Back to Journals » Drug, Healthcare and Patient Safety » Volume 7
Safety profile of dalfampridine extended release in multiple sclerosis: 5-year postmarketing experience in the United States
Authors Jara M, Aquilina T, Aupperle P, Rabinowicz A
Received 25 September 2015
Accepted for publication 3 November 2015
Published 15 December 2015 Volume 2015:7 Pages 169—174
DOI https://doi.org/10.2147/DHPS.S97113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chul Ahn
Michele Jara, Thomas Aquilina, Peter Aupperle, Adrian L Rabinowicz
Acorda Therapeutics, Inc., Ardsley, NY, USA
Background: Dalfampridine extended release tablets (dalfampridine-ER; prolonged-, modified, or sustained-release fampridine outside the US), 10 mg twice daily, was approved by the US Food and Drug Administration (FDA) in January 2010 to improve walking in people with multiple sclerosis, as determined by an increase in walking speed.
Objective: To provide a descriptive analysis of reported adverse events (AEs) for commercially available dalfampridine-ER from March 2010 through March 31, 2015.
Methods: Five-year postmarketing data for dalfampridine-ER were available from the exposure of approximately 107,000 patients in the US (103,700 patient-years). Commonly reported AEs (≥2% of all reported AEs) and serious AEs were determined. The incidence of reported seizures was determined and the events were further investigated.
Results: Among the 107,000 patients exposed to dalfampridine-ER (70% female; mean age 52.1), the most common AEs were dizziness (3.7%), insomnia (3.2%), balance disorder (3%), fall (2.4%), headache (2.4%), nausea (2.1%), and urinary tract infection (2%). Other common AEs were drug ineffectiveness (5.8%), gait disturbance (4.6%), and inappropriate dosing (3.1%). Serious AEs included rare anaphylactic reactions (five cases) and drug hypersensitivity reactions (eight cases). A total of 657 seizure cases were reported (6.3/1,000 patient-years); of these, 324 were medically confirmed (3.1/1,000 patient-years). Incidence of reported seizures was stable over time. Duration of treatment prior to a seizure ranged from a single dose to >4 years; 12% of the seizures occurred within a week of starting treatment.
Conclusion: The 5-year US postmarketing safety data of dalfampridine-ER is consistent with the safety profile observed in clinical trials. Incidence of reported seizures remained stable over time. Since commercial availability in March 2010, a warning regarding the risk of anaphylaxis and severe allergic reactions was added to the US prescribing information.
Keywords: dalfampridine, fampridine, multiple sclerosis, adverse events, postmarketing safety
Introduction
Mobility impairment is common among patients with multiple sclerosis (MS), and walking difficulty is often considered the most challenging aspect of the disease.1 Based on consistent changes in walking speed in clinical studies,2,3 dalfampridine extended release tablets (dalfampridine-ER; known as prolonged-release fampridine in Europe and as fampridine modified- or sustained-release elsewhere) 10 mg twice daily, were approved by the US Food and Drug Administration (FDA) to improve walking in people with MS.4 Dalfampridine-ER is a broad spectrum blocker of voltage-gated potassium channels that enhances nerve conduction in demyelinated axons, which is the proposed mechanism of action for its clinical effects.5
In the clinical trials, dalfampridine-ER at the recommended dosage of a 10 mg tablet taken twice daily, approximately 12 hours apart, had a tolerability profile with adverse events (AEs) that were generally of mild or moderate severity. The most commonly reported AEs were balance disorders, paresthesia, dizziness, anxiety, headache, insomnia, and confusion.2,3 Since people with MS have an approximately 3-fold higher risk of seizure relative to the general population,6,7 and because dalfampridine-ER has a dose-dependent risk of seizure,8,9 it was important to monitor for seizure-related events. In the double-blind clinical studies, seizures were reported in 0.2% of patients,2,3 and in the pooled extensions of these trials the seizure rate was 0.8% (corresponding to 4.1 per 1,000 patient-years) among those who had up to 5 years of dalfampridine-ER exposure.10 These rates were generally similar to the 3.49 per 1,000 patient-years reported by Eriksson et al11 for a first seizure in a Swedish MS population. Although dalfampridine-ER is contraindicated in patients with a history of seizures,4 a Risk Evaluation and Mitigation Strategy was instituted to inform health care providers and patients about seizure risk with use of dalfampridine-ER. The Risk Evaluation and Mitigation Strategy was subsequently discontinued, since the program met its goals and was no longer required.12 However, postmarketing surveillance for seizure events continues to be an important component of monitoring the safety of dalfampridine-ER in clinical practice.
A previous analysis evaluated the safety data of dalfampridine-ER in the US over the first year postmarketing.13 The findings were consistent with the safety profile observed during clinical development, including a seizure incidence that was not substantially different from that observed in clinical trials. The current study was undertaken to extend this safety evaluation of dalfampridine-ER in the US based on AEs reported from 2010 through 2015, encompassing the first 5 years postmarketing. Furthermore, data from a post-approval program of enhanced surveillance and assessment were evaluated to further characterize the seizure risk potential.
Methods
AE reporting and database
In the US, AEs can be reported to the drug manufacturer by consumers as well as by health care professionals. The manufacturer is then required to send all reported AEs to the FDA.14 The current analysis was prospectively designed to descriptively evaluate all AEs of dalfampridine-ER, regardless of causality, that were reported and recorded in the manufacturer’s product safety database from March 1, 2010 through March 31, 2015; the product safety database is an Argus (Oracle® Argus Safety 7.0; Redwood Shores, CA, USA) AE management system. All AEs in the database were classified using the Medical Dictionary for Regulatory Activities (MedDRA; version 18.0). As this research was conducted on deidentified data from electronic databases containing federally mandated AE information, no ethics committee approval was sought.
AEs
The proportion of AE reports was estimated by event, as classified at the MedDRA preferred term level. The most commonly reported AEs were those with a frequency ≥2% of all reported AEs.
Reports of seizures
Patients with events reported under the MedDRA High Level Group Term Seizures, including subtypes, were defined as seizure cases. Any seizure case reported or confirmed by a health care practitioner was considered as medically confirmed; whereas, those reported only by consumers were considered as potential seizure cases. The fact that dalfampridine-ER is only available for dispensing via specialty pharmacies in the US allowed accurate estimates of patient exposure. The incidence rate of seizure cases was calculated using the estimate of patient exposure of 103,700 person-years. All reports of confirmed and potential seizures were followed-up using a structured questionnaire designed to collect information on dose, regimen, usage, seizure history, concomitant medications, and any prior use of pharmacy-compounded formulations of dalfampridine. All cases of seizure were reviewed to ascertain patient demographics, time to event from treatment onset, and the presence of any additional seizure-related risk factors.
Results
Demographics
Surveillance data were available from approximately 107,000 patients who filled a prescription for dalfampridine-ER during the 5 years of product availability, representing an overall exposure of approximately 103,700 patient-years. Of these patients for whom demographic data were available (n=87,815), 70% were female, and the mean age was 52.1 years; the proportion of patients aged ≥65 years was 13%. Among the 107,000 patients exposed to dalfampridine-ER, 23.9% (25,526) reported at least one AE. Of the patients who reported AEs, 75% were female, and the mean age was 55.3; the proportion of patients aged ≥65 years was 19% (Table 1).
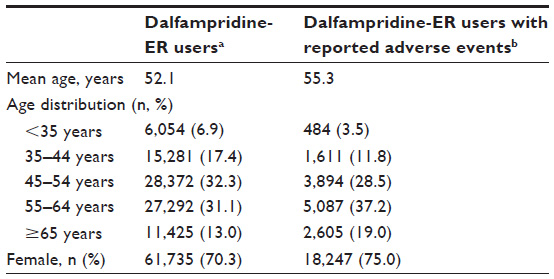 | Table 1 Demographic characteristics of dalfampridine-ER users |
AEs
A total of 60,949 AEs were reported that were generally consistent with those reported in double-blind clinical studies and comparable to those in the FDA-approved prescribing information for dalfampridine-ER. The most commonly reported AEs (Table 2) included dizziness (3.7%), insomnia (3.2%), balance disorder (3%), fall (2.4%), headache (2.4%), nausea (2.1%), and symptoms of urinary tract infection (UTI; 2%). Other common AEs were drug ineffectiveness (5.8%), gait disturbance (4.6%), and inappropriate dosing (3.1%). Inappropriate dosing was reported as an AE when a patient failed to take the prescribed dose, including accidental overdose; in the majority of cases, this code represented 10 mg once daily dosing.
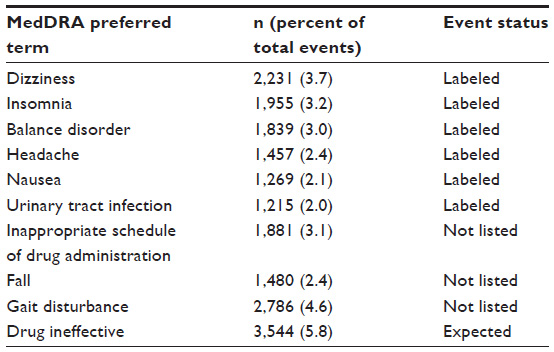 | Table 2 Most commonly reported adverse events (≥2% of all reported events; N=60,949) |
Commonly reported serious AEs (SAEs) included dyspnea (n=48), suicidal ideation (n=32), and cerebrovascular events (n=37). Rare SAEs included anaphylactic reactions (n=5) and drug hypersensitivity reactions (n=8). At least two of the anaphylaxis events were considered positive dechallenge cases, ie, the events resolved after withdrawal of dalfampridine-ER. At least 4 non-serious positive rechallenge drug hypersensitivity cases were observed (ie, signs and symptoms of the event occurred again following reinitiation of the drug).
Seizures
Of the 107,000 patients exposed to dalfampridine-ER, 657 cases reported seizures, representing a rate of approximately 6.3 per 1,000 patient-years of use (Table 3); a total of 685 seizure events were reported among these cases, indicating that some had more than one seizure. Of these, 324 cases were medically confirmed, yielding an estimated incidence rate of 3.1 per 1,000 patient-years of use (Table 3). The demographic characteristics of patients who reported seizures showed that approximately two thirds were women (449 women, 171 men, 37 unknown), and the mean patient age was 53 years; 168 cases were missing age information.
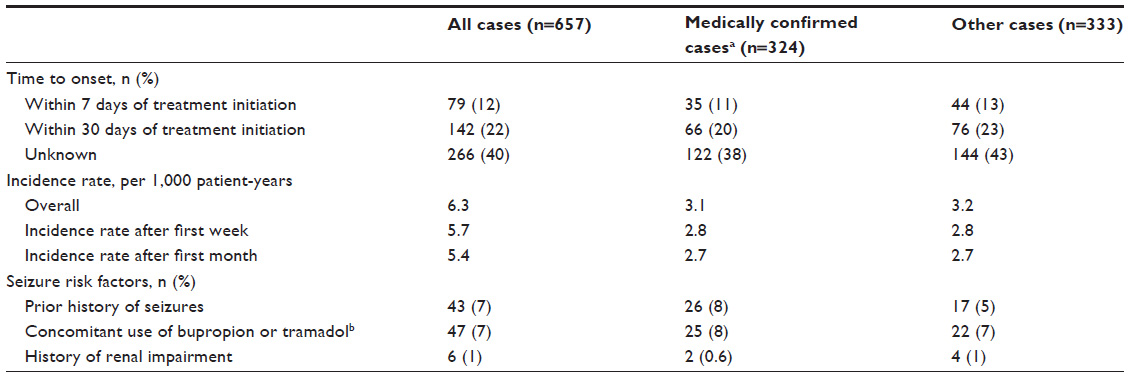 | Table 3 Reported seizure cases in the first 5 years of postmarketing surveillance of dalfampridine-ER |
Duration of dalfampridine-ER treatment prior to the earliest seizure event ranged from one dose to >4 years. A total of 79 seizures (12%) occurred within a week of starting treatment, and the incidence rate of reported seizure cases was approximately 5.7 per 1,000 patient-years of use after the first week (Table 3).
In addition to MS, other seizure-related risk factors that were present in these patients with reported seizures were a prior history of seizure, including febrile seizure (7%); concomitant use of bupropion or tramadol (7%); and a history of renal impairment (1%; severity not known) (Table 3). Inappropriate dosing was also a risk factor, as 5% of all reported seizure cases (n=32) had dosing intervals that were shorter than 8 hours. For both the total and medically confirmed seizure cases, the cumulative incidence was stable over time (Figure 1), with only month-to-month fluctuations in reporting.
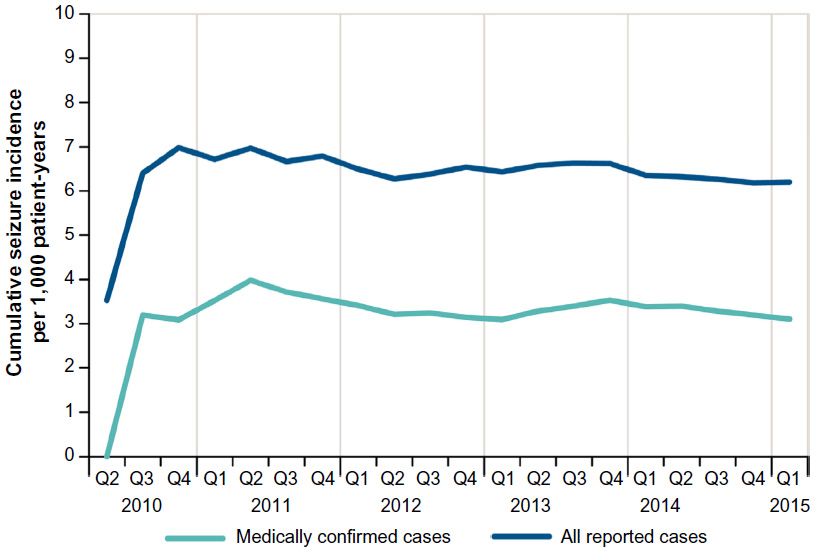 | Figure 1 Cumulative incidence of seizure events. |
Discussion
This 5-year postmarketing safety evaluation of dalfampridine-ER shows that the reported AEs were generally consistent with expectations based on experience from the dalfampridine clinical development program. The findings were similar to those of previously reported 1- and 2-year US postmarketing surveillance periods,13,15 and comparable to dalfampridine postmarketing surveillance data in France, using the European Database for Multiple Sclerosis.16
In the first 5 years of marketing, the incidence rate of medically confirmed seizures was 3.1 per 1,000 patient-years of use, and 11% of the cases occurred within the first week of exposure. The incidence of any reported seizure was higher than the incidence of medically confirmed seizure, 6.3 per 1,000 patient-years of use, with 12% of those events occurring within the first week of use. Considering that there are different symptoms that can be confused with a seizure disorder such as confusion, loss of consciousness, incoherence, muscle spasms, and syncope, the actual incidence of seizure associated with dalfampridine-ER is likely between the overall observed rate and that of medically confirmed cases. Further, the actual seizure rate in real-world users of dalfampridine-ER has been stable over time and comparable to the incidence rate from the long-term, open-label trials (4.1 per 1,000 patient-years).10
A number of factors may have contributed to the reported seizures in this postmarketing patient population. In addition to the known seizure risk associated with MS, a notable proportion of patients who reported seizures had one or more other risk factors, including prior seizure history, concomitant use of medications that are known to lower seizure threshold, or renal impairment. The severity of renal impairment in this postmarketing population was not known. Since a history of seizure, or moderate or severe renal impairment are contraindications for dalfampridine-ER use,4 these findings suggest that patients may be either inadequately evaluated for contraindications or treated despite the presence of contraindications. In this regard, we previously reported that 3% of dalfampridine-ER users had a history of seizures and 5% had some level of renal impairment at the time of prescription.17 Another factor that may have contributed to seizures is the use of concomitant medications that are known to lower seizure threshold. Use of such medications is not a specific contraindication of dalfampridine. However, the fact that 7% of all patients who reported seizures had concomitant use of either bupropion or tramadol suggests the need for careful monitoring. Inappropriate dosing of dalfampridine-ER also led to seizures in some patients; there is a dose-dependent increase in the risk of seizures with dalfampridine-ER.8,9 Given that there is only one approved dose (10 mg, twice daily, approximately 12 hours apart), reports of inappropriate dosing are seemingly high, thus highlighting the importance of adhering to the prescribing guidelines as well as to the approved dosing schedule for dalfampridine-ER.
Overall, the most frequently reported postmarketing AEs were those that had already been identified in the clinical trials as common events including dizziness, insomnia, balance disorder, headache, nausea, and UTI.2,3 People with MS have an increased risk for neurogenic bladder and UTI;18 however, recent studies that used confirmatory laboratory analyses showed that dalfampridine-ER does not increase the risk of confirmed bacterial infections but only of urinary symptom reports.18,19 Although falls were commonly reported in this postmarketing population, the rate of falls was lower than expected for an MS population, in which the underlying pathology of gait disturbance, balance impairment, spasticity, and muscle weakness contributes to the fall frequency and a higher fall incidence than in the general population.20,21 For perspective, a meta-analysis that focused on a 3-month observation period reported falls in 56% of people with MS.22 In clinical studies, the frequency of falls was not higher with dalfampridine-ER relative to placebo.2,3,19 Similarly, while gait disturbance was frequently reported as an AE, this event was generally reported with a similar incidence in placebo- and dalfampridine-ER-treated patients in clinical trials.2,3,19
Although the reporting of SAEs did not indicate additional safety signals, there were several clinically relevant safety issues that should be noted, including the occurrence of dyspnea in 48 patients. It is likely that these reports are related to disease rather than to treatment with dalfampridine-ER, as respiratory complications may occur in any stage of MS owing to respiratory muscle weakness, bulbar weakness, impaired voluntary control and impaired automatic control.23–25 Similarly, cerebrovascular events were reported in some patients and these may also relate to the disease, as the prevalence of ischemic heart disease, congestive heart failure, and stroke are higher in MS relative to the general population.26,27 Suicidal behavior was also reported in the postmarketing population. It is likely these events are not related to treatment, as patients with MS have been reported to have a greater than 2-fold higher suicide rate relative to the general population.28,29
There were a few incidences of rare SAEs, such as hypersensitivity reactions and anaphylaxis reported in a small number of patients. Anaphylaxis is a clinical diagnosis based on systemic manifestations, often following acute exposure to a causative agent. The true incidence of anaphylaxis in people with MS is not known, but the prevalence for the US general population is at least 1.6% and is likely to be higher.30 A warning regarding the risk of anaphylaxis and severe allergic reaction was added to the dalfampridine-ER prescribing information.4 Although cases of serious liver injury or cases of AEs related to abuse potential were of special interest to the FDA at the time of dalfampridine-ER approval,31 neither of these issues was identified as a safety concern during the postmarketing surveillance period.32
Limitations of this analysis include that it is dependent on spontaneous reporting of AEs. Thus, it is possible that some events may have been underreported. On the other hand, AEs were evaluated regardless of causality, and some of the events may reflect the disease state and may not be specific to treatment with dalfampridine.
Conclusion
Findings from 5-year postmarketing surveillance suggest that the safety profile of dalfampridine-ER in real-world clinical practice is generally consistent with the safety profile established in the clinical trials. The seizure incidence in medically confirmed cases was similar to that of previous reports, and the overall incidence of reported seizures remained stable over time. Since product launch, a warning regarding the risk of anaphylaxis and severe allergic reactions has been added to the US prescribing information. While postmarketing surveillance safety data will continue to be collected, data collected to date do not indicate any new safety signals after 5 years on the market.
Acknowledgments
This study was funded by Acorda Therapeutics, Inc. Editorial assistance was provided by E Jay Bienen, PhD, of The Curry Rockefeller Group, LLC, and was funded by Acorda Therapeutics, Inc.
Disclosure
Authors are employees and stock holders of Acorda Therapeutics, Inc. The authors confirm that this paper is an accurate representation of the study results.
References
LaRocca NG. Impact of walking impairment in multiple sclerosis: perspectives of patients and care partners. Patient. 2011;4(3):189–201. | |
Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373(9665):732–738. | |
Goodman AD, Brown TR, Edwards KR, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68(4):494–502. | |
Ampyra® (dalfampridine) extended release (ER) tablets [prescribing information]. New York: Acorda Therapeutics, Inc.; 2014. | |
Dunn J, Blight A. Dalfampridine: a brief review of its mechanism of action and efficacy as a treatment to improve walking in patients with multiple sclerosis. Curr Med Res Opin. 2011;27(7):1415–1423. | |
Nicoletti A, Sofia V, Biondi R, et al. Epilepsy and multiple sclerosis in Sicily: a population-based study. Epilepsia. 2003;44(11):1445–1448. | |
Poser CM, Brinar VV. Epilepsy and multiple sclerosis. Epilepsy Behav. 2003;4(1):6–12. | |
Haut SR, Bienen EJ, Miller A. Clinical overview of the seizure risk of dalfampridine. Expert Opin Drug Saf. 2012;11(4):651–657. | |
Cornblath DR, Bienen EJ, Blight AR. The safety profile of dalfampridine extended release in multiple sclerosis clinical trials. Clin Ther. 2012;34(5):1056–1069. | |
Goodman AD, Bethoux F, Brown TR, et al. Long-term safety and efficacy of dalfampridine for walking impairment in patients with multiple sclerosis: results of open-label extensions of two Phase 3 clinical trials. Mult Scler. 2015;21(10):1322–1331. | |
Eriksson M, Ben-Menachem E, Andersen O. Epileptic seizures, cranial neuralgias and paroxysmal symptoms in remitting and progressive multiple sclerosis. Mult Scler. 2002;8(6):495–499. | |
Food and Drug Administration. Supplemental Approval, Release REMS Requirement, REMS Assessment Acknowledgment. Silver Spring, MD: Food and Drug Administration; 2013. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2013/022250Orig1s008ltr.pdf. Accessed November 4, 2015. | |
Jara M, Barker G, Henney HR 3rd. Dalfampridine extended-release tablets: one year of post-marketing safety experience in the United States. Neuropsychiatr Dis Treat. 2013;9:365–370. | |
US Food and Drug Administration [homepage on the Internet]. FDA Adverse Event Reporting System (FAERS). Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/. Accessed November 4, 2015. | |
Jara M, Adera M, Adedeji A, Henney H, Carrazana E. Dalfampridine Extended Release Tablets: Safety Profile After 2 Years of Postmarketing Experience in the United States [abstract]. Mult Scler. 2012;18(Suppl):476–477. | |
Berthe C, Ongagna JC, Courtois S, et al. Prolonged-release Fampridine post-marketing experience in Alsace, France [abstract]. Neurology. 2015;84 Suppl:P1.126. | |
Jara M, Sidovar MF, Henney HR 3rd. Prescriber utilization of dalfampridine extended release tablets in multiple sclerosis: a retrospective pharmacy and medical claims analysis. Ther Clin Risk Manag. 2014;11:1–7. | |
Kantor D, Chancellor MB, Snell CW, Henney HR 3rd, Rabinowicz AL. Assessment of confirmed urinary tract infection in patients treated with dalfampridine for multiple sclerosis. Postgrad Med. 2015; 127(2):218–222. | |
Hupperts R, Lycke J, Short C, et al. Prolonged-release fampridine and walking and balance in multiple sclerosis: randomized MOBILE study. Mult Scler. Epub 2015 Apr 28. | |
Matsuda PN, Shumway-Cook A, Bamer AM, Johnson SL, Amtmann D, Kraft GH. Falls in multiple sclerosis. PM R. 2011;3(7):624–632. | |
Gunn HJ, Newell P, Haas B, Marsden JF, Freeman JA. Identification of risk factors for falls in multiple sclerosis: a systematic review and meta-analysis. Phys Ther. 2013;93(4):504–513. | |
Nilsagard Y, Gunn H, Freeman J, et al. Falls in people with MS-an individual data meta-analysis from studies from Australia, Sweden, United Kingdom and the United States. Mult Scler. 2015;21(1):92–100. | |
Howard RS, Wiles CM, Hirsch NP, Loh L, Spencer GT, Newsom-Davis J. Respiratory involvement in multiple sclerosis. Brain. 1992;115(Pt 2):479–494. | |
van Klaveren R, Buyse T, Van De Gaer L, Meekers J, Rochette F, Demedts M. Micturitional disturbances are associated with impaired breathing control in multiple sclerosis. Chest. 1999;115(6):1539–1545. | |
Farhat MR, Loring SH, Riskind P, Weinhouse G. Disturbance of respiratory muscle control in a patient with early-stage multiple sclerosis. Eur Respir J. 2013;41(6):1454–1456. | |
Marrie RA, Reider N, Cohen J, et al. A systematic review of the incidence and prevalence of cardiac, cerebrovascular, and peripheral vascular disease in multiple sclerosis. Mult Scler. 2015;21(3):318–331. | |
Marrie RA, Rudick R, Horwitz R, et al. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology. 2010;74(13):1041–1047. | |
Siegert RJ, Abernethy DA. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469–475. | |
Manouchehrinia A, Tanasescu R, Tench CR, Constantinescu CS. Mortality in multiple sclerosis: meta-analysis of standardised mortality ratios. J Neurol Neurosurg Psychiatry. Epub 2015 May 2. | |
Wood RA, Camargo CA Jr, Lieberman P, et al. Anaphylaxis in America: the prevalence and characteristics of anaphylaxis in the United States. J Allergy Clin Immunol. 2014;133(2):461–467. | |
Food and Drug Administration. NDA Approval. Silver Spring, MD: Food and Drug Administration; 2010. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2010/022250s000ltr.pdf. Accessed November 4, 2015. | |
Jara M, Gauvin DV, Caggiano AO, Walter B, Parry TJ, Henney H. Evaluation of the Abuse Potential of Dalfampridine Extended Release Tablets (P565). 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis; October 10–13; 2012; Lyon, France. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.