Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Safety, tolerability, and pharmacokinetics of single and repeat ascending doses of CHF6001, a novel inhaled phosphodiesterase-4 inhibitor: two randomized trials in healthy volunteers
Authors Mariotti F, Govoni M, Lucci G, Santoro D, Nandeuil MA ![]()
Received 14 May 2018
Accepted for publication 3 September 2018
Published 18 October 2018 Volume 2018:13 Pages 3399—3410
DOI https://doi.org/10.2147/COPD.S174156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Fabrizia Mariotti,1 Mirco Govoni,1 Germano Lucci,1 Debora Santoro,1 Marie Anna Nandeuil2
1Global Clinical Development, Chiesi Farmaceutici SpA, Parma, Italy; 2Global Clinical Development, Chiesi S.A.S., Courbevoie, France
Purpose: The purpose of this study was to evaluate safety, tolerability, and pharmacokinetics (PK) of CHF6001, an inhaled phosphodiesterase-4 inhibitor.
Materials and methods: Two healthy volunteer, randomized, double-blind, placebo-controlled studies were conducted. In each, Part 1 evaluated single ascending doses, with PK sampling up to 48 hours post-dose; Part 2 evaluated multiple ascending doses (Study 1, 7 days; Study 2, 14 days), with PK sampling up to 24 hours post-dose on first and last day of each period. In Study 1, treatments were administered via single-dose dry-powder inhaler (SDDPI; Aerolizer): Part 1, 20, 100, 200, 400, 800, 1,600, and 2,000 µg or placebo; Part 2, 100, 300, 600, 1,200, and 1,600 µg or placebo once daily (OD). In Study 2, treatments were administered via multi-dose dry-powder inhaler (MDDPI; NEXThaler): Part 1, 2,400, 4,000, and 4,800 µg or placebo; Part 2, 1,200, 2,000, or 2,400 µg twice daily (BID) or placebo. Modeling and simulation then compared OD and BID dosing via MDDPI.
Results: There was a clear correlation between CHF6001 dose and plasma concentration, following single and multiple doses and using SDDPI and MDDPI. CHF6001 plasma concentration area under the curve (AUC) was dose proportional, with steady state slopes of the fitted line of 0.95 (90% CI: 0.86, 1.04) for AUC0–24 h in Study 1, and 0.85 (90% CI: 0.38, 1.32) for AUC0–12 h in Study 2. Bioavailability was ~30% higher with MDDPI than SDDPI. The PK simulation confirmed dose proportionality; the same total daily dose OD or BID via MDDPI resulted in similar 24 hours exposure, with BID dosing providing smaller fluctuation and lower maximum concentration. CHF6001 was well tolerated with no relationship between dose and adverse events.
Conclusion: CHF6001 demonstrated a good safety profile. There was a clear dose proportionality for systemic exposure, with higher bioavailability via MDDPI, suggesting that the MDDPI provides better pulmonary drug deposition. BID dosing was associated with a better exposure profile.
Keywords: healthy volunteers, chronic obstructive pulmonary disease, phosphodiesterase-4 inhibitors, pharmacokinetics
Introduction
Anti-inflammatory agents have an established role in the management of COPD, especially for the prevention of exacerbations.1 Two main classes of anti-inflammatory agents are currently approved and used in COPD – inhaled corticosteroids (ICSs) and oral phosphodiesterase-4 (PDE4) inhibitors.1 In addition to the prevention of exacerbations, the combination of ICSs with inhaled long-acting bronchodilators has been shown to improve lung function and quality of life, although not all patients with COPD benefit from treatment.1 Similarly, roflumilast, the only PDE4 inhibitor currently marketed, reduces the risk of exacerbations and improves lung function in combination with inhaled long-acting bronchodilators, but only in a subgroup of patients with COPD.1–4 However, roflumilast is taken orally and is associated with a higher incidence of systemic adverse events (AEs) than inhaled COPD maintenance medication, with some of these events (including diarrhea, nausea, weight loss, and abdominal pain) resulting in high withdrawal from clinical trials.1–4 Since many patients with COPD continue to exacerbate despite therapy,5 an effective anti-inflammatory treatment with a good tolerability profile thanks to an inhaled delivery route (ie, a wide therapeutic window) and that might also be suitable for co-formulation with other established inhaled therapies would be welcome.
CHF6001 is a novel PDE4 inhibitor currently in clinical development. Unlike roflumilast, CHF6001 has been specifically designed and formulated to be delivered via inhalation, a route of administration that has the potential to avoid the gastrointestinal adverse effects that are characteristic of oral therapy in this class. Indeed, in preclinical studies in rats and ferrets (the latter representing an established model for evaluation of such effects), following intratracheal administration at doses compatible with those used in humans, CHF6001 exerted anti-inflammatory effects with no evidence of emesis or nausea.6 Furthermore, the combination of CHF6001 and a LAMA (either tiotropium or glycopyrrolate) was more effective than either compound alone in terms of inhibition of airway smooth muscle remodeling in a guinea pig lung slice model.7 In addition, following rhinovirus challenge of human bronchial epithelial cells, CHF6001 reduced the levels of a range of cytokines in a concentration-dependent manner, both alone and in combination with low-dose corticosteroids, indicating anti-inflammatory activity against virus-induced inflammatory mediators.8 CHF6001 is highly plasma protein bound (>99% in humans),9 with low systemic bioavailability after intratracheal administration and very low bioavailability after oral administration suggesting that any portion ingested following inhaled administration is likely to have a negligible contribution to overall systemic exposure.6 It is metabolically stable in human liver, with the main metabolites, which are pharmacologically inactive, being related to hydrolysis, and is rapidly eliminated following intravenous administration, predominantly via the fecal route.10 Two Phase II studies are ongoing in patients with COPD.
This manuscript describes the pharmacokinetics (PK) and safety profile of CHF6001 from two single and repeat ascending-dose studies in healthy volunteers. In the first study, CHF6001 was administered via a capsule-based single-dose dry-powder inhaler (SDDPI). In the second study, a reservoir-based multi-dose dry-powder inhaler (MDDPI) was used (the NEXThaler® – the device that is being used for the ongoing clinical development of CHF6001). Modeling and simulation were then used to compare the PK profiles of once daily (OD) and twice daily (BID) dosing via the MDDPI.
Materials and methods
Trial design
Both studies were randomized, double-blind, and placebo controlled and were conducted at a single center. Study 1 comprised two parts. Part 1 was single ascending dose, crossover in design. After a screening visit, eligible subjects were randomized into one of two cohorts attending alternating visits, with each visit separated by 7–14 days; Cohort A attended four visits and Cohort B attended three. Subjects remained at the study center from the evening of day–2 until the morning of day 2, and returned on day 3 for 48 hours post-dose assessments. Blood samples were taken for PK analysis pre-dose and at 5, 10, 15, and 30 minutes, and 1, 1.5, 2, 4, 6, 8, 12, 18, 24, and 48 hours post-dose.
Part 2 of Study 1 was repeat, ascending dose (7 day dosing), in a parallel group. After a screening visit, subjects were randomized to one of five cohorts, with subjects in each cohort receiving one of the CHF6001 doses or placebo (the CHF6001 dose escalated with subsequent cohorts). The next cohort did not start treatment until the safety and PK assessments were completed for the previous cohort, and an independent safety advisory committee had reviewed all data. Subjects remained at the study center from the evening of day–2 until the morning of day 8, returning on day 9 for the 48 hours post-dose assessments. Blood samples were taken for PK analysis up to 24 hours post-dose on days 1 and 7 at the same time points as Part 1 and pre-dose on days 2–6.
Study 2 also comprised two parts. Part 1 was three period, single ascending dose in design. After a screening visit, eligible subjects were randomized to one of four treatment sequences. Subjects then attended three visits, receiving CHF6001 or placebo at each visit, with the administered CHF6001 dose increasing at each visit. Subsequent visits did not take place until the acceptable safety and tolerability profile of the lower CHF6001 dose was confirmed (a minimum of 18 days between subsequent doses). Subjects remained at the study center from the evening of day–2 until the morning of day 3, with blood samples taken for PK analysis pre-dose and at 30 minutes and 1, 1.5, 2, 3, 4, 6, 8, 12, 18, 24, and 48 hours post-dose.
Part 2 of Study 2 was repeat, ascending dose (14 day dosing), parallel group. Eligible subjects were randomized to one of three cohorts, with subjects in each cohort receiving either one of the CHF6001 doses or placebo (the CHF6001 dose escalated with subsequent cohorts). The next cohort did not start treatment until the safety assessments had been completed for the previous cohort. Subjects remained at the study center from the evening of day–2 until the morning of day 15, with blood samples taken for PK analysis up to 12 hours post-dose on days 1 and 14 at the same time points as Part 1, and before the morning dose on days 2–13.
In both studies, blood samples (4 mL at each time point) were collected in lithium heparin tubes and immediately chilled in an ice bath. Plasma was prepared by centrifugation at 2,000 ×g for 15 minutes at ~4°C and was transferred into polypropylene tubes pre-filled with 1 M citric acid (100 μL per 1 mL plasma) to stabilize CHF6001. All the plasma samples were stored at ≤−65°C until analysis. The plasma concentration of CHF6001 was measured using a validated liquid chromatography with tandem mass spectrometry (LC-MS/MS) method with a lower limit of quantification of 10 pg/mL, using deuterium-labeled CHF6001 as internal standard. Following solid phase extraction of the plasma samples using 96-well plates (Oasis HLB; Waters Corp., Milford, MA, USA), samples were injected onto the LC-MS/MS (Study 1: AB Sciex API 5500; AB Sciex LLC, Framingham, MA, USA; Study 2: Mass Spectrometer; Waters Xevo, Milford, MA, USA) using heated electrospray ionization. Quality control samples spiked with CHF6001 were analyzed together with the study samples, to demonstrate the accuracy and precision of the study. The method was originally validated for a CHF6001 concentration range of 10–1,000 pg/mL and was used in Study 1. Subsequently, a wider concentration range (10–3,000 pg/mL) was validated and applied to Study 2. The linearity of the analytical method was validated for both CHF6001 concentration ranges. The precision of the analytical method as characterized by the coefficient of variation was ≤11.7%, with an accuracy (based on the relative error for the given concentrations) of ≥88%. The method was specific in human plasma since no significant interference was observed in either the blank plasma samples or in the zero samples (control plasma plus internal standard).
AEs were captured in both studies, with investigators being asked to record the relationship to study medication. In addition, clinical hematology and blood chemistry were assessed at baseline and at the end of dosing, with vital signs (systolic and diastolic blood pressure) and electrocardiogram (ECG) and 24 hours Holter ECG data captured at regular intervals. Lung function (FEV1) was also assessed as a safety parameter in both parts of both studies to exclude the occurrence of post-dose bronchospasm.
All subjects provided written informed consent prior to any study-related procedure. The studies were conducted at the same institution, were approved by the following independent ethics committees: Study 1, Copenhurst Independent Research Ethics Committee, Wirral, UK (25 July 2011); Study 2, Wales Research Ethics Committee, Cardiff, UK (6 November 2014), and were performed in accordance with the principles of the declaration of Helsinki and the International Conference on Harmonization notes for guidance on Good Clinical Practice (ICH/CPMP/135/95). Both studies are registered in ClinicalTrials.gov (Study 1: NCT01703052; Study 2: NCT02386761). There were no substantial amendments to Study 2. Study 1 required two substantial amendments, both to Part 2 (one to permit escalation to the 1,200 μg dose, and one to permit escalation to the 1,600 μg dose).
Participants
Both studies recruited healthy volunteers aged 18–55 years, non- or ex-smokers (<5 pack-years), and in good physical and mental health status. Study 1 recruited male subjects only, whereas Study 2 recruited both male and female subjects. The main exclusion criteria for both studies were clinically significant abnormal 24 hours Holter ECGs at screening, clinically relevant abnormal laboratory values or clinically relevant and uncontrolled disorders that could have impacted completion of the study, and use of medication (whether over-the-counter or prescribed) in the 14 days prior to screening. The full inclusion and exclusion criteria are listed in the Supplementary material. Subjects recruited for Part 1 of each study were not recruited for Part 2.
Interventions
In both studies, subjects were assigned to treatment sequence using a randomization list prepared by the sponsor. All subjects, investigators, site staff, and employees of the sponsor were blinded to treatment assignment. The only exception was a qualified person in the pharmacy of the clinical center who performed the staggered dosing in Study 1, and who therefore had a copy of the randomization list (but did not take part in any of the study assessments).
Treatments administered were:
- Study 1, Part 1: Single doses of CHF6001: 20, 100, 200, 400, 800, 1,600, and 2,000 μg or matching placebo, administered via SDDPI
- Study 1, Part 2: CHF6001 100, 300, 600, 1,200, and 1,600 μg or matching placebo OD for 7 days, administered via SDDPI
- Study 2, Part 1: Single doses of CHF6001: 2,400, 4,000, and 4,800 μg or matching placebo, administered via MDDPI
- Study 2, Part 2: CHF6001 1,200, 2,000, or 2,400 μg BID (total daily doses of 2,400, 4,000, or 4,800 μg) or matching placebo for 14 days, administered via MDDPI
Outcomes
The primary objective of both studies was to investigate the safety and tolerability of CHF6001, with the secondary objective being to investigate its PK profile. PK endpoints calculated included maximum plasma concentration (Cmax), time to maximum concentration (tmax), terminal elimination half-life (t½; calculated following single doses only), and clearance (CL) determined at steady state, adjusted for absolute bioavailability (F). Areas under the plasma concentration–time curve were computed using the trapezoidal rule, from administration to last measurable concentration (AUC0-t) in Part 1 of the two studies and from administration to the end of the dosing interval in Part 2 (ie, to 24 hours post-dose for OD regimens [AUC0–24 h] and to 12 hours post-dose for BID regimens [AUC0–12 h]). The accumulation ratio (Racc) was calculated following multiple dosing.
Sample size and statistical methods
No formal sample size calculations were performed for either study. The following sample sizes were considered adequate: Study 1, Part 1, 20 subjects (10 per cohort); Study 1, Part 2, 48 subjects (12 per cohort); Study 2, Part 1, 12 subjects; Study 2, Part 2, 36 subjects (12 per cohort). All PK parameters were determined using non-compartmental methods, with Study 1 employing WinNonlin v6.1 and Study 2 employing WinNonlin v6.3.0.395.
In both studies, the safety population was defined as all randomized subjects who had received at least one dose of study medication, whereas the PK population was all subjects in the safety population who had at least one valid PK measurement and who had no major PK-related protocol deviations. Data were summarized using SAS (Version 8.2 or higher; SAS Institute Inc., Cary, NC, USA).
A model was developed to compare the PK characteristics of OD and BID dosing of CHF6001 administered via MDDPI. A two-compartment disposition model was found to best fit the data, with three-way parallel absorption pathways and first-order elimination from the central compartment. The model was built using NONMEM (Version 7.3), supplemented with the PsN toolkit (Version 4.4.8). Simulations were run based on 100 subjects replicated 10 times per dose, performed using NONMEM Version 7.4.1 and processed using Phoenix WinNonlin (Version 7.0) and GraphPad Prism (Version 7.0). NONMEM data sets were prepared using SAS 9.4.
Results
Participants
In Study 1, 20 subjects were randomized into Part 1, with 17 completing all treatment periods (one subject withdrew due to an AE after dosing with CHF6001 200 μg, and two were lost to follow-up). For Part 2, 54 subjects were randomized, one of whom was withdrawn prior to receiving any study medication; the remaining 53 completed the study. In Study 2, 12 subjects were randomized into Part 1, with 11 completing (one withdrew due to an AE after receiving CHF6001 2,400 μg), and 36 subjects were randomized into Part 2 with 33 completing (one withdrew due to an AE in the CHF6001 2,400 μg BID group, and two withdrew consent). The baseline demographics of the included subjects are presented in Table 1.
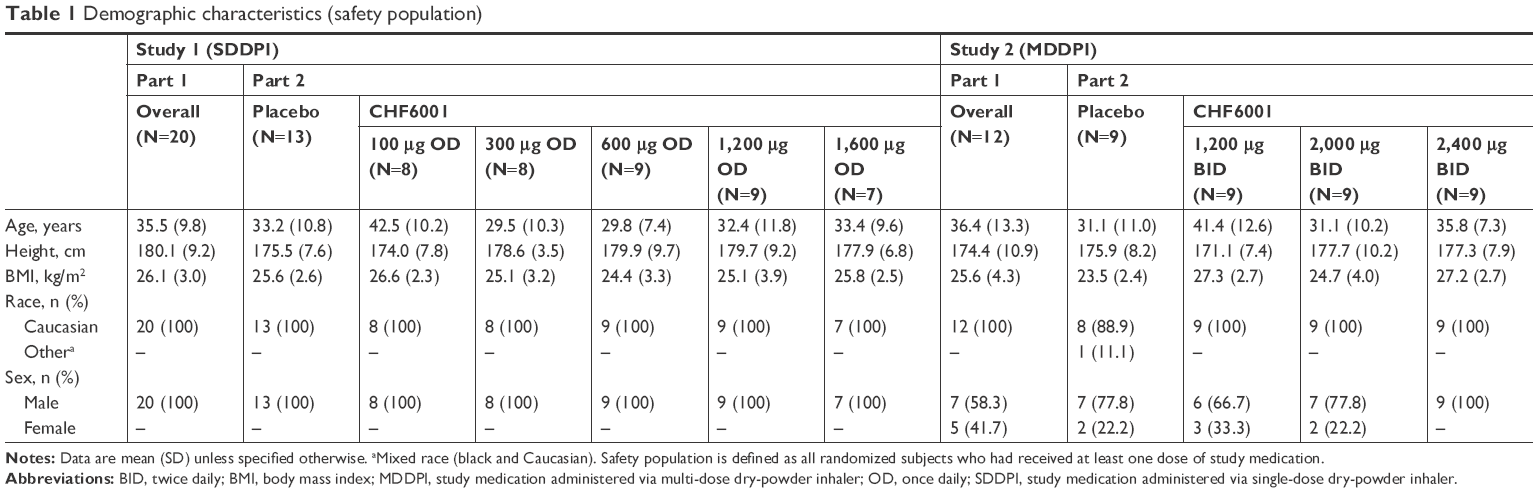 | Table 1 Demographic characteristics (safety population) |
PK
Single dose
Across the two studies, there was a clear correlation between the dose inhaled and CHF6001 plasma concentration, both via the SDDPI and the MDDPI (Figure 1 and Table 2). With the exception of the 20 μg dose (for which plasma concentration was below the limit of quantification from 8 hours post-dose), tmax was achieved for CHF6001 at ~2 hours post-dose with SDDPI and at 3 hours post-dose with MDDPI. The t½ varied between 30 and 45 hours for the SDDPI administration and between 40 and 50 hours for the MDDPI administration.
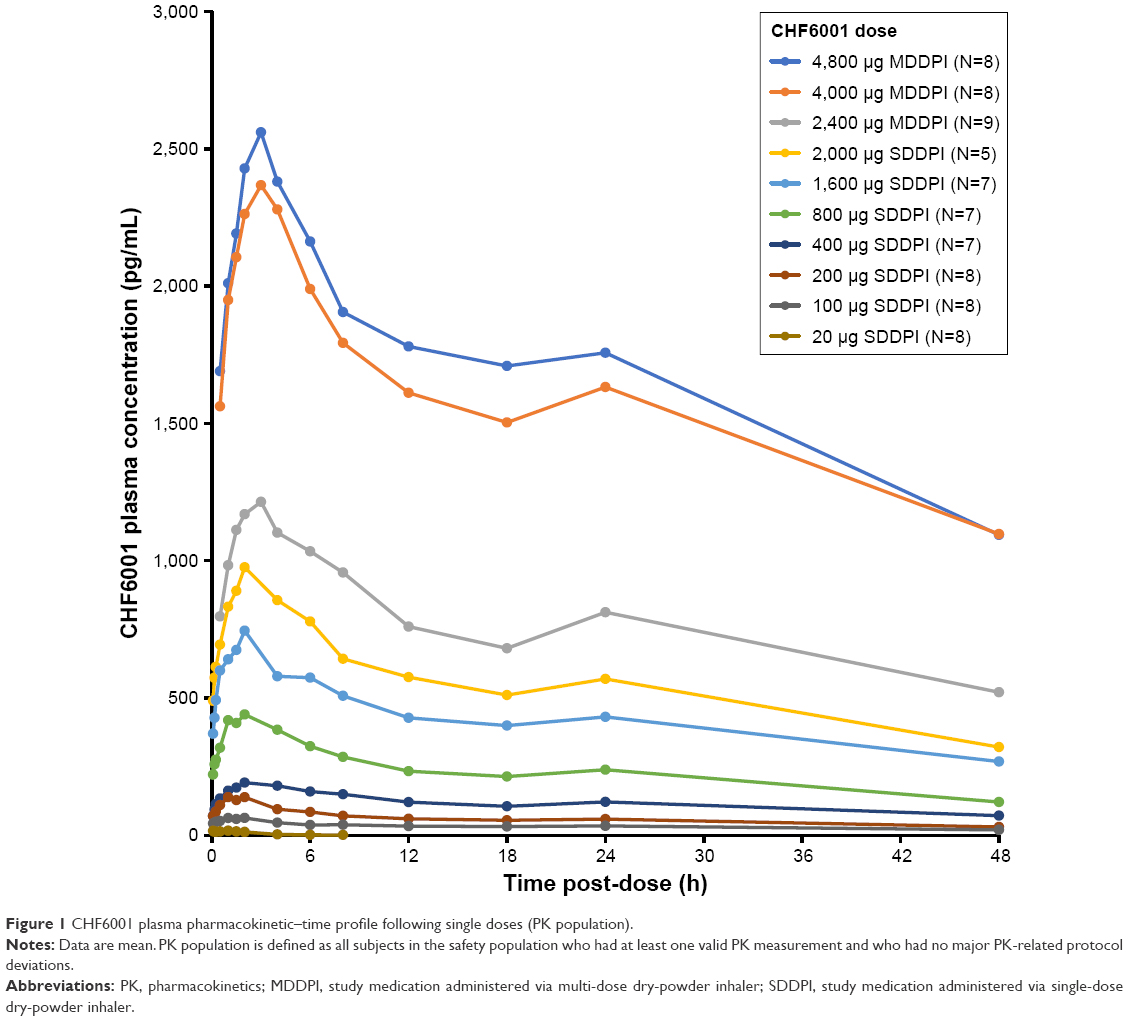 | Figure 1 CHF6001 plasma pharmacokinetic–time profile following single doses (PK population). |
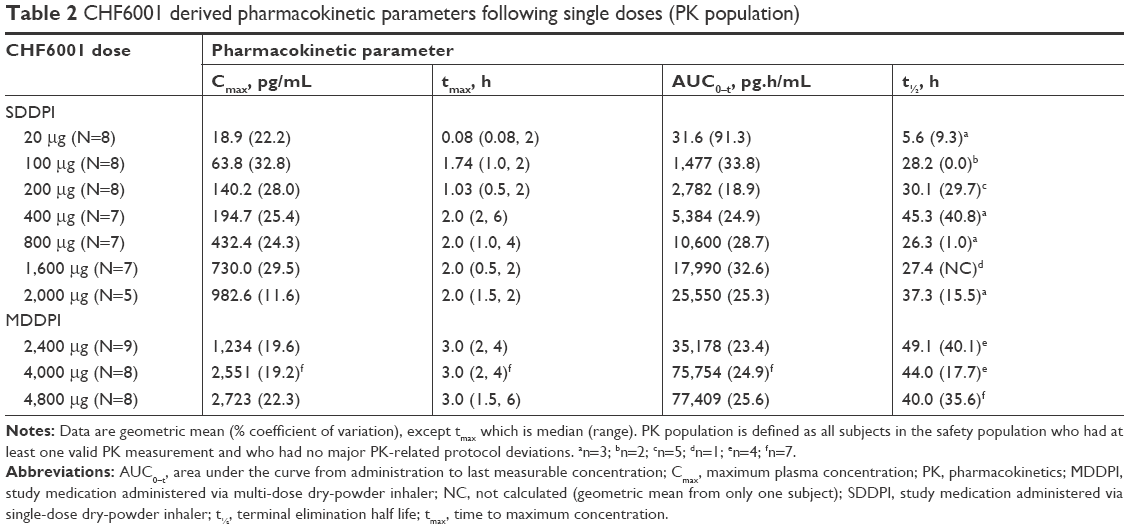 | Table 2 CHF6001 derived pharmacokinetic parameters following single doses (PK population) |
Multiple dose
As with single-dose administration, following administration of multiple doses of CHF6001, plasma concentration increased with increasing dose, both on OD (Figure 2 and Table 3) and BID administration (Figure 3 and Table 3). The increase in AUC was dose proportional, with slopes of the fitted line values of 0.96 (90% CI: 0.89, 1.03) and 0.95 (90% CI: 0.86, 1.04) for AUC0–24 h on Days 1 and 7 of Study 1, and 1.13 (90% CI: 0.80, 1.46) and 0.85 (90% CI: 0.38, 1.32) for AUC0–12 h on Days 1 and 14 of Study 2. At steady-state, median tmax occurred between 1 and 2 hours following OD administration, and between 1.5 and 3 hours following BID dosing. CL/F and Racc were unaffected by dose. The average weighted AUC for the SDDPI device was 19.9 pg.h/mL.μg, compared with 26.6 pg.h/mL.μg for the MDDPI, indicating ~30% higher bioavailability with the latter.
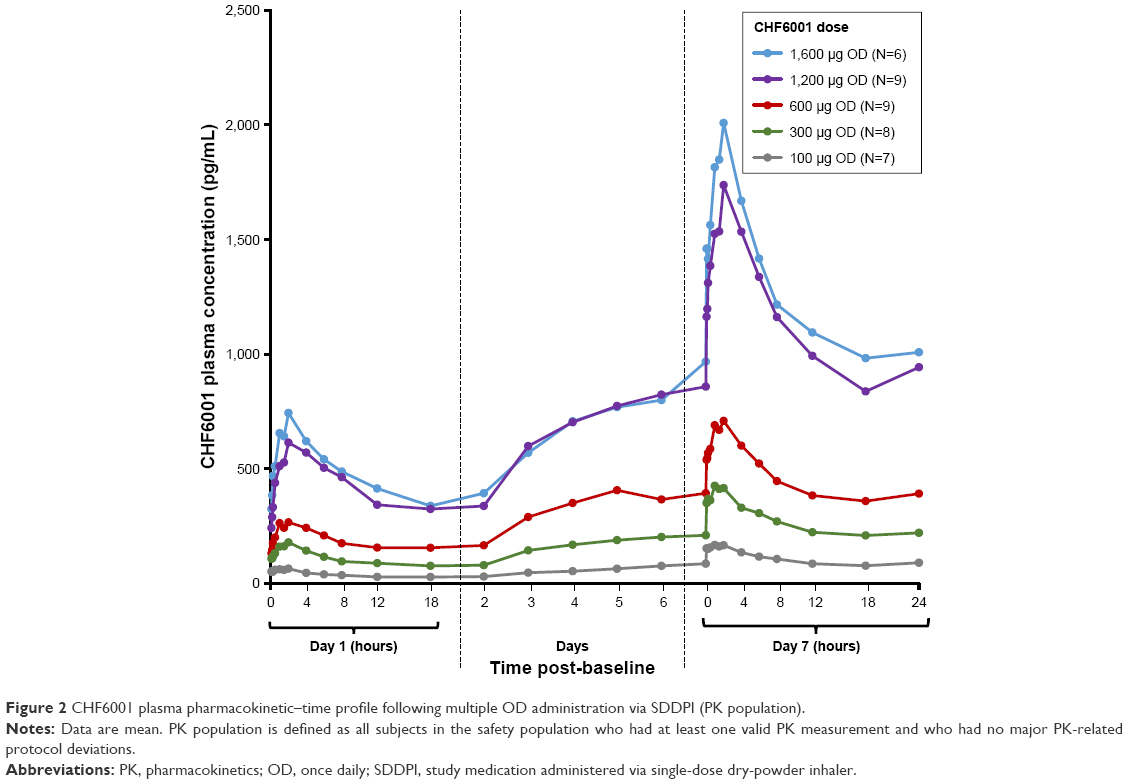 | Figure 2 CHF6001 plasma pharmacokinetic–time profile following multiple OD administration via SDDPI (PK population). |
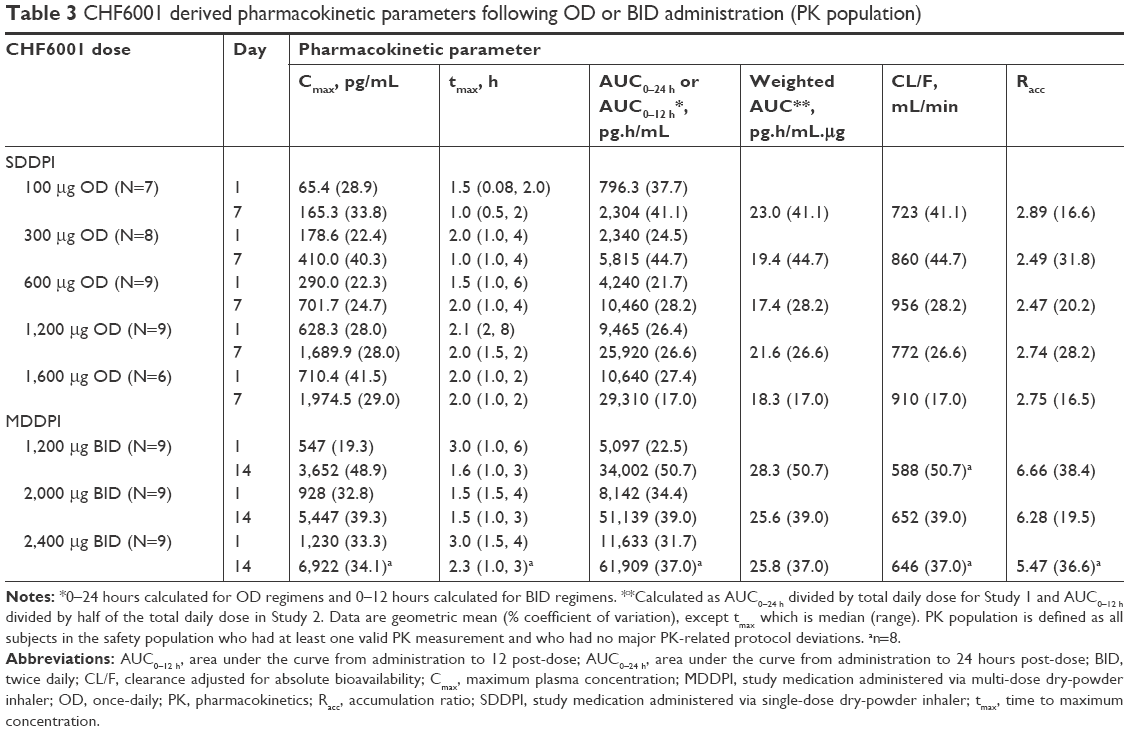 | Table 3 CHF6001 derived pharmacokinetic parameters following OD or BID administration (PK population) |
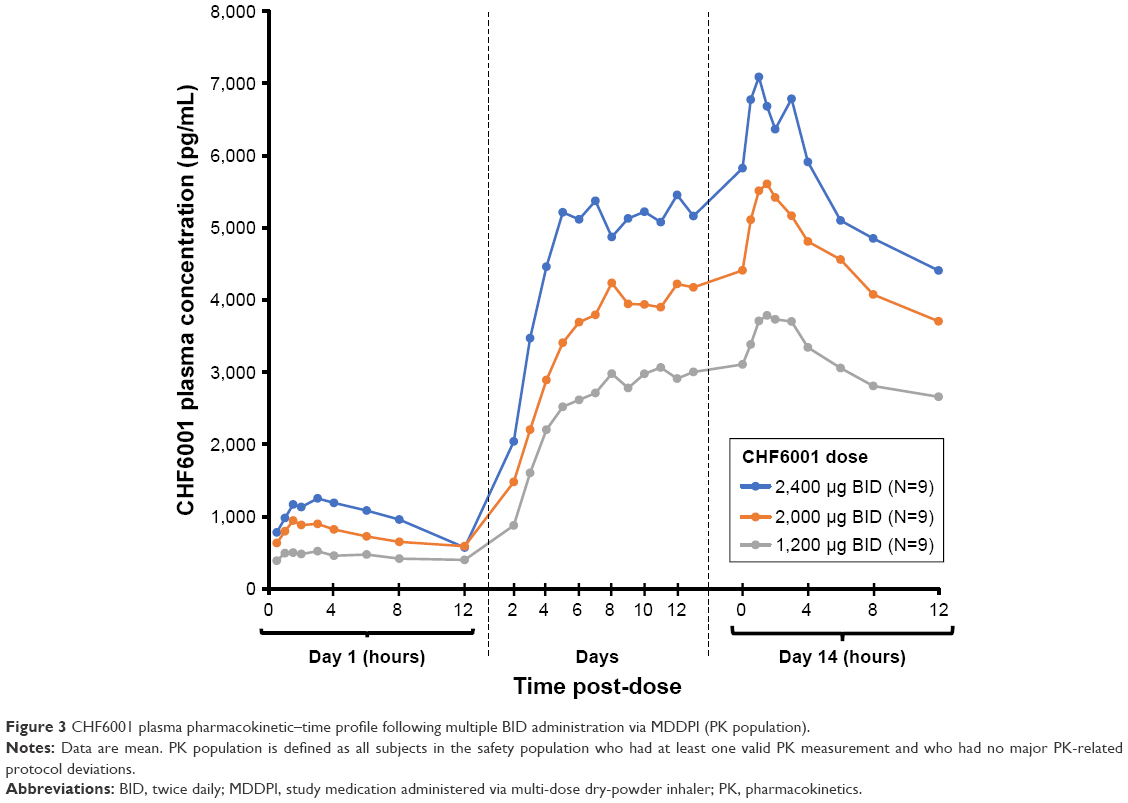 | Figure 3 CHF6001 plasma pharmacokinetic–time profile following multiple BID administration via MDDPI (PK population). |
PK modeling and simulation
The PK modeling and simulation confirmed dose-proportionality for the BID dosing regimen via the MDDPI, in terms of maximum, minimum, and total systemic exposure at steady state (Table 4), which had been reached for all CHF6001 doses by day 14 (Figure 4). As shown in Figure 5 and Table 4, simulated systemic exposure of the same total daily dose given OD or BID via MDDPI (ie, 1,200 μg BID and 2,400 μg OD) resulted in similar exposure over 24 hours at steady state, but with BID dosing resulting in smaller fluctuation and a lower Cmax.
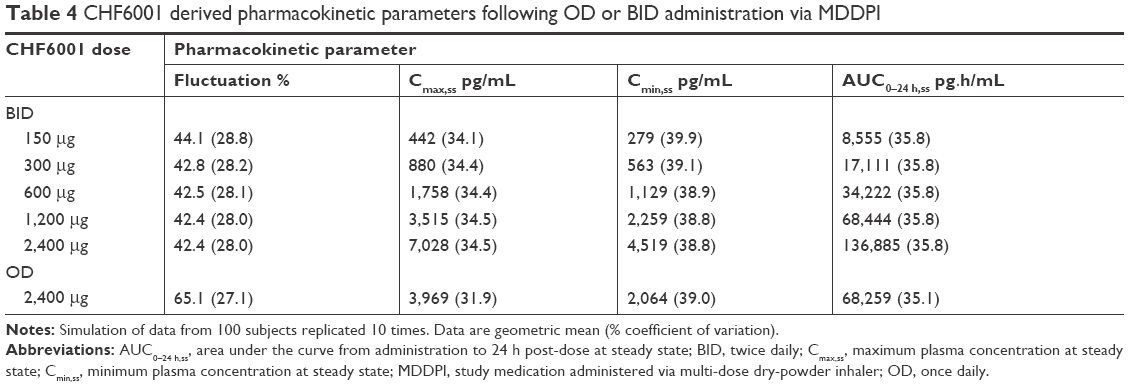 | Table 4 CHF6001 derived pharmacokinetic parameters following OD or BID administration via MDDPI |
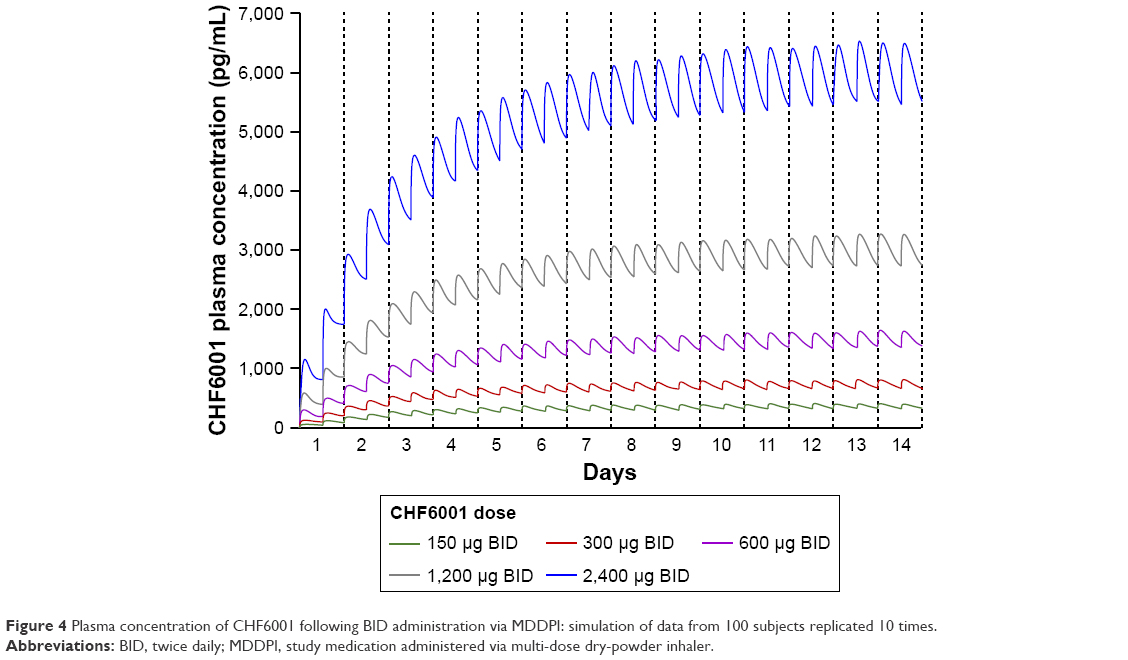 | Figure 4 Plasma concentration of CHF6001 following BID administration via MDDPI: simulation of data from 100 subjects replicated 10 times. |
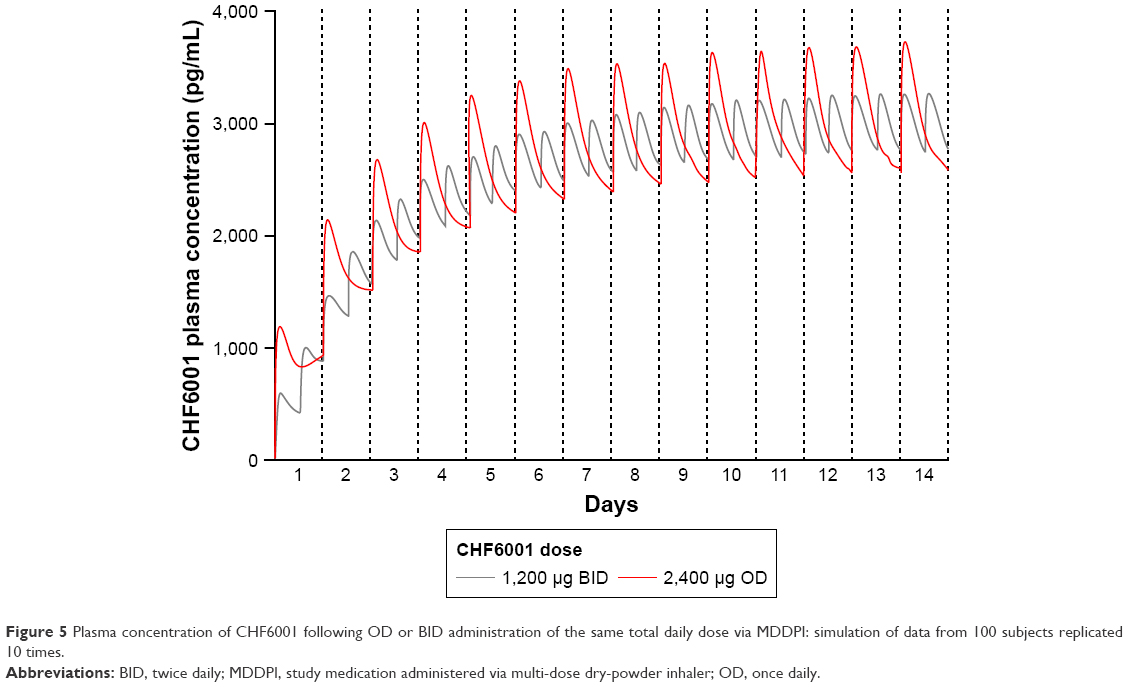 | Figure 5 Plasma concentration of CHF6001 following OD or BID administration of the same total daily dose via MDDPI: simulation of data from 100 subjects replicated 10 times. |
Safety
There was no relationship between CHF6001 dose and the occurrence of AEs or serious adverse events, either on single (Table 5) or multiple dosing (Table 6). When grouped by preferred term, no AE occurred in more than two subjects with any treatment.
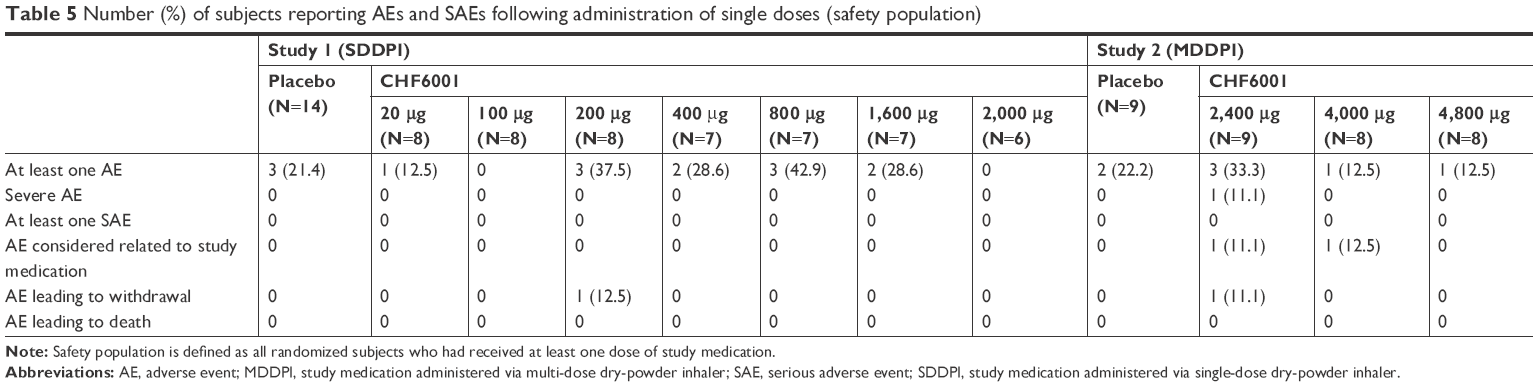 | Table 5 Number (%) of subjects reporting AEs and SAEs following administration of single doses (safety population) |
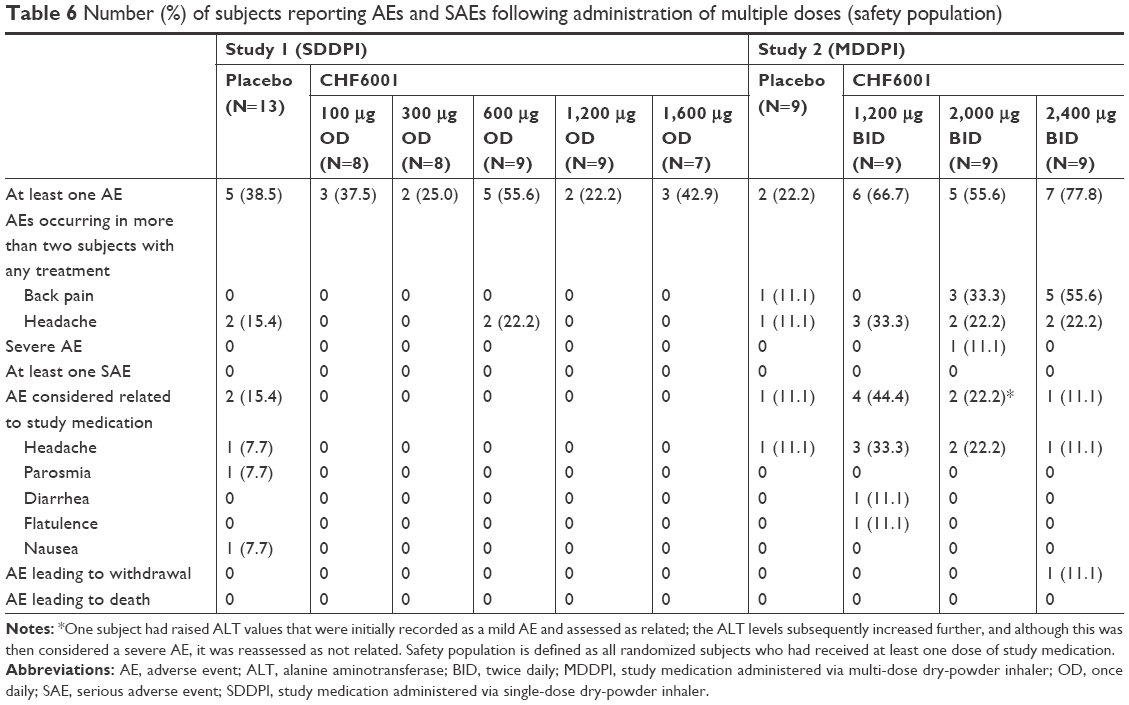 | Table 6 Number (%) of subjects reporting AEs and SAEs following administration of multiple doses (safety population) |
Following single doses of CHF6001 in both studies, there were no nausea events; one subject reported diarrhea, abdominal pain, and constipation following administration of CHF6001 200 μg, but these events were not considered related to study medication. One subject withdrew from Study 1 due to an asymptomatic, non-sustained ventricular tachycardia detected during 24 hours Holter ECG monitoring after receiving CHF6001 200 μg, an event that was assessed as mild in severity and not related to study medication. One subject withdrew from Study 2 due to abnormal liver function tests (increases in alanine aminotransferase [ALT] and gamma glutamyltransferase [GGT]) after receiving CHF6001 2,400 μg. However, both ALT and GGT values were already high at screening, and the AE was therefore considered not related to study medication. Two events were considered potentially related to study medication – dizziness following the 2,400 μg dose and cough following the 4,000 μg dose, but neither subject withdrew prematurely from the study.
On multiple dosing, although in Study 2 a higher proportion of subjects experienced AEs with CHF6001 than placebo, only one event was considered severe (increased ALT, 10 days after completing treatment with CHF6001 2,000 μg BID); this event was not considered related to study medication (Table 6). Two subjects had nausea as AEs, one with CHF6001 300 μg OD that was not considered related to treatment and one with placebo. One subject experienced diarrhea and one experienced flatulence, both with CHF6001 1,200 μg BID, and both considered related to treatment although mild in nature, and one subject experienced abdominal pain with CHF6001 2,000 μg BID, not considered related to treatment. One subject withdrew from the CHF6001 2,400 μg BID group prior to completing the study, due to moderate otitis media that was not considered related to study treatment.
In both studies, mean laboratory values were within normal ranges, with few individual values outside the normal range and no relationship with CHF6001 dose. All mean values for systolic and diastolic blood pressure were within the normal range following both single and multiple doses, with no notable changes from baseline and no notable dose-related trend in the number of subjects with substantial changes. All mean ECG values were within the normal range following both single and multiple doses, with no notable changes from baseline and no marked difference between CHF6001 doses, and no clinically significant individual findings. Following multiple dosing in Study 2, there were a small number of statistically significant differences between CHF6001 and placebo in mean heart rate, PR interval and QT interval corrected for heart rate using Fridericia’s formula (QTcF) interval, none of which were considered clinically significant. There were no clinically significant post-dose decreases in FEV1, indicating that none of the subjects experienced bronchospasm.
Discussion
A significant dose range with a large number of doses were studied: CHF6001 was well tolerated even up to high doses above the predicted therapeutic dose. There were few AEs considered related to treatment and no relationship between overall AE incidence and CHF6001 dose, either following single doses or multiple doses. Importantly, gastrointestinal AEs were rare, with only two events considered related to CHF6001 treatment (one event each of diarrhea and flatulence), both of which were mild in severity and did not result in study withdrawal. These results suggest that CHF6001 given via the inhaled route of administration may be more acceptable to patients than roflumilast given via the oral route, although this needs to be confirmed in larger, longer-term studies. There were no clinically significant findings from the other safety data, with no evidence of any dose-related trend.
A clear relationship was shown between the inhaled dose of CHF6001 and plasma exposure (total and maximum) following administration of single and multiple doses, and when delivered via both devices. The weighted AUC systemic bioavailability of CHF6001 was ~30% higher following administration via the MDDPI than via the SDDPI. Since the systemic bioavailability of CHF6001 is mainly due to absorption from the lungs rather than oral ingestion;6 this suggests that the MDDPI device provides more efficient pulmonary drug deposition than the SDDPI device.
The PK modeling and simulation confirm the findings from the two studies, clearly demonstrating dose-proportionality. One of the strengths of modeling these data is that by simulating PK exposure, it is possible to generate data from many more time points than is the case from clinical data, especially on the intermediate days between PK sampling periods (days 2–13). This means that a more detailed characterization of the PK profiles is possible, confirming that steady state has been reached by day 14 for all doses when administered BID via the MDDPI. The PK modeling also supports BID administration of CHF6001, since although the model suggests that both OD and BID administration via the MDDPI would result in similar plasma exposure of CHF6001, the Cmax from BID dosing was lower than that from OD dosing, with smaller fluctuation in plasma levels. In addition to reducing the risk of systemic adverse effects, this lower variability could potentially result in tighter engagement at the target receptors. Further larger and longer-term studies are being conducted to evaluate the effects of CHF6001 in patients.
Conclusion
Overall, CHF6001 demonstrated a good safety profile, with tolerability similar to that of placebo. There was a clear relationship between the inhaled dose of CHF6001 and systemic exposure, with dose proportionality demonstrated for both devices. The higher systemic bioavailability of CHF6001 suggests that the NEXThaler MDDPI device provides better pulmonary drug deposition than the SDDPI, with the BID regimen providing the most appropriate exposure profile.
Data availability
The data of this study are available on request, following submission of a valid research protocol to the corresponding author.
Acknowledgments
The authors would like to thank the investigators and subjects at the investigative site for their support of this study, and QuotientSciences for performing both studies. PK modeling support was provided by Koen Jolling and Andreas Lindauer, SGS Exprimo NV (Belgium). Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici SpA. These studies were funded by Chiesi Farmaceutici SpA. An abstract summarizing the results of Study 1 was presented at the European Respiratory Society Annual Conference in 2013 as a poster presentation. The abstract was published in the European Respiratory Journal, Volume 42 (Supplement 57). Abstracts summarizing the results of Study 2 were presented at the American Thoracic Society Annual Conference in 2016 and the European Respiratory Society Annual Conference in 2016 as poster presentations. The abstracts were published in the American Journal of Respiratory and Critical Care Medicine, Volume 193, and the European Respiratory Journal, Volume 48 (Supplement 60).
Author contributions
FM, DS, and MAN contributed to the design of the studies, GL oversaw their clinical conduct and data acquisition, DS oversaw the data analysis, and MG performed the PK simulations and related data analyses. All authors contributed to the interpretation of the results, critically revised the manuscript for intellectual content, approved the final version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors are employees of Chiesi, the sponsor of the study. The authors report no other conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease; 2018. Available from: https://goldcopd.org/wp-content/uploads/2017/11/GOLD-2018-v6.0-FINAL-revised-20-Nov_WMS.pdf. Accessed September 25, 2018. | ||
Calverley PM, Rabe KF, Goehring UM, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374(9691):685–694. | ||
Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374(9691):695–703. | ||
Martinez FJ, Rabe KF, Sethi S, et al. Effect of Roflumilast and Inhaled Corticosteroid/Long-Acting β2-Agonist on Chronic Obstructive Pulmonary Disease Exacerbations (RE(2)SPOND). A Randomized Clinical Trial. Am J Respir Crit Care Med. 2016;194(5):559–567. | ||
Agusti A, Edwards LD, Celli B, et al. Characteristics, stability and outcomes of the 2011 GOLD COPD groups in the ECLIPSE cohort. Eur Respir J. 2013;42(3):636–646. | ||
Villetti G, Carnini C, Battipaglia L, et al. CHF6001 II: a novel phosphodiesterase 4 inhibitor, suitable for topical pulmonary administration – in vivo preclinical pharmacology profile defines a potent anti-inflammatory compound with a wide therapeutic window. J Pharmacol Exp Ther. 2015;352(3):568–578. | ||
Moretto N, Caruso P, Bosco R, et al. CHF6001 I: a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration. J Pharmacol Exp Ther. 2015;352(3):559–567. | ||
Edwards MR, Facchinetti F, Civelli M, Villetti G, Johnston SL. Anti-inflammatory effects of the novel inhaled phosphodiesterase type 4 inhibitor CHF6001 on virus-inducible cytokines. Pharmacol Res Perspect. 2016;4(1):e00202. | ||
Armani E, Amari G, Rizzi A, et al. Novel class of benzoic acid ester derivatives as potent PDE4 inhibitors for inhaled administration in the treatment of respiratory diseases. J Med Chem. 2014;57(3):793–816. | ||
Cenacchi V, Battaglia R, Cinato F, et al. In vitro and in vivo metabolism of CHF 6001, a selective phosphodiesterase (PDE4) inhibitor. Xenobiotica. 2015;45(8):693–710. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.