measured by an EdU assay. (D) Clonogenic assay of LDN or free SAL or CDN in PLC/PRF/5, HCC-LM3, and Hep 3B cells. Data are expressed as the following: **P < 0.01, ***P < 0.001 vs control. |
Furthermore, the coefficient of drug interaction (CDI) was used to evaluate the drug interaction between LDN and SAL. The results suggested that the CDI was <1 in all cell lines, which represents a synergistic effect between LDN+ SAL. And we found that the degree of synergistic effect varies in different cells. However, the synergistic effect increases as the concentration of SAL increases in all the three cell lines (Figure 4B).
In addition, the antitumor effects of the two drugs were further investigated using enhanced BrdU (EdU) staining and Colony formation assay. HCC cells treated with LDN+ SAL presented with a lower EdU-positive cell ratio than compared to LDN and free SAL at equivalent drug concentration (Figure 4C). Meanwhile, LDN, free SAL and CDN all induced significantly decreased colonies compared with control (Figure 4D). These results indicate that combining LA-SN38 and SAL significantly decreases the cell mitosis rate.
Codelivery of Prodrug Nanoparticles Promotes Apoptosis of HCC Cells in vitro
Many studies have shown that CSCs play a key role in chemotherapeutic resistance.23,24 Therefore, we hypothesized that the CSC-inhibition drug SAL might overcome the resistance of chemotherapeutic drugs. Here, we tested the apoptosis-inducing effect of LDN, free SAL, and CDN via Annexin V-FITC/PI staining to detect apoptosis and cell death by flow cytometry. As shown in Figure 5, apoptosis of PLC/PRF/5 and LM3 cells treated with CDN significantly increased; the apoptotic ratio was 62.05%±4.46 and 62.43±2.52%, respectively. These results suggest that CDN has a stronger apoptotic-inducing effect on HCC cells than LDN (26.39±2.92% in PLC/PRF/5 and 36.31%±3.75 in LM3 cells) or free SAL (22.27±5.11% in PLC/PRF/5 cells and 19.19%±6.91 in LM3 cells).
 |
Figure 5 CDN significantly induces HCC cell apoptosis in vitro. Apoptosis of PLC/PRF/5 and HCC-LM3 cells, treated with different drug formulations, was detected by Annexin V and PI double staining. The CDN has a stronger apoptotic-inducing effect on HCC cells than LDN (62.05%±4.46 vs 26.39±2.92% in PLC/PRF/5 and 62.43±2.52% vs 36.31%±3.75% in LM3 cells) or free SAL (62.05%±4.46 vs 22.27±5.11% in PLC/PRF/5 cells and 62.43±2.52% vs 19.19%±6.91 in LM3 cells). Data are expressed as the following: **P < 0.01, ***P < 0.001 vs control. |
Codelivery of SAL and LA-SN38 Effectively Inhibits the HCC Cell Migration and Invasion
Over the past few decades, accumulating evidence has suggested that CSCs comprise a small percentage of total cells in tumor tissues; however, CSCs are considered the primary causes of tumor recurrence and metastasis.25–27 Furthermore, cancer cells can acquire CSC characteristics by inducing epithelial–mesenchymal transition (EMT), which is critical for tumor invasion and metastasis.28 Here, the inhibitory effect of different therapeutic formulations on HCC cell migration and invasion cells was examined by using the transwell and wound-healing assays. As shown in Figure 6A, the ability of invasion was indicated by the number of cells in the upper chamber that travelled through the membrane to the lower chamber. The results suggested that cells treated with free SAL (327.3 ± 26.1 in LM3 cells and 337.3 ± 40 in PLC/PRF/5 cells) and CDN (208.3 ± 15.3 in LM3 cells and 147.7 ± 21.2 in PLC/PRF/5 cells) exhibited decreased invasive capabilities compared with those treated with DMSO (810 ± 36.1 in LM3 cells and 1046.7 ± 65.8 in PLC/PRF/5 cells) and LDN (612.7 ± 16.8 in LM3 cells and 816.7 ± 33.9 in PLC/PRF/5 cells) groups (Figure 6A, P < 0.01). The wound-healing assay and scratch areas of HCC cells treated with SAL, LDN, and CDN were significantly smaller than those of the control group (Figure 6B, P < 0.05). This result was most robust for the CDN group, where representative HCC cells migrated more slowly after being treated with CDN. These data show that CDN effectively inhibit HCC cell invasion and migration.
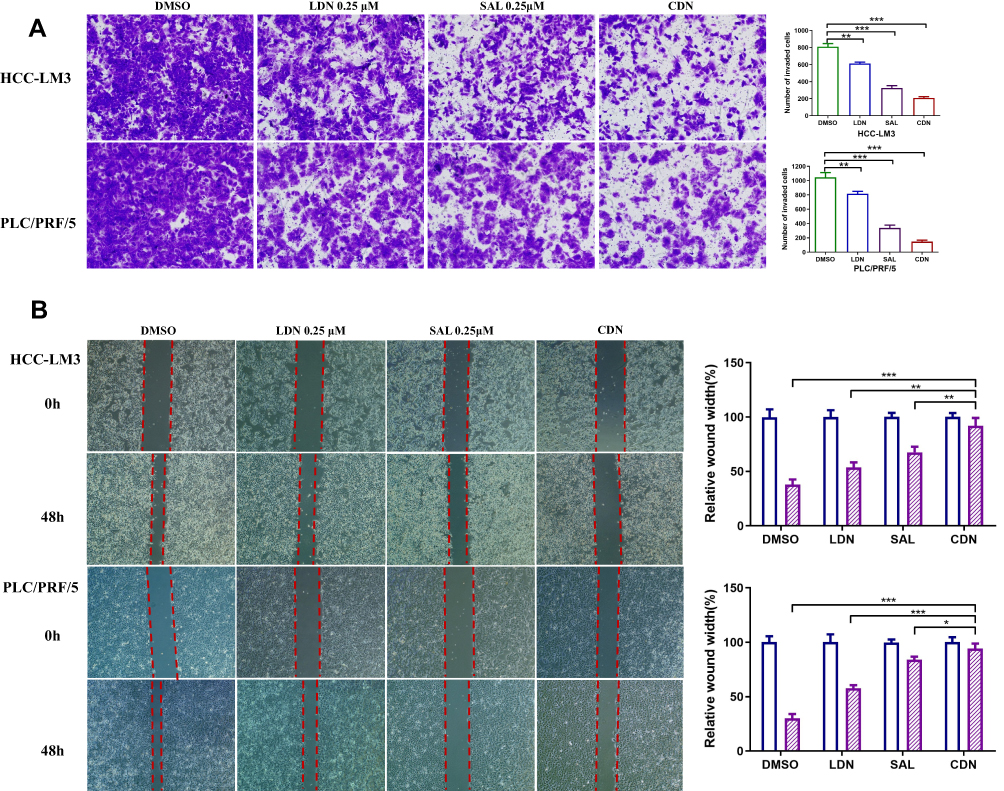 |
Figure 6 The effect of different formulations on HCC cell invasion and migration. (A) Representative photographs of invasive HCC cells treated with different formulation. (B) Wound repair analysis at 0 h and 48 h after scratch wounding. The CDN effectively inhibit HCC cell invasion and migration compared to control group. Data are expressed as the following: *P < 0.05, **P < 0.01, ***P < 0.001. |
Codelivered Nanoparticles Effectively Inhibit HCC Cells
Previous studies have identified CD133 as a cell-surface marker of CSCs in HCC patients,29 and CD133 overexpression leads to a shorter HCC survival period and a higher risk of recurrence.30,31 In addition, many studies have shown that CD133+ HCC cells are resistant to conventional chemotherapy and radiation.32–34 In the present study, we examined the ability of different formulations to inhibit HCC-CSCs. PLC cells were treated with free SAL, LDN, or CDN at a concentration of 0.25 μM for 48 h, and then the CD133+ subpopulation percentage was determined by FACS. The percentage of CD133+ cells in the SAL and CDN groups significantly decreased compared to the control group (5.15%±0.49 vs 8.71%±0.51 and 3.99%±1.33 vs 8.71%±0.51, respectively) (Figure 7A, p< 0.05)(). We further investigated the inhibitory effect of different formulations on CSC spheroid formation in HCC cells. LM3 and PLC cells were incubated with 0.25 μM free SAL, LDN, or CDN for 10 days, and free SAL and CDN significantly decreased spheroid-formation capacity (Figure 7B). These data suggest that CDN inhibit HCC-CSCs in vitro.
 |
Figure 7 The effect of CSC inhibition on HCC cells. (A) The proportion of the CD133+ subpopulation of PLC/PRF/5 cells in different treatment groups. The percentage of CD133+ cells in the SAL and CDN groups were significantly decreased compared to the control group. (B) Tumor-sphere formation of HCC-LM3 and PLC/PRF/5 cells. Free SAL and CDN significantly decreased spheroid-formation capacity of HCC-LM3 and PLC/PRF/5 cells compared to the control group. Data are shown as means ± SD, **P < 0.01, ***P < 0.001 for SAL or CDN vs control. |
Dual-Therapy Treatment Strategies Efficiently Inhibit in vivo Tumor Growth
To explore the anti-tumor effect of these nano-formulations in vivo, we generated an LM3-CDX preclinical model. Given the in vitro therapeutic results, we use a 1:1 dose of SAL and LA-SN38 (both 10 mg/kg) for in vivo treatment. However, tumor-bearing nude mice died after intravenous injection of SAL, suggesting that the mice could not tolerate this dosage because of systemic SAL toxicity. Therefore, we tested several different concentrations of SAL in tumor-bearing mice and used 4 mg/kg for further studies. To investigate the in vivo antitumor efficacy of these experimental formulations, once the tumors reached approximately 100 mm3, the xenograft mice were randomly divided into four groups and were then treated with NS, free SAL (4 mg/kg), LDN (10 mg/kg), and CDN every two days. Tumors were fast-growing in the saline and SAL group, while LDN and CDN treatments significantly inhibited tumor growth. Notably, CDN showed a greater antitumor effect in the CDX model (Figure 8A). The most potent CDN antitumor was likely due to following: (i) the smaller particle size of CDN increases the penetration of nanoparticles in tumor tissues via the EPR effect, and (ii) the codelivery of SAL and LA-SN38 has a significant synergistic anti-tumor effect. Additionally, we noticed that SAL monotherapy exhibited no obvious inhibition of tumor growth.
 |
Figure 8 Therapeutic activity of different drug formulations in cell- and patient-derived xenografts. Animals were randomized (five animals per group), and treated with saline, free SAL (4 mg/kg), LDN (10 mg/kg), and CDN (10 mg/kg + 4 mg/kg) for two weeks. (A) Photographic images of tumors from the xenograft tumor-bearing nude mice with in response to different treatments. (B) The tumor growth curve in CDX and PDX models. Tumor volume are presented as the mean ± SD (n = 5). **p < 0.01 for CDN (703.37±170.34) vs LDN (1598.92±542.44) and ***P < 0.001 for CDN vs SAL (2083.50±308.15) or Saline (2443.42±643.42) in CDX models, *p < 0.05 for CDN (458.26±207.44) vs LDN (846.39±297.11) and **P < 0.01 for CDN vs SAL (4280.74±1746.87) or Saline (2798.65±886.85) in PDX models. (C) Bodyweight change of the CDX and PDX tumor-bearing nude mice in each group. |
Recently, increasing numbers of researchers have claimed that CDX models are not ideal for predicting therapeutic clinical efficacy because the behaviors, genomics, and heterogeneity of tumor cells are different from those of primary tumor tissues.32 However, PDX models may preserve the heterogeneity and microenvironment of the original patient tumors, suggesting that drug metabolism results obtained in these models are more clinically predictive than those obtained in CDX models.5,35–38 Consequently, we investigated the anti-tumor effect of these formulations in a PDX model. Following treatment with NS, free SAL (4 mg/kg), LDN (10 mg/kg), and CDN (SAL 4 mg/kg + LA-SN38 10 mg/kg), the tumor-bearing mice treated with CDN showed a significant decrease in tumor volume (Figure 8B). In addition, SAL monotherapy failed to suppress tumor growth. Taken together, these results imply that CDN are efficient anticancer agent for HCC. Furthermore, no obvious body weight changes were observed in the mice following the different treatments (Figure 8C). This demonstrates that the PDX model results may provide valuable information for the clinical translation of CDN.
Histological Analysis of the Sections of Tumor Tissues
PDX and CDX tumors were harvested at the end of the treatment period and used for further histological analysis. H&E staining was performed to evaluate tumor histomorphology, and we observed necrosis in CDN-treated tumors. Compared to that in the control group, the protein expression of the cell proliferation marker Ki67 was markedly decreased in tumor tissues treated with LDN or CDN, with CDN exhibiting the strongest inhibition of tumor proliferation. TUNEL staining was used to detect apoptosis in tumor tissues, and the CDN group showed greater levels of intratumoral apoptosis than the other groups (Figure 9).
 |
Figure 9 Tumor section analysis by H&E staining and immunohistochemical imaging. Representative images of histological, Ki-67, and TUNEL staining of the tumor sections are shown. The ratio of Ki67-positive cells significantly reduced and the TUNEL staining cells significantly increased in the LDN and CDN group compared with saline or free SAL group. The results are expressed as the means ± SD of triplicate samples. **p < 0.01, ***P < 0.001. |
Discussion
The overall five-year survival rate of HCC is less than 10%. This is mainly due to the high incidence of postoperative recurrence and chemo-resistance, which is often attributed to CSCs. Therefore, it is crucial to develop novel therapeutic strategies targeting CSCs. In the present study, we developed a SAL-loaded LA-SN38 nanoprodrug for HCC treatment. Our results suggest that the SAL-loaded nanoprodrug formulation has superior therapeutic efficacy against HCC by effectively restraining CSCs and the malignant phenotypes of tumors.
SAL has been shown to inhibit CSCs in various cancer types; several studies have indicated that SAL combined with chemotherapeutic drugs, such as doxorubicin, tamoxifen, and cisplatin, enables synergistic anticancer activity in gastric cancer, breast cancer, and cholangiocarcinoma.39–41 However, SAL possesses poor aqueous solubility and unfavorable properties in terms of toxicity. Thus, to overcome these disadvantages, nanoparticles have been used to deliver SAL. SAL-loaded DSPE-PEG-methotrexate nanoparticles significantly suppress tumor growth in head and neck squamous cell carcinoma.42 In addition, codelivery of SAL and doxorubicin using nanoliposomes significantly decreases the percentage of HCC-CSCs in vivo.43
Conventional drug carriers consist of phospholipid-, cholesterol-, or amphiphilic-polymers. Extensive use of these excipients may lead to problems of carrier toxicity and metabolism and can increase patient excretory burden;44 substantial effort has been devoted to resolving this these issues. Liang et al developed a liposome-like nanocapsule by coupling two hydrophobic CPT molecules and two hydrophilic-floxuridine molecules. This nanocapsule exhibited the ability to codeliver two therapeutics without the need for a carrier.45 Wang et al employed PEGylated C16-ceramide (PEG-ceramide) as an effective and safe therapeutic drug carrier to deliver SAL in liver cancer, which increased apoptosis-inducing activity.46 In our previous studies, SN-38 was modified with a lipophilic tail and, allowing it to self-assemble in aqueous solutions. Here, we applied LA-SN38 as a drug carrier loaded with SAL to achieve combinational therapeutic effects.
Previous studies have reported that SAL inhibits proliferation and induce apoptosis of HCC cells.47 Zhou et al demonstrated that the combination of doxorubicin and SAL for targeting HCC decreases doxorubicin resistance by activating FOXO3a, thereby inhibiting the expression of β-catenin/TCF target genes.48 Here, we found that SAL enhanced HCC SN38 sensitivity. The adopted drug formulations—including SAL, LDN, and CDN—effectively inhibited HCC cell proliferation and induced apoptosis. Combined treatment exhibited stronger effects compared to that of separate treatments. The inhibitory effect of CDN was significantly higher than that of the control, LDN, or SAL alone. Furthermore, we evaluated the effects of the different drug formulations on cell migration and invasion. Our results suggest that CDN inhibits HCC cell migration and invasion. These results may be due to the reduction in the CSC population induced by SAL. However, there was no significant difference between SAL and CDN. We suspected that SN38 plays broad-spectrum antitumor activity by inhibiting DNA synthesis; however, CSCs have the ability to induce cell cycle arrest (quiescent state) that support them to become resistant to SN38. Therefore, in our codelivery system, SAL plays a major role in stem cell inhibition, and the two drugs had little synergistic effect on stem cell inhibition.
The therapeutic efficacy of these drug formulations was tested in two tumor xenograft-bearing mice models: CDX and PDX. Our data suggested that CDN markedly inhibits tumor proliferation compared with PBS, SAL, or LDN. The significantly enhanced therapeutic efficacy of CDN may have been due to codelivery multiple therapeutic agents by one nanoparticle that coordinates pharmacokinetics and CSC inhibition by SAL. These results demonstrate that although low-dose SAL monotherapy is unable to suppress tumor growth, low-dose SAL delivery by a prodrug-based nanosystem increases synergistic antitumor effects likely due to the EPR effect and increased sensitivity to drugs with different therapeutic mechanisms.
Conclusion
In summary, our results show that a codelivery system using self-assembled small-molecule prodrugs represents a potentially effective treatment strategy for improving the survival of HCC patients. The advantage of this codelivery strategy is its ability to encapsulate two drugs with different mechanisms into a single nanoparticle with a simple process. Furthermore, PEGylation prolongs nanoprodrug circulation half-life and increases the blood stability of nanoprodrugs to reduce systemic toxicity. Here, both in vitro and in vivo results suggest that therapeutic strategies using this codelivery nanoformulation may be beneficial for HCC treatment in humans. This nanoprodrug platform enables exploration of new strategies for combination therapy to overcome monotherapy resistance. We propose a new type of prodrug-based codelivery system that exhibits a synergistic anti-tumor effect via elimination of CSCs.
Funding
This work was supported by the National S&T Major Project (No. 2017ZX10203205), the National Natural Science Foundation of China (No. 81801824), the Key Research & Development Plan of Zhejiang Province (No. 2019C03050) and the Major Science and Technology Project of Zhejiang Province (No. 2018C04010).
Disclosure
The authors declare no conflicts of interest.
References
1. Zhang Y, Guan DX, Shi J, et al. All-trans retinoic acid potentiates the chemotherapeutic effect of cisplatin by inducing differentiation of tumor initiating cells in liver cancer. J Hepatol. 2013;59(6):1255–1263. doi:10.1016/j.jhep.2013.07.009
2. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911–1918. doi:10.1172/JCI66024
3. Yamashita T, Kaneko S. Orchestration of hepatocellular carcinoma development by diverse liver cancer stem cells. J Gastroenterol. 2014;49(7):1105–1110. doi:10.1007/s00535-014-0951-1
4. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt–β-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67(22):10831–10839. doi:10.1158/0008-5472.CAN-07-0908
5. Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8(14):3737–3750. doi:10.7150/thno.25487
6. Mendes LP, Sarisozen C, Luther E, Pan J, Torchilin VP. Surface-engineered polyethyleneimine-modified liposomes as novel carrier of siRNA and chemotherapeutics for combination treatment of drug-resistant cancers. Drug Deliv. 2019;26(1):443–458. doi:10.1080/10717544.2019.1574935
7. Khan MW, Zhao P, Khan A, et al. Synergism of cisplatin-oleanolic acid co-loaded calcium carbonate nanoparticles on hepatocellular carcinoma cells for enhanced apoptosis and reduced hepatotoxicity. Int J Nanomedicine. 2019;14:3753–3771. doi:10.2147/IJN.S196651
8. Wang HX, Xie HY, Wang JG, et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor efficacy. Adv Funct Mater. 2015;25(31):4956–4965. doi:10.1002/adfm.201501953
9. Chen F, Zeng Y, Qi X, et al. Targeted salinomycin delivery with EGFR and CD133 aptamers based dual-ligand lipid-polymer nanoparticles to both osteosarcoma cells and cancer stem cells. Nanomedicine. 2018;14:2115–2127.
10. Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. doi:10.1016/j.cell.2009.06.034
11. Fuchs D, Daniel V, Sadeghi M, Opelz G, Naujokat C. Salinomycin overcomes ABC transporter-mediated multidrug and apoptosis resistance in human leukemia stem cell-like KG-1a cells. Biochem Biophys Res Commun. 2010;394(4):1098–1104. doi:10.1016/j.bbrc.2010.03.138
12. Resham K, Patel PN, Thummuri D, et al. Preclinical drug metabolism and pharmacokinetics of salinomycin, a potential candidate for targeting human cancer stem cells. Chem Biol Interact. 2015;240:146–152. doi:10.1016/j.cbi.2015.08.007
13. Mao X, Liu J, Gong Z, et al. iRGD-conjugated DSPE-PEG2000 nanomicelles for targeted delivery of salinomycin for treatment of both liver cancer cells and cancer stem cells. Nanomedicine. 2015;10(17):2677–2695. doi:10.2217/nnm.15.106
14. Xie F, Zhang S, Liu J, et al. Codelivery of salinomycin and chloroquine by liposomes enables synergistic antitumor activity in vitro. Nanomedicine. 2016;11(14):1831–1846. doi:10.2217/nnm-2016-0125
15. Wang Q, Wu P, Ren W, et al. Comparative studies of salinomycin-loaded nanoparticles prepared by nanoprecipitation and single emulsion method. Nanoscale Res Lett. 2014;9(1):351. doi:10.1186/1556-276X-9-351
16. Jin JL, Gong J, Yin TJ, et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur J Pharmacol. 2011;654(1):17–25. doi:10.1016/j.ejphar.2010.12.004
17. Wang H, Lu Z, Wang L, et al. New generation nanomedicines constructed from self-assembling small-molecule prodrugs alleviate cancer drug toxicity. Cancer Res. 2017;77(24):6963–6974. doi:10.1158/0008-5472.CAN-17-0984
18. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi:10.1016/S0065-2571(00)00013-3
19. Mei L, Rao J, Liu Y, Li M, Zhang Z, He Q. Effective treatment of the primary tumor and lymph node metastasis by polymeric micelles with variable particle sizes. J Control Release. 2018;292:67–77. doi:10.1016/j.jconrel.2018.04.053
20. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi:10.1038/nnano.2011.166
21. Stylianopoulos T. EPR-effect: utilizing size-dependent nanoparticle delivery to solid tumors. Ther Deliv. 2013;4(4):421–423. doi:10.4155/tde.13.8
22. Perry JL, Reuter KG, Luft JC, Pecot CV, Zamboni W, DeSimone JM. Mediating passive tumor accumulation through particle size, tumor type, and location. Nano Lett. 2017;17(5):2879–2886. doi:10.1021/acs.nanolett.7b00021
23. Chen D, Wu M, Li Y, et al. Targeting BMI1(+) cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. 2017;20(5):621–634 e6. doi:10.1016/j.stem.2017.02.003
24. Dianat-Moghadam H, Heidarifard M, Jahanban-Esfahlan R, et al. Cancer stem cells-emanated therapy resistance: implications for liposomal drug delivery systems. J Control Release. 2018;288:62–83. doi:10.1016/j.jconrel.2018.08.043
25. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi:10.1038/35102167
26. McClements L, Annett S, Yakkundi A, et al. FKBPL and its peptide derivatives inhibit endocrine therapy resistant cancer stem cells and breast cancer metastasis by downregulating DLL4 and Notch4. BMC Cancer. 2019;19(1):351. doi:10.1186/s12885-019-5500-0
27. Wang X, Cao Y, Zhang S, et al. Stem cell autocrine CXCL12/CXCR4 stimulates invasion and metastasis of esophageal cancer. Oncotarget. 2017;8(22):36149–36160. doi:10.18632/oncotarget.15254
28. Wang C, Shao L, Pan C, et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res Ther. 2019;10(1):175. doi:10.1186/s13287-019-1265-2
29. Jang JW, Song Y, Kim SH, et al. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer Lett. 2017;389:1–10. doi:10.1016/j.canlet.2016.12.023
30. Bai HY, Liao YJ, Cai MY, et al. Eukaryotic initiation factor 5A2 contributes to the maintenance of CD133(+) hepatocellular carcinoma cells via the c-Myc/microRNA-29b axis. Stem Cells. 2018;36(2):180–191. doi:10.1002/stem.2734
31. Tang H, Jin Y, Jin S, Tan Z, Peng Z, Kuang Y. Arsenite inhibits the function of CD133(+) CD13(+) liver cancer stem cells by reducing PML and Oct4 protein expression. Tumour Biol. 2016;37(10):14103–14115. doi:10.1007/s13277-016-5195-7
32. Yang T, Chen Y, Zhao P, et al. Enhancing the therapeutic effect via elimination of hepatocellular carcinoma stem cells using Bmi1 siRNA delivered by cationic cisplatin nanocapsules. Nanomedicine. 2018;14(7):2009–2021. doi:10.1016/j.nano.2018.05.012
33. Wu R, Murali R, Kabe Y, et al. Baicalein targets GTPase-mediated autophagy to eliminate liver tumor-initiating stem cell-like cells resistant to mTORC1 inhibition. Hepatology. 2018;68(5):1726–1740. doi:10.1002/hep.30071
34. Takahashi K, Yan IK, Kogure T, Haga H, Patel T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio. 2014;4:458–467. doi:10.1016/j.fob.2014.04.007
35. Fong ELS, Toh TB, Lin QXX, et al. Generation of matched patient-derived xenograft in vitro-in vivo models using 3D macroporous hydrogels for the study of liver cancer. Biomaterials. 2018;159:229–240. doi:10.1016/j.biomaterials.2017.12.026
36. Coussy F, de Koning L, Lavigne M, et al. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. Int J Cancer. 2019. doi:10.1002/ijc.32266
37. Wang W, Iyer NG, Tay HT, et al. Microarray profiling shows distinct differences between primary tumors and commonly used preclinical models in hepatocellular carcinoma. BMC Cancer. 2015;15:828. doi:10.1186/s12885-015-1814-8
38. Zhang X, Claerhout S, Prat A, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73(15):4885–4897. doi:10.1158/0008-5472.CAN-12-4081
39. Zhang Z, Zhao J, Pang Q, Wang A, Chen M, Wei X. An in vitro study on the effects of the combination of salinomycin with cisplatin on human gastric cancer cells. Mol Med Rep. 2017;16(2):1031–1038. doi:10.3892/mmr.2017.6731
40. Yu Z, Cheng H, Zhu H, et al. Salinomycin enhances doxorubicin sensitivity through reversing the epithelial-mesenchymal transition of cholangiocarcinoma cells by regulating ARK5. Braz J Med Biol Res. 2017;50(10):e6147. doi:10.1590/1414-431x20176147
41. Sommer AK, Hermawan A, Mickler FM, et al. Salinomycin co-treatment enhances tamoxifen cytotoxicity in luminal A breast tumor cells by facilitating lysosomal degradation of receptor tyrosine kinases. Oncotarget. 2016;7(31):50461–50476. doi:10.18632/oncotarget.10459
42. Zhu M, Chen S, Hua L, et al. Self-targeted salinomycin-loaded DSPE-PEG-methotrexate nanomicelles for targeting both head and neck squamous cell carcinoma cancer cells and cancer stem cells. Nanomedicine. 2017;12(4):295–315. doi:10.2217/nnm-2016-0382
43. Gong Z, Chen D, Xie F, et al. Codelivery of salinomycin and doxorubicin using nanoliposomes for targeting both liver cancer cells and cancer stem cells. Nanomedicine. 2016;11(19):2565–2579. doi:10.2217/nnm-2016-0137
44. Zhang T, Huang P, Shi L, et al. Self-assembled nanoparticles of amphiphilic twin drug from floxuridine and bendamustine for cancer therapy. Mol Pharm. 2015;12(7):2328–2336. doi:10.1021/acs.molpharmaceut.5b00005
45. Liang X, Gao C, Cui L, Wang S, Wang J, Dai Z. Self-assembly of an amphiphilic janus camptothecin-floxuridine conjugate into liposome-like nanocapsules for more efficacious combination chemotherapy in cancer. Adv Mater. 2017;29(40):1703135. doi:10.1002/adma.201703135
46. Wang M, Xie F, Wen X, et al. Therapeutic PEG-ceramide nanomicelles synergize with salinomycin to target both liver cancer cells and cancer stem cells. Nanomedicine. 2017;12(9):1025–1042. doi:10.2217/nnm-2016-0408
47. Wang F, He L, Dai WQ, et al. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS One. 2012;7(12):e50638. doi:10.1371/journal.pone.0050638
48. Zhou Y, Liang C, Xue F, et al. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the β-catenin/TCF complex association via FOXO3a activation. Oncotarget. 2015;6(12):10350–10365. doi:10.18632/oncotarget.3585
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.