Back to Journals » Drug Design, Development and Therapy » Volume 12
Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways
Authors Feng J ![]() , Chen K
, Chen K ![]() , Xia Y, Wu L
, Xia Y, Wu L ![]() , Li J, Li S, Wang W, Lu X, Liu T, Guo C
, Li J, Li S, Wang W, Lu X, Liu T, Guo C ![]()
Received 18 January 2018
Accepted for publication 3 April 2018
Published 22 June 2018 Volume 2018:12 Pages 1837—1853
DOI https://doi.org/10.2147/DDDT.S162950
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Jiao Feng, Kan Chen, Yujing Xia, Liwei Wu, Jingjing Li, Sainan Li, Wenwen Wang, Xiya Lu, Tong Liu, Chuanyong Guo
Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, China
Purpose: Liver fibrosis is commonly seen and a necessary stage in chronic liver disease. The aim of this study was to explore the effect of salidroside on liver fibrosis in mice and its potential mechanisms.
Materials and methods: Two mouse liver fibrosis models were established by intraperitoneal injection of carbon tetrachloride (CCl4) for 8 weeks and bile duct ligation for 14 days. Salidroside was injected intraperitoneally at doses of 10 and 20 mg/kg once a day. Gene and protein expression levels were determined by quantitative real-time polymerase chain reaction, enzyme-linked immunosorbent assay, Western blot, immunohistochemistry, and immunofluorescence.
Results: Salidroside inhibited the production of extracellular matrix (ECM) and regulated the balance between MMP2 and TIMP1 and, therefore, alleviated liver fibrosis in the two fibrosis models. Salidroside reduced the production of transforming growth factor (TGF)-β1 in Kupffer cells and hepatic stellate cells (HSCs) via the nuclear factor-κB signaling pathway and, therefore, inhibited the activation of HSCs and autophagy by downregulation of the TGF-β1/Smad3 signaling pathway.
Conclusion: Salidroside can effectively attenuate liver fibrosis by inhibiting the activation of HSCs in mice.
Keywords: liver fibrosis, salidroside, hepatic stellate cells, autophagy, TGF-β1/Smad3
Introduction
Liver fibrosis is a chronic but reversible process in liver diseases, characterized by the overdeposition of extracellular matrix (ECM), especially collagen, in liver tissues. It represents a necessary stage in liver cirrhosis and may progress to primary hepatocellular carcinoma.1–3 Chronic liver injuries involving viral infection, alcohol abuse, nonalcoholic steatohepatitis, drug use, autoimmune and cholestatic liver diseases, and metabolic disorders may damage hepatocytes, resulting in activation of the wound-healing response and consequent liver fibrosis.1,4–6 Current treatments for liver fibrosis include immunosuppressive agents, anti-inflammatory agents, antioxidants, and antiviral therapies. However, although liver transplantation is the most effective option, it is limited by a shortage of donor organs and high costs.7–9 Alternative therapeutic or preventive strategies for liver fibrosis are therefore urgently required.
The mechanisms of liver fibrosis are complicated, but the process is initiated by the activation of hepatic stellate cells (HSCs).4–6,10 Under normal conditions, HSCs remain in the stationary phenotype and maintain a balance between ECM production and degradation. However, following activation by various factors, such as mechanical stimulation and inflammatory cytokines, especially transforming growth factor-β1 (TGF-β1), quiescent HSCs are transformed into myofibroblasts and produce ECM.11 The secretion of TGF-β1 and other inflammatory cytokines is mainly regulated by the nuclear factor-kappa B (NF-κB) signaling pathway.12–14 TGF-β1 can not only cause continuous activation of HSCs but can also interact with TGF-β receptors (TβRs) to phosphorylate Smad3 and promote the translocation of phospho-Smad3 (p-Smad3) to the nucleus, leading to the production of ECM.6,15 TGF-β1 has also been reported to mediate autophagy, which is an intracellular self-digestion reaction that contributes to the activation of HSCs.16
Salidroside is a phenylpropanoid glycoside extracted from the plant Rhodiola rosea L. Salidroside has demonstrated antiautophagy, anti-inflammatory, antioxidant, antihypoxia, antitumor, antiaging, antidepressive, and neuro- and cardioprotective properties.17–19 Our previous study has demonstrated that salidroside could inhibit apoptosis and autophagy during hepatic ischemia–reperfusion, which will also lead to the activation of HSCs.19 Ouyang et al20 recently showed that salidroside could improve liver fibrosis by acting synergistically with rat mesenchymal stem cell transplantation. Furthermore, Tang et al demonstrated that salidroside could attenuate bleomycin-induced lung fibrosis by inhibiting the NF-κB and TGF-β1/Smad2/3 pathways.17 These indicate that salidroside may be a potential therapeutic agent to treat liver fibrosis possibly through the regulation of NF-κB and TGF-β1/Smad pathways.
Therefore, the present study aimed to investigate the effect of salidroside on liver fibrosis using two different animal models. We hypothesized that salidroside may reduce liver fibrosis via downregulation of the TGF-β1/Smad3 and NF-κB pathways.
Materials and methods
Reagents
Salidroside and pentobarbital sodium salt were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Carbon tetrachloride (CCl4) was purchased from China Sinopharm International Corporation (Shanghai, China). The kits for determining alanine aminotransferase (ALT) (cat no C009-2), hydroxyproline (cat no A030-2), and aspartate aminotransferase (AST) (cat no C010-2) were purchased from Jiancheng Bioengineering Institute (Nanjing, China). The primers were obtained from Generay (Shanghai, China). The primary antibodies used in the study were as follows: alpha smooth muscle actin (α-SMA), tissue inhibitor of matrix metalloproteinases (TIMP1), matrix metalloproteinase 2 (MMP2), NF-κB, nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor alpha (IκBα), Beclin-1, LC3, and p62 (all 1:1,000; Proteintech, Chicago, IL, USA), collagen-I (Col-1), TGF-β1, F4/80, Smad3, and p-Smad3 (all 1:500; Abcam, Cambridge, MA, USA), and β-actin (1:1,000; Cell Signaling Technology, Danvers, MA, USA). The PrimeScript™ RT Reagent Kit and SYBR Premix Ex Taq were purchased from TaKaRa Biotechnology (Dalian, China).
Animals
The experiment was approved by the Animal Care and Use Committee of Shanghai Tongji University, and was executed following the National Institutes of Health Guidelines. Six to eight weeks’ old male C57 mice (24±2 g) were acquired from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed in a clean room with free access to food and water at a room temperature of 24°C±2°C and 60% humidity.
Experimental models and drug treatment
We established two different mouse liver fibrosis models.
To create the CCl4-induced liver fibrosis model, mice were injected with 10% CCl4 (1.0 mL/kg, diluted in peanut oil) three times a week for 8 weeks. Salidroside was diluted with normal saline and injected intraperitoneally at dosages of either 10 or 20 mg/kg once a day for 8 weeks. Thirty-two mice were randomly divided into the following four groups: 1) vehicle group: n=8, mice were injected with peanut oil intraperitoneally; 2) CCl4 group: n=8, mice were injected with CCl4 intraperitoneally; 3) CCl4 + Sal 10 mg/kg group: n=8, mice were injected with CCl4 and 10 mg/kg salidroside intraperitoneally; and 4) CCl4 + Sal 20 mg/kg group: n=8, mice were injected with CCl4 and 20 mg/kg salidroside intraperitoneally.
In the bile duct ligation (BDL)-induced liver fibrosis model, all mice were fasted for 12 h and anesthetized intraperitoneally by 1.25% pentobarbital sodium salt (40 mg/kg). After opening the abdomen via linea alba, the bile duct was exposed and isolated over a certain length. Two surgical knots were tied in the isolated bile duct, which was then cut between the knots. The abdomen was then closed. Salidroside was administered on the second day. For the BDL model, 32 mice were divided into the following four groups: 1) sham group: n=8, all mice underwent laparotomy without BDL; 2) BDL group: n=8, mice underwent BDL surgery; 3) BDL + Sal 10 mg/kg group: n=8, mice were injected intraperitoneally with 10 mg/kg salidroside once a day for 14 days after BDL; and 4) BDL + Sal 20 mg/kg group: n=8, mice were injected intraperitoneally with 20 mg/kg salidroside once a day for 14 days after BDL.
Vehicle and sham groups were used as controls in both models. At the end of the experiment, blood samples and liver tissues were collected with diethyl ether anesthesia. Serum was acquired by centrifugation (4,500 rpm, 4°C, 10 min) and kept at −80°C. Liver tissues were stored at −80°C.
Biochemical assays
Serum ALT and AST levels were measured by spectrophotometry using AU1000 (Olympus Corporation, Tokyo, Japan). Liver collagen concentrations were reflected by measuring hydroxyproline levels. All these procedures were implemented following manufacturers’ instructions.
Histopathology
Liver tissues were excised and first fixed in 4% paraformaldehyde, dehydrated in ethyl alcohol, and embedded in paraffin. Then, the 3 μm thick sections were stained with hematoxylin and eosin (H&E) to observe liver injury. The procedure for Masson’s trichrome staining to detect collagen deposition was as follows. The paraffin sections were deparaffinized and rehydrated with xylene and a descending alcohol series. Then, the sections were treated with Bouin’s solution at 58°C for 15 min and washed with running water and, then, stained with modified Weigert’s iron hematoxylin for 5 min and washed with running water. The sections were placed in 0.5% hydrochloric acid in 70% alcohol for 5 s and washed again. Then, the sections were stained with the Biebrich scarlet-acid fuchsin solution for 5 min, washed again, and stained with phosphotungstic acid solution for 5 min and then with aniline blue for 20 min. Finally, the sections were placed in acetic acid solution for 10 s, dehydrated in graded alcohols, cleared in xylene, and mounted with synthetic resin.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time PCR (qPCR)
The total RNA of liver tissues was extracted by TRIzol (Tiangen Biotech, Beijing, China) and reverse transcribed into cDNA by manufacturer’s kit. The expression of target genes was determined by qPCR using a 7900HT Fast PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Table 1 shows the primer sequences used in the process.
 | Table 1 The primers used in the study |
Transmission electron microscopy (TEM)
Liver tissues were cut into 1 mm3 fragments and fixed in 5% glutaraldehyde buffer. After embedded in Epon, tissues were cut into 1.5 μm sections and stained with toluidine blue. The cells were observed by TEM (JEM 1230; JEOL, Tokyo, Japan).
Western blot analysis
The total protein was extracted from liver tissues using radioimmunoprecipitation assay buffer. Proteins were electrophoresed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes. The 5% nonfat milk was used to block nonspecific binding sites for 1 h, followed by incubation with primary antibodies overnight and secondary antibodies for 1 h. Results were detected using an Odyssey two-color infrared laser imaging system (LI-COR, Lincoln, NE, USA).
Immunohistochemistry
Paraffin-embedded liver slides were dewaxed and rehydrated. Antigen retrieval process was as follows: 1) heating to 95°C for 10 min, 2) cooling to room temperature, 3) and repeated for four cycles. Then, we used 3% hydrogen peroxide and 5% bovine serum albumin to block endogenous peroxidase activity and nonspecific binding sites, respectively. Sections were then incubated with Col-1, α-SMA, TGF-β1, p-Smad3, NF-κB, Beclin-1, and LC3 primary antibodies (1:500) at 4°C overnight. After incubation with secondary antibody, the slides were then stained with H&E.
Double-immunofluorescence staining
Paraffin-embedded liver sections were dewaxed and rehydrated first. Antigen retrieval process was performed using the EDTA (elhylene diamine tetraacetic acid) antigen repair buffer. Then, 3% bovine serum albumin was used to block nonspecific binding sites. Sections were then incubated with α-SMA (red) or F4/80 (red) and TGF-β1 (green) or NF-κB (green) primary antibodies (1:500) at 4°C overnight. After incubation with fluorescence secondary antibody, the nucleus was counter-stained blue with DAPI (4′,6-diamidino-2-phenylindole) and mounted with antifluorescence quenching sealant. Slides were viewed with inverted fluorescence microscope (DMIRB; Leica Microsystems, Wetzlar, Germany).
Statistical analysis
All quantitative data were represented as mean ± standard deviation (SD). Serum levels of ALT and AST, liver levels of hydroxyproline, and standard analysis of Western blot and immunohistochemistry were analyzed by Student’s t-test. The qPCR data were analyzed by one-way analysis of variance. P-values <0.05 were considered to be statistically significant.
Results
Salidroside alleviated liver fibrosis in CCl4- and BDL-induced liver fibrosis models
Elevated levels of ALT and AST are considered to reflect liver injuries. We therefore measured serum ALT and AST levels to evaluate liver injuries in the two mouse models. Liver enzyme levels were significantly increased in model groups compared with the vehicle or sham control groups (Figure 1A). Liver hydroxyproline levels were also increased in both CCl4 and BDL groups. These results indicated that liver dysfunction occurred in both models. However, serum liver enzymes and liver hydroxyproline levels were reduced by salidroside (10 or 20 mg/kg), with 20 mg/kg salidroside having a greater beneficial effect. We also evaluated the protective effects of salidroside histologically. CCL4 administration was associated with the rearrangement of liver lobular structures, marked fatty degeneration, ballooning or necrosis of hepatocytes, accumulation of inflammatory cells, and formation of pericellular collagen deposition according to H&E staining. BDL resulted in the necrosis of liver tissues, inflammatory cell infiltration, proliferation of the bile duct, and deposition of collagen around the portal areas. Masson stain showed increased collagen staining in both model groups compared with the control groups. However, salidroside ameliorated the histopathological lesions in both models significantly (Figure 1B and C). These results indicated the successful establishment of two mouse liver fibrosis models and demonstrated that salidroside could effectively alleviate liver fibrosis in both models.
 | Figure 1 Effect of salidroside on liver function and pathology in CCl4- and BDL-induced liver fibrosis. |
Salidroside ameliorated autophagy in mouse liver fibrosis models
Autophagy has been reported to provide energy for HSCs in liver fibrosis,16 which was characterized by the formation of autophagosomes. In Figure 2A, TEM was used to observe the formation of autophagosomes in liver cells, which were indicated by red arrows. There was more autophagosomes formation in model groups, while salidroside could reduce the formation of autophagosomes. The effect of salidroside on autophagic flux is shown in Figure S1. The expression levels of Beclin-1, LC3, and p62 were measured by Western blot, qPCR, and immunohistochemistry to assess the autophagy process in liver fibrosis mouse models. Protein levels of Beclin-1 and LC3, which promote the formation of autophagosomes during the autophagy process, were increased in both model groups compared with the control groups (Figure 2B), while levels of p62, which is an ubiquitin-binding protein that can be degraded in autophagosome formation, were decreased in the CCl4 and BDL groups.25 These results suggested that autophagy was advanced in liver fibrosis. However, salidroside reduced Beclin-1 and LC3 levels and increased p62 in a dose-dependent manner (Figure 2B). The qPCR and immunohistochemistry demonstrated similar results to those of Western blotting (Figure 2C and D). The results suggested that salidroside could ameliorate autophagy in mouse liver fibrosis models.
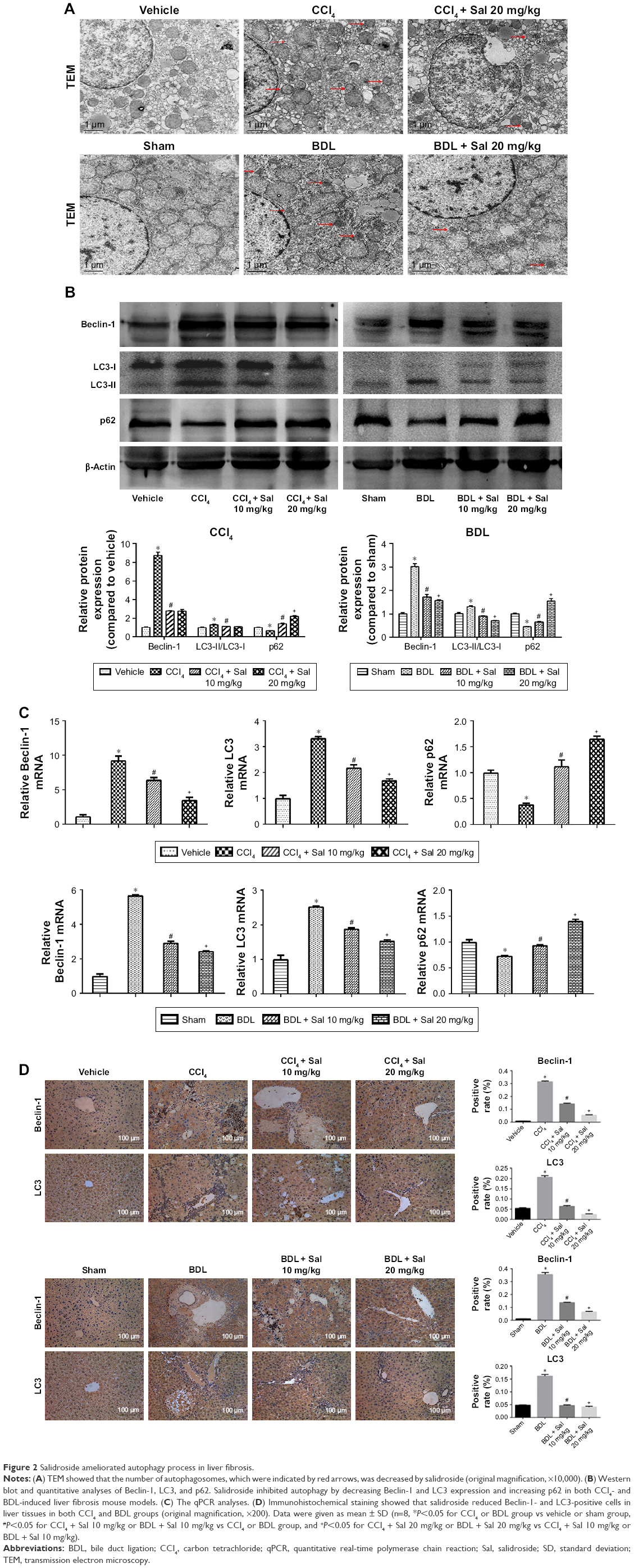 | Figure 2 Salidroside ameliorated autophagy process in liver fibrosis. |
Salidroside inhibited the production of ECM and regulated the balance between MMP2 and TIMP1
Liver fibrosis is characterized by the overdeposition of ECM containing collagen, fibronectin, laminin, and glycosaminoglycan in liver tissues.26 ECM is mainly produced by HSCs, and the production and degradation of ECM are counterbalanced mainly by MMPs and TIMPs. α-SMA (also known as alpha-actin-2) is commonly used as a marker of the activation of HSCs, the formation of myofibroblasts, and the production of ECM. We therefore assessed ECM production in the liver fibrosis mouse models by measuring liver tissue levels of Col-1, α-SMA, MMP2, and TIMP1. Col-1, α-SMA, and TIMP1 protein levels were significantly higher in both model groups than in the vehicle and sham groups, and this increase was attenuated by salidroside in a dose-dependent manner (Figure 3A). In contrast, MMP2 expression was reduced in both model groups but increased by salidroside. The mRNA levels were consistent with the protein levels determined by Western blotting (Figure 3B). The protein levels of Col-1 and α-SMA determined by immunohistochemistry were also in accordance with the Western blot and qPCR analyses (Figure 3C). These findings indicated that salidroside could inhibit the production of ECM, as well as upregulate the expression of MMP2 and downregulate TIMP1, leading to the reduced deposition of ECM in both liver fibrosis mouse models.
 | Figure 3 Salidroside inhibited ECM production and regulated the balance between MMP2 and TIMP1. |
Salidroside inhibited HSC activation by downregulating NF-κB and TGF-β1/Smad3 signaling pathways
HSCs are the major source of ECM, and HSCs’ activation by TGF-β1 is known to play an important role in the progression of liver fibrosis. The secretion of TGF-β1 and other inflammatory cytokines is mainly regulated by the NF-κB signaling pathway.12 We therefore examined the NF-κB and TGF-β1/Smad3 signaling pathways. Liver levels of NF-κB, TGF-β1, and p-Smad3/Smad3 proteins were upregulated in both model groups compared with the control groups. These increases were attenuated by 10 or 20 mg/kg salidroside. Meanwhile, expression of IκBα, which inhibits the transcription factor NF-κB, was downregulated by CCl4 and BDL but upregulated following salidroside treatment (Figures 4A and 5A). At the transcriptional level, NF-κB, TGF-β1, and Smad3 mRNA levels were also increased by CCl4 and BDL, but these increases were attenuated by salidroside (Figures 4B and 5B). Expression levels of TGF-β1, NF-κB, and p-Smad3 in liver tissues determined by immunohistochemistry were in accordance with those determined by Western blot and qPCR analyses (Figures 4C and 5C). Since HSCs and Kupffer cells (KCs) are two main kinds of cells giving rise to fibrosis, double-immunofluorescence staining was used to detect whether TGF-β1 and NF-κB signaling pathways were enhanced in these cells. As shown in Figures 4D and 5D, there were positive areas in CCl4 and BDL groups, which confirmed the activation of TGF-β1 and NF-κB signaling pathways in KCs and HSCs. However, salidroside treatment could effectively inhibit both the pathways in them. These results demonstrated that salidroside could downregulate the NF-κB and TGF-β1/Smad3 signaling pathways to inhibit the activation of HSCs.
 | Figure 4 Salidroside downregulated the NF-κB pathway in liver fibrosis. |
 | Figure 5 Salidroside inhibited the TGF-β1/Smad3 pathway in liver fibrosis. |
Discussion
Liver fibrosis is a scarring response to chronic liver injury,5,27 which can result in hepatic dysfunction and may even progress to liver cirrhosis and primary hepatocellular carcinoma.28 Liver transplantation is currently the only curative treatment for liver fibrosis, and novel safe and effective antifibrotic agents are urgently needed to tackle this problem.
Salidroside is considered to be a liver-friendly plant derivative.20,29,30 Our previous study has also demonstrated that salidroside could inhibit apoptosis and autophagy during hepatic ischemia–reperfusion.19 These suggest that salidroside may be a promising protective agent in liver fibrosis. Therefore, in our study, we established two liver fibrosis models to mimic chemical substances’ induced fibrosis and cholestatic-induced periportal biliary fibrosis and explored the protective effect and mechanisms of salidroside in liver fibrosis. CCl4 can mimic human chronic disease associated with toxic damage and is widely used for studying liver fibrosis and cirrhosis in mice.21 After intraperitoneal administration, the liver metabolizes CCl4 via cytochrome P450 2E1 to yield free radicals, leading to necrosis of centrilobular hepatocytes, activation of KCs, and induction of the inflammatory response, resulting in liver fibrosis.1,22 Common BDL can also be used to induce cholestatic injury and mimic periportal biliary fibrosis. Obstruction of the bile duct increases biliary pressure, resulting in inflammation, generation of reactive oxygen species, and proliferation of biliary epithelial cells that can secrete fibrogenic markers, including TIMP1, α-SMA, collagen, and TGF-β1.23,24 Our results showed that both 10 and 20 mg/kg salidroside could improve liver fibrosis.
Liver fibrosis is characterized by the progressive accumulation of ECM and a failure of ECM degradation.5,27,31–33 ECM is mainly produced by HSCs and also by hepatocytes, liver sinusoidal endothelial cells (SECs), KCs, and other fibroblasts.6,26 Under normal conditions, HSCs exist in a quiescent phenotype in Disse’s space in the liver and contribute to the metabolism and storage of vitamin A,34 ECM production and degradation, and hepatic sinusoidal blood flow regulation.35 However, when the liver is injured, KCs are initially activated, resulting in the secretion of various cytokines, especially TGF-β1.4,10,36,37 HSCs express TGF-β1 receptors and can secrete TGF-β1 as well, leading to persistent activation of HSCs. Activated HSCs can transform into myofibroblasts, characterized by enhanced proliferative ability, loss of vitamin A and lipid droplets, expression of α-SMA, and increased ECM synthesis.4,5,27,38 Furthermore, the balance of ECM levels, which is mainly regulated by MMPs and TIMPs, is disrupted in activated HSCs, with increased ECM production and reduced degradation, leading to accumulation and deposition of ECM, especially Col-1, in liver tissues.11 Liver fibrosis is thus initiated and promoted by activation of HSCs. In our study, we investigated HSCs’ activation and ECM levels in liver fibrosis mouse models. Our results indicated that salidroside could inhibit activation of HSCs and deposition of ECM in liver tissues by inhibiting ECM production and promoting ECM degradation.
The TGF-β1/Smad3 signaling pathway is known to play a critical role in the pathogenesis of liver fibrosis.6,13,15,31 As noted earlier, TGF-β1 is an important factor in HSCs’ activation. Our results proved that TGF-β1 was increased in liver tissues after injury. It then activates TβR type II, which can further phosphorylate and activate TβR type I and interact with it.15 TGF-β1 then propagates the signal via phosphorylation of its downstream signaling molecule Smad3, leading to the accumulation of p-Smad3 in the nucleus, DNA binding, and gene transcription.15,38,39 Activation of the TGF-β1/Smad3 signaling pathway can thus result in the production of ECM components that facilitate fibrosis pathogenesis. The current results showed that the enhancement of TGF-β1 and p-Smad3 was reduced by salidroside treatment, indicating that salidroside inhibited the TGF-β1/Smad3 signaling pathway in activated HSCs.
Inflammation is thought to be the first step in fibrogenesis.6 NF-κB is a key transcriptional regulator of the inflammatory response and also plays an important role in the process of liver fibrosis. The NF-κB pathway can induce the transcription of many proinflammatory mediators, including TGF-β1, to stimulate the activation of HSCs and the production of ECM to promote liver fibrosis.12 Pradere et al40 reported that hepatic macrophages (or KCs) enhanced myofibroblast survival via the NF-κB pathway to promote liver fibrosis. Moreover, Feng et al found that NF-κB inhibited TNF-α-induced apoptosis to protect activated HSCs from cell death.12 The NF-κB inhibitor IκBα can bind to NF-κB subunits to form the IκBα/p50/p65 complex, which hides NF-κB’s nuclear localization site and prevents NF-κB translocation into the nucleus.41 However, TGF-β1 was reported to induce the degradation of IκBα, resulting in the enhancement of NF-κB.13 TGF-β1 can also promote the activation of NF-κB by TGF-β-activated kinase TAK1 and the IκB kinase.42 In turn, both NF-κB and Smad3 can increase the expression of Smad7, which competes for TGF-β1 receptors and downregulates the TGF-β1/Smad3 pathway, providing negative feedback.43 Our results showed that NF-κB expression was increased while IκBα expression was reduced in fibrotic liver tissues. However, salidroside reversed these situations, indicating that it could inhibit the NF-κB pathway in fibrogenesis in mice.
Autophagy is an intracellular material recycling and energy production process during energy restriction, stress, or inflammation.16,44–47 Autophagy is thus beneficial to the survival of cells but is also involved in many disorders, including liver fibrosis. Intracellular lipid droplets are lost during HSCs’ activation, and Hernandez-Gea et al48 found that lipid droplets could be digested via autophagy to supply energy for the activation of HSCs, thereby promoting liver fibrosis. Thoen et al49 reported that an autophagy inhibitor could ameliorate HSCs’ activation by inhibiting autophagy. These two studies also demonstrated that autophagy contributed to HSCs’ activation via increased LC3-II and decreased p62 expression.48,49 A close connection between autophagy and the TGF-β1/Smad3 pathway has been reported, whereby TGF-β1/Smad3 pathway can lead to the transcription of Beclin-1, which plays a critical role in the nucleation of the autophagy process.10,50 Therefore, there is extensive evidence to support a role for autophagy in the activation of HSCs. Yin et al51 reported that salidroside could protect cortical neurons from glutamate-induced cytotoxicity by inhibiting autophagy. The results of the current study suggest that salidroside can also relieve liver fibrosis by inhibiting autophagy of HSCs.
In summary, this study demonstrated that salidroside can attenuate liver fibrosis in mice. Salidroside inhibits the production of TGF-β1 in KCs and HSCs via the NF-κB signaling pathway. Insufficient TGF-β1 results in reduced activation of HSCs and downregulation of the TGF-β1/Smad3 pathway, including decreased ECM production and inhibition of autophagy (Figure 6). Salidroside may thus be a promising agent to attenuate liver fibrosis and TGF-β1 may be the probable target. Since liver fibrosis may progress into hepatocarcinoma and several studies have demonstrated that natural products with antifibrotic property are effective to hepatocarcinoma as well,52–54 it is probably that salidroside is promising for hepatocarcinoma treatment. However, further studies are needed to investigate the safety of salidroside for clinical applications and its role in hepatocarcinoma.
 | Figure 6 Protective mechanisms of salidroside against liver fibrosis. |
Conclusion
We confirmed that salidroside could effectively attenuate liver fibrosis in mice. Salidroside reduced the production of TGF-β1 in KCs and HSCs via inhibition of the NF-κB pathway, thereby inhibiting the activation and autophagy of HSCs through downregulation of the TGF-β1/Smad3 pathway.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant numbers 81670472, 81700502, and 81500466).
Disclosure
The authors report no conflicts of interest in this work.
References
Crespo Yanguas S, Cogliati B, Willebrords J, et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90(5):1025–1048. | ||
Schuppan D. Liver fibrosis: common mechanisms and antifibrotic therapies. Clin Res Hepatol Gastroenterol. 2015;39(suppl 1):S51–S59. | ||
Wu L, Zhang Q, Mo W, et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-beta1/Smads and PI3K/Akt pathways. Sci Rep. 2017;7(1):9289. | ||
Mao Y, Zhang S, Yu F, Li H, Guo C, Fan X. Ghrelin attenuates liver fibrosis through regulation of TGF-beta1 expression and autophagy. Int J Mol Sci. 2015;16(9):21911–21930. | ||
Toosi AE. Liver fibrosis: causes and methods of assessment, a review. Rom J Intern Med. 2015;53(4):304–314. | ||
Su TH, Kao JH, Liu CJ. Molecular mechanism and treatment of viral hepatitis-related liver fibrosis. Int J Mol Sci. 2014;15(6):10578–10604. | ||
Eom YW, Shim KY, Baik SK. Mesenchymal stem cell therapy for liver fibrosis. Korean J Intern Med. 2015;30(5):580–589. | ||
Czaja AJ. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J Gastroenterol. 2014;20(10):2515–2532. | ||
Berardis S, Dwisthi Sattwika P, Najimi M, Sokal EM. Use of mesenchymal stem cells to treat liver fibrosis: current situation and future prospects. World J Gastroenterol. 2015;21(3):742–758. | ||
Li J, Chen K, Li S, et al. Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the TGF-beta1/Smad pathway-mediated inhibition of extracellular matrix and autophagy. Drug Des Devel Ther. 2016;10:619–630. | ||
De Minicis S, Seki E, Uchinami H, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132(5):1937–1946. | ||
Feng X, Tan W, Cheng S, et al. Upregulation of microRNA-126 in hepatic stellate cells may affect pathogenesis of liver fibrosis through the NF-kappaB pathway. DNA Cell Biol. 2015;34(7):470–480. | ||
Freudlsperger C, Bian Y, Contag Wise S, et al. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32(12):1549–1559. | ||
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. | ||
Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. | ||
Mao YQ, Fan XM. Autophagy: A new therapeutic target for liver fibrosis. World J Hepatol. 2015;7(16):1982–1986. | ||
Tang H, Gao L, Mao J, et al. Salidroside protects against bleomycin-induced pulmonary fibrosis: activation of Nrf2-antioxidant signaling, and inhibition of NF-kappaB and TGF-beta1/Smad-2/-3 pathways. Cell Stress Chaperones. 2016;21(2):239–249. | ||
Zhang Z, Ding L, Wu L, Xu L, Zheng L, Huang X. Salidroside alleviates paraquat-induced rat acute lung injury by repressing TGF-beta1 expression. Int J Clin Exp Pathol. 2014;7(12):8841–8847. | ||
Feng J, Zhang Q, Mo W, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting the mitogen-activated protein kinase pathway in mice. Drug Des Devel Ther. 2017;11:1989–2006. | ||
Ouyang J, Gao Z, Ren Z, Hong D, Qiao H, Chen Y. Synergistic effects of rMSCs and salidroside on the experimental hepatic fibrosis. Pharmazie. 2010;65(8):607–613. | ||
Domenicali M, Caraceni P, Giannone F, et al. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J Hepatol. 2009;51(6):991–999. | ||
Constandinou C, Henderson N, Iredale JP. Modeling liver fibrosis in rodents. Methods Mol Med. 2005;117:237–250. | ||
Chang ML, Yeh CT, Chang PY, Chen JC. Comparison of murine cirrhosis models induced by hepatotoxin administration and common bile duct ligation. World J Gastroenterol. 2005;11(27):4167–4172. | ||
Park KC, Park JH, Jeon JY, et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br J Pharmacol. 2014;171(21):4820–4830. | ||
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. | ||
Bruckner-Tuderman L, Bruckner P. Genetic diseases of the extracellular matrix: more than just connective tissue disorders. J Mol Med. 1998;76(3–4):226–237. | ||
Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. 2013;305(8):C789–C799. | ||
Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364(1–2):33–60. | ||
Yang ZR, Wang HF, Zuo TC, Guan LL, Dai N. Salidroside alleviates oxidative stress in the liver with non-alcoholic steatohepatitis in rats. BMC Pharmacol Toxicol. 2016;17:16. | ||
Hu B, Zou Y, Liu S, et al. Salidroside attenuates concanavalin A-induced hepatitis via modulating cytokines secretion and lymphocyte migration in mice. Mediators Inflamm. 2014;2014:314081. | ||
Samarakoon R, Overstreet JM, Higgins PJ. TGF-beta signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25(1):264–268. | ||
Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–841. | ||
Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20(23):7312–7324. | ||
Shirakami Y, Lee SA, Clugston RD, Blaner WS. Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta. 2012;1821(1):124–136. | ||
Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis. 2001;21(3):337–349. | ||
Shen M, Chen K, Lu J, et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-beta1 expression and autophagy. Mediators Inflamm. 2014;2014:954502. | ||
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. | ||
Tang LX, He RH, Yang G, et al. Asiatic acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One. 2012;7(2):e31350. | ||
Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. | ||
Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–1473. | ||
Luedde T, Schwabe RF. NF-kappaB in the liver – linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–118. | ||
Sakurai H, Miyoshi H, Toriumi W, Sugita T. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J Biol Chem. 1999;274(15):10641–10648. | ||
Attisano L, Lee-Hoeflich ST. The Smads. Genome Biol. 2001;2(8):REVIEWS3010. | ||
Go KL, Lee S, Zendejas I, Behrns KE, Kim JS. Mitochondrial dysfunction and autophagy in hepatic ischemia/reperfusion injury. Biomed Res Int. 2015;2015:183469. | ||
Xu S, Wu L, Zhang Q, et al. Pretreatment with propylene glycol alginate sodium sulfate ameliorated concanavalin A-induced liver injury by regulating the PI3K/Akt pathway in mice. Life Sci. 2017;185:103–113. | ||
Chen K, Li J, Li S, et al. 15d-PGJ2 alleviates ConA-induced acute liver injury in mice by up-regulating HO-1 and reducing hepatic cell autophagy. Biomed Pharmacother. 2016;80:183–192. | ||
Liu T, Zhang Q, Mo W, et al. The protective effects of shikonin on hepatic ischemia/reperfusion injury are mediated by the activation of the PI3K/Akt pathway. Sci Rep. 2017;7:44785. | ||
Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. | ||
Thoen LF, Guimaraes EL, Dolle L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55(6):1353–1360. | ||
Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3(1):17–27. | ||
Yin WY, Ye Q, Huang HJ, et al. Salidroside protects cortical neurons against glutamate-induced cytotoxicity by inhibiting autophagy. Mol Cell Biochem. 2016;419(1–2):53–64. | ||
Li S, Wu L, Feng J, et al. In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity. Sci Rep. 2016;6:28479. | ||
Li J, Chen K, Wang F, et al. Methyl jasmonate leads to necrosis and apoptosis in hepatocellular carcinoma cells via inhibition of glycolysis and represses tumor growth in mice. Oncotarget. 2017;8(28):45965–45980. | ||
Xu L, Dai W, Li J, et al. Methylation-regulated miR-124-1 suppresses tumorigenesis in hepatocellular carcinoma by targeting CASC3. Oncotarget. 2016;7(18):26027–26041. |
Supplementary material
 | Figure S1 Effect of Sal on autophagic flux in rat HSC-T6 cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.