Back to Journals » Journal of Blood Medicine » Volume 16
Safety of Recombinant von Willebrand Factor in the Treatment of von Willebrand Disease: Real-World Data from an EU Post-Authorization Safety Study
Authors Sinclair SM, Ba Y, Badejo K ![]()
Received 19 December 2024
Accepted for publication 2 August 2025
Published 2 October 2025 Volume 2025:16 Pages 457—467
DOI https://doi.org/10.2147/JBM.S512634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Susan M Sinclair, Yi Ba, Kayode Badejo
Takeda Development Center Americas, Inc., Cambridge, MA, USA
Correspondence: Susan M Sinclair, Takeda Development Center Americas, Inc., 650 E Kendall Street, Cambridge, MA, 02142, USA, Email [email protected]
Introduction: In Europe, recombinant von Willebrand factor (rVWF) is approved for the prevention and treatment of hemorrhage or surgical bleeding in adults with von Willebrand disease (VWD) for whom desmopressin alone is ineffective or contraindicated. Real-world data on rVWF safety are limited.
Aim: To assess the safety of rVWF in real-world European clinical practice.
Methods: EU post-authorization safety study (NCT05265078, EUPAS45617) was a multicenter, retrospective, non-interventional study conducted in adults with VWD who received rVWF at 1 of 30 participating sites in Europe (January 2019–March 2023). Data were collected retrospectively for ≥ 7 days and ≤ 6 months after the first rVWF infusion, and similarly, after each subsequent rVWF course. Primary outcomes were the risk of hypersensitivity reactions, thromboembolic events, and VWF/factor VIII (FVIII) inhibitor formation when used for hemorrhage treatment or prevention/treatment of surgical bleeding; and the association of thromboembolic events with concurrent use of FVIII for hemorrhage treatment or prevention/treatment of surgical bleeding.
Results: In the primary analysis, 87 patients received 203 rVWF treatment courses. In total, 2 hypersensitivity-related AEs of mild severity occurred in 1 patient who received rVWF (0.00068 events per person-day at risk), and 1 thromboembolic AE of moderate severity (venous thrombosis) was reported in 1 patient (0.00127 events per person-week at risk). There were no reports of VWF or FVIII inhibitor formation. The association between thromboembolic events and the concurrent use of rVWF and FVIII could not be assessed because no patient received rVWF in conjunction with FVIII.
Conclusion: In this EU post-authorization safety study, the risks of hypersensitivity reactions and thromboembolic events with rVWF were low and there were no reports of VWF or FVIII inhibitor formation. Overall, no new safety signals were identified in this European real-world study when rVWF was used for the prevention or treatment of hemorrhage or surgical bleeding in VWD.
Keywords: von Willebrand disease, recombinant von Willebrand factor, real-world data
Introduction
von Willebrand disease (VWD) is an inherited condition caused by a deficiency or dysfunction of the multimeric plasma glycoprotein, von Willebrand factor (VWF). VWF mediates platelet adhesion and supports platelet coagulation by stabilizing coagulation factor VIII (FVIII).1 The prevalence of VWD is estimated to range from 0.6% to 1.3%; VWD leading to clinically relevant bleeding has been estimated in ~1 per 10,000 individuals.1–3 VWD is classified into 6 categories: 1 (partial VWF deficiency); 2A, 2B, 2M, 2N (qualitative defects); and 3 (complete/near-complete absence of VWF).4,5
Treatment of bleeding in patients with VWD includes the use of desmopressin and VWF replacement therapy.5 In Europe, recombinant VWF (rVWF, vonicog alfa; VEYVONDI®) is approved for the prevention and treatment of hemorrhage and surgical bleeding in adults with VWD when desmopressin treatment alone is ineffective or contraindicated.6 rVWF is manufactured by recombinant DNA technology, and therefore does not contain any plasma-derived components.6 This prevents potential adverse events (AEs) associated with plasma-derived ingredients. However, there are special warnings and precautions listed in the EU Summary of Product Characteristics (SmPC) that include hypersensitivity reactions, thrombosis and embolism, and neutralizing antibodies (inhibitors).6
While the safety and efficacy of rVWF were established in phase 3 clinical trials,7–9 there was a need at that time for real-world safety data to confirm these findings in a larger sample of patients, and to investigate safety in real-world clinical practice. Phase 3 clinical trial safety populations have ranged from 15 to 37 participants,7–9 while post-authorization safety studies could potentially draw on a larger database of patients and may therefore enable more accurate investigation of the incidences of AEs including hypersensitivity, thrombosis and embolism, and development of neutralizing antibodies.
In this analysis, we report the results of a multicenter, retrospective, non-interventional post-authorization safety study, which was conducted in six European countries. This study aimed to investigate the real-world safety of rVWF in European clinical practice, focusing on the risks of hypersensitivity reactions, thromboembolic events, and VWF/FVIII inhibitor formation.
Methods
Study Design
This was a non-interventional, retrospective cohort study (NCT05265078, EUPAS45617) using secondary data obtained from the medical records of adults with VWD who received rVWF at one of the participating study sites during the site-specific data window. Participating study sites were hematology specialty institutions and treatment centers in European countries where rVWF was commercially available. The site investigators selected consecutive patients for the study in chronological order to avoid selection bias and to optimize retrospective data collection. There were 30 study sites across Austria, Denmark, France, Germany, the Netherlands, and the UK.
The site-specific observation period commenced from the date of rVWF approval in that country (01 January 2019 for the first country), and continued up to one day before activation of that site (the last activation date was 29 March 2023). The study protocol and informed consent form were reviewed and approved by institutional review boards or independent ethics committees (Supplementary Table 1) in accordance with national and local regulations at each study site.
The study entry point for each patient was the index date, defined as the date of the earliest rVWF infusion in the medical record. Medical record data were abstracted for a minimum of 7 days and a maximum of 6 months after the index date, and similarly, after each subsequent course of rVWF within the data window. Patients with multiple treatment courses had multiple observation periods, which were summed to obtain the total person-time at-risk for each of the adverse events of special interest (AESIs; hypersensitivity reactions, thromboembolic events, and inhibitor formation). AESIs occurring beyond the at-risk period were not included in the exposure-adjusted incidence rates in the primary analysis, but were summarized separately. A concomitant medication was defined as any treatment administered between the first and last rVWF treatment course. rVWF was defined as being used in conjunction with FVIII if the two products were used together, ie, the FVIII start date fell within the rVWF start and end dates. FVIII administration during the study was considered concomitant if it occurred outside the period of an active rVWF treatment course. Parameters abstracted from the medical records included indication and exposure variables, demographic and baseline characteristics, and covariates and risk factors (a full list of variables collected is provided in Supplementary Methods).
Patients
Eligible patients were adults (age ≥18 years) diagnosed with congenital VWD who had received commercially available rVWF, with or without a FVIII concentrate, and had ≥7 days of observation time in the medical record after the index dose. Patients were excluded if they had a history of any other coagulation or platelet disorder, including acquired VWD, or if they had a history of neutralizing antibodies/inhibitors to VWF or FVIII.
Study Objectives
The primary objective was to measure the risk of hypersensitivity reactions, thromboembolic events, and VWF/FVIII inhibitor formation among adults with VWD receiving rVWF for hemorrhage treatment or prevention/treatment of surgical bleeding; and the association of thromboembolic events with the concurrent use of FVIII and rVWF in this study population.
Exploratory objectives were the risks of hypersensitivity reactions, thromboembolic events, and VWF/FVIII inhibitor formation after treatment with rVWF in the participants prescribed rVWF outside of the approved indications at the time of the study (eg, prophylaxis).
Statistical Analysis
Due to the relatively small potential patient population, the target sample size was up to 100 patients; however, based on precision estimates for AESIs, the minimum sample size required to meet the primary study objective was 80 patients. With 80 patients and an assumed event rate of 2.4% (incidence of thrombotic event of any type among patients with VWD10), the event rate was expected to have a precision of ±3.936%. With 100 patients and an event rate of 2.4%, the expected precision was ±3.430.
The full analysis set (FAS) included all patients who fulfilled the study inclusion and exclusion criteria and who received at least one dose of rVWF. The primary analysis set included all patients in the FAS who received rVWF for an indication that was approved at the time of the study for an individual treatment course. The exploratory analysis set included all patients in the FAS who received rVWF for an indication that was not approved at the time of the study for that treatment course.
For the primary and exploratory analyses, risks of hypersensitivity reactions, thromboembolic events, and inhibitor formation were estimated for patients in the primary analysis set and exploratory analysis set, respectively, using time at-risk, exposure-adjusted incidence rates, and 95% confidence intervals (CI) calculated for each AESI. This was calculated for each AESI at-risk period as:

The at-risk period for each AESI was measured from the first dose in each treatment course until the following number of days after infusion: hypersensitivity reactions up to 7 days; thromboembolic events up to 30 days; VWF inhibitor or FVIII inhibitor formation up to 180 days. Further details on person-time at-risk calculations are provided in the Supplementary Methods.
Results
Patients
In total, 101 patients were enrolled (Figure 1). The FAS included 100 patients, as one patient did not receive a dose of rVWF and was therefore excluded. The primary analysis set included 87 patients and the exploratory analysis set included 35 patients. Most patients were from France (n=39; 39.0%) or the UK (n=36; 36.0%). The exploratory analysis set included 10 pregnant patients; however, data on pregnancy outcomes were not collected because while not an exclusion criterion, the focus of this study was the risk of the three AESIs of interest.
 |
Figure 1 Patient disposition. Abbreviation: rVWF, recombinant von Willebrand factor. |
Baseline demographics and disease characteristics are shown in Table 1, with medical history shown in Supplementary Table 2. The most common comorbidities were hypertension (n=18; 18.0%) and osteoarthritis (n=7; 7.0%). Seven patients (7.0%) had a history of arterial or venous thromboembolism. Overall, 50 patients (50.0%) received concomitant medications during the study, including 26 patients (26.0%) who received blood coagulation factors and 25 (25.0%) who received tranexamic acid (Supplementary Table 3).
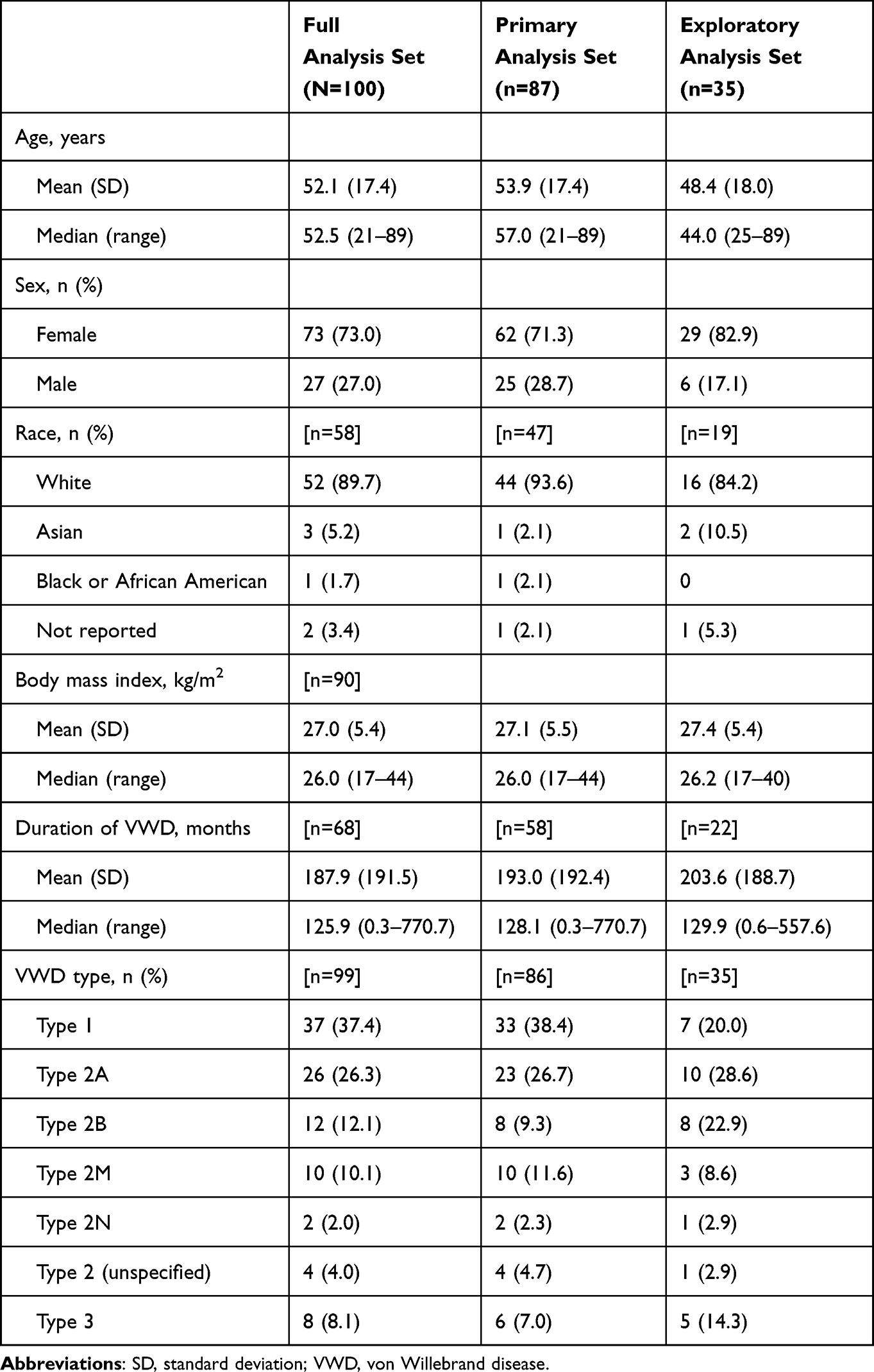 |
Table 1 Baseline Characteristics |
In the FAS, patients received 273 treatment courses (Table 2). The mean (standard deviation; SD) number of treatment courses per patient was 2.7 (3.1), with 48 patients (48.0%) reporting >1 treatment course. Prevention of surgical bleeding was the most common treatment indication (54.9% of treatment courses). The median duration of treatment courses was 2.0 days (range: 1–974 days). The total number of infusions administered to patients in the FAS was 595 and the mean (SD) dose per infusion was 3025 (8569) IU. The mean (SD) cumulative dose per treatment was 6453 (19655) IU. In the primary analysis set, 87 patients received a total of 203 rVWF treatment courses during the follow-up period, almost all of whom received at least one rVWF treatment course (n=86; 98.9%) (Table 2). In the exploratory analysis set, 35 patients received a total of 70 rVWF treatment courses during the follow-up period, most of whom received at least one rVWF treatment course (n=24; 68.6%) (Table 2).
 |
Table 2 Summary of rVWF Treatment Details |
Details of bleeds and surgeries reported during the study are shown in Supplementary Table 4. Among spontaneous bleeds, the most common reasons for rVWF infusion were other spontaneous bleeds (31 treatment courses [11.4%]), followed by epistaxis (20 treatment courses [7.3%]) and gastrointestinal bleeds (19 treatment courses [7.0%]). Among trauma-induced bleeds, laceration and “other” traumatic bleed were the only bleeds reported for more than one treatment course (two treatment courses [0.7%] each). The bleed type was “other reason” for 40 treatment courses (14.7%), and bleed type was missing for 164 treatment courses. Primary surgery type was minor for 78 treatment courses (28.6%), major for 71 treatment courses (26.0%), and as dental or oral for 13 treatment courses (4.8%). Primary surgery type was missing for 134 treatment courses.
Bleed outcomes are shown in Supplementary Table 5. For 99 bleeds for which the duration was given, the mean (SD) duration of bleed was 12.6 (16.1) days. The outcome of bleed in most treatment courses was “resolved” (135 of 229 bleeds; 59.0%), followed by “other” (n=24; 10.5%) and “ongoing” (n=11; 4.8%). The outcome was missing for 58 treatment courses.
Hypersensitivity Reactions
In the primary analysis set, a total of two hypersensitivity-related AEs of mild severity (nausea and dizziness) occurred in one patient with Type 2A VWD who received rVWF. The exposure-adjusted incidence rate of hypersensitivity reactions up to 7 days after the first dose in each treatment course (95% CI) was 0.00068 (0.00010–0.00486) events per person-day during a total at-risk person-time of 1371 days (Table 3). No serious hypersensitivity-related AEs occurred during the study.
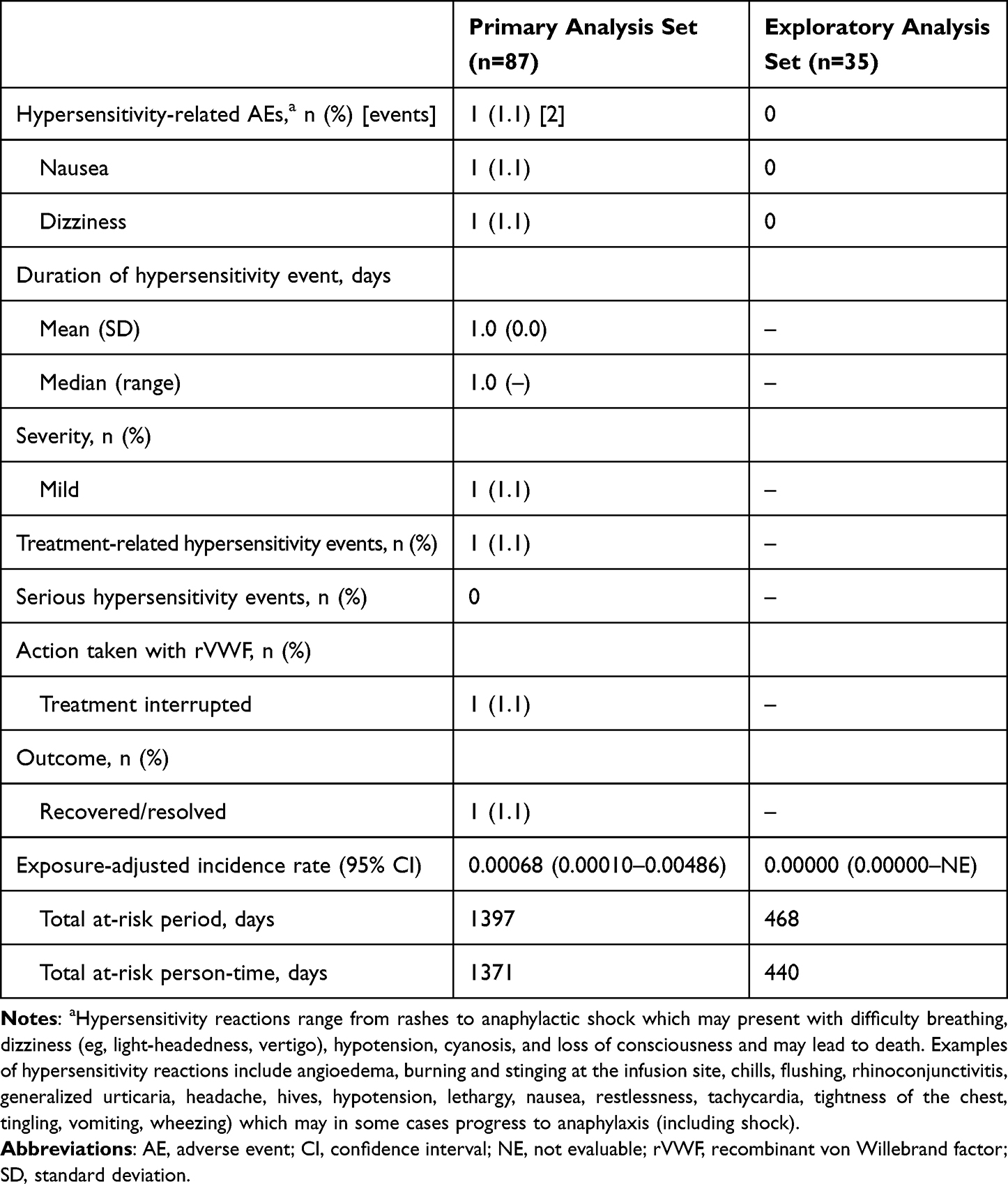 |
Table 3 Exposure-Adjusted Incidence of Hypersensitivity Reactions up to 7 days After the First Dose in Each Treatment Course |
In the exploratory analysis set, no hypersensitivity-related AEs were reported (Table 3). One patient (1.1%) with Type 3 VWD who received rVWF experienced eczema as an AE of moderate severity. As the dates of the event and the causality were missing, the patient was not included in the risk calculations.
Thromboembolic Events
In the primary analysis set, one thromboembolic AE of moderate severity (venous thrombosis) occurred in one patient (1.1%) with Type 2N VWD who received rVWF. The exposure-adjusted incidence rate of thromboembolic events up to 30 days after the first dose in each treatment course (95% CI) was 0.00127 (0.00018–0.00904) events per person-week during a total at-risk person-time of 737 weeks (Table 4). There were no serious thromboembolic events in either the primary or exploratory analysis sets (Table 4).
 |
Table 4 Exposure-Adjusted Incidence of Thromboembolic Events up to 30 days After the First Dose in Each Treatment Course |
The patient with venous thrombosis received a loading dose of rVWF 2600 IU followed by rVWF 1300 IU for the prevention of bleeding during total knee replacement surgery. Eight days after starting rVWF, the patient experienced venous thrombosis in the deep calf vein and was treated with enoxaparin. The thromboembolic event resolved after 59 days. The medical history of this patient included myocardial infarction, angiodysplasia, hypertension, and chronic kidney disease. Additional risk factors were dyslipidemia, immobility, and an age of 87 years.
No patient in the primary analysis set used rVWF in conjunction with FVIII, and thus the association between thromboembolic events and the concurrent use of rVWF and FVIII could not be assessed.
Inhibitor Formation
There were no reports of VWF or FVIII inhibitor AEs among the 87 patients included in the primary analysis set during a total at-risk period of 3094 weeks and total at-risk person-time of 3080 weeks, but these were not routinely screened for in the absence of clinical suspicion (Table 5).
 |
Table 5 Exposure-Adjusted Incidence of VWF or FVIII Inhibitor Formation up to 180 days After the First Dose in Each Treatment Course |
For the exploratory analysis set, the total at-risk period for inhibitor formation was 704 weeks and total at-risk person-time was 697 weeks (Table 5). No VWF or FVIII inhibitor formation AEs were reported in the exploratory analysis set.
Discussion
This multicenter, retrospective, non-interventional post-authorization safety study presents real-world data on the safety of rVWF in European clinical practice. When used for treatment of hemorrhage or prevention/treatment of surgical bleeding in VWD, the risk of developing a hypersensitivity reaction was 0.00068 events per person-day at-risk, and the risk of developing a thromboembolic event was 0.00127 events per person-week at-risk. There were no reports of VWF or FVIII inhibitor formation.
These data extend prior clinical and real-world reports on the low incidence of hypersensitivity reactions, thromboembolic events, and inhibitor formation with rVWF for treatment of hemorrhage or prevention/treatment of surgical bleeding in VWD. In the primary analysis, there were two mild hypersensitivity-related AEs in one patient. This low incidence aligns with prior phase 3 clinical trials,7,8 which reported no severe hypersensitivity-related AEs in patients with severe VWD treated with rVWF (n=37 and n=15 patients, respectively7,8), as well as a real-world study from France that also reported no hypersensitivity-related AEs among 55 patients with VWD of any severity (Type 1: 40%, Type 2: 56%, Type 3: 4%) undergoing surgery.11 There was one thromboembolic event in the present study; similarly Peyvandi et al 20198 reported one thromboembolic event,11 and Gill et al 20157 and Desprez et al 202111 reported no thromboembolic events. In our study, there were no instances of rVWF or FVIII inhibitor formation, consistent with these previous reports.7,8,11 In comparison, inhibitor formation has been reported in patients receiving plasma-derived VWF.12 Notably, the present study had a higher percentage of female patients (73%) compared with previous rVWF studies (48–55%).7–9,11 This may suggest that there are no sex-based differences in the incidences of these selected AEs. This study could not provide further information on any potential association between thromboembolic events and the concurrent use of rVWF and FVIII, as no patient used rVWF in conjunction with FVIII.
In the exploratory analysis where rVWF was used for prophylaxis in VWD, there were no reports of hypersensitivity AEs, thromboembolic events, or events of VWF or FVIII inhibitor formation, resulting in exposure-adjusted incidence rates of zero. This is consistent with a phase 3 trial of rVWF prophylaxis in 23 patients with VWD, where one patient experienced a non-serious hypersensitivity reaction, one patient experienced a non-serious thromboembolic event, and no patients developed antibodies to rVWF or FVIII.9 The present findings therefore align with the low AE incidence observed in the clinical trial setting, and support the safety of rVWF for prophylaxis, as well as for the treatment of hemorrhage or prevention/treatment of surgical bleeding, in VWD.
This study has several strengths and limitations. A strength was that the data represent real-world use of rVWF in Europe. The study included sites across six countries in Europe, and incorporated data from approved use and indications not approved at the time of the study. Although the study sites were hematology specialty institutions and treatment centers, there was a lower percentage of patients with Type 3 VWD (8%) versus the phase 3 trials that focused on patients with severe VWD (53–78%) and who may be more likely to develop inhibitors.7–9 The population in the present analysis is similar to a previous real-world study of VWD, where 4% of patients had Type 3 VWD,11 suggesting that the present analysis may be more reflective of the real-world population of patients with VWD than the phase 3 studies.
The precision of the risk estimates in this study was limited by the small number of patients. This was expected as VWD is a rare disease, and the trial fulfilled the calculated minimum sample size required to meet the primary study objective. Sample size was maximized by including sites across multiple countries, and by including prophylactic use of rVWF (not an approved indication at the time of the study). Another limitation is that data quality and completeness were dependent on the quality of information in the medical records, as well as the accuracy and consistency of data abstraction and data entry. To address this, feasibility analyses were conducted prior to study initiation to ensure selected sites were routinely collecting the data required to meet the study objectives, and a quality control plan was in place to monitor data quality during the study. Finally, it is possible that the incidence of inhibitor formation could have been underestimated because testing for inhibitors would not typically have been performed unless clinically indicated, primarily through failure to respond adequately to treatment with VWF.
In conclusion, in this EU post-authorization safety study, the risks of hypersensitivity reactions and thromboembolic events with rVWF were low, with two mild hypersensitivity-related AEs in one patient, and one moderate severity thromboembolic event in one patient. There were no reports of VWF or FVIII inhibitor formation. Overall, no new safety signals were identified in this European real-world study when rVWF was used for the prevention or treatment of hemorrhage or surgical bleeding in VWD.
Data Sharing Statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within 3 months from the initial request to researchers who provide a methodologically sound proposal. The data will be provided after its deidentification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. The study sponsor analyzed the data and conducted the statistical analyses.
Ethics Approval and Informed Consent
The study was conducted in accordance with the Declaration of Helsinki. All medical record data were abstracted retrospectively; therefore, an exemption or waiver of informed consent was sought from all associated independent ethics committees (Supplementary Table 1). If informed consent was required, it was collected by the site investigators in accordance with local ethical and institutional requirements.
Acknowledgments
The authors thank the investigators and study staff involved in data collection at the participating centers. The authors also acknowledge Nirjhar Chatterjee from Takeda Development Center Americas Inc., Cambridge, MA, USA (at the time of the study), for valuable contributions to the interpretation of the study results.
Medical writing support was provided by Nasser Malik, PhD, and Susan Tan, PhD, employees of Excel Medical Affairs (Fairfield, CT, USA), and was funded by Takeda Development Center Americas, Inc.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by Baxalta US Inc., a Takeda company, Lexington, MA, USA.
Disclosure
Susan M Sinclair, Yi Ba, and Kayode Badejo are Takeda employees and stock owners. The authors report no other conflicts of interest in this work.
References
1. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84(2):160–174. doi:10.1055/s-0037-1613992
2. Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69(2):454–459. doi:10.1182/blood.V69.2.454.454
3. Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123(6):893–898. doi:10.1016/S0022-3476(05)80384-1
4. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–2114. doi:10.1111/j.1538-7836.2006.02146.x
5. Connell NT, Flood VH, Brignardello-Petersen R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021;5(1):301–325. doi:10.1182/bloodadvances.2020003264
6. 2024 Veyvondi [Summary of product characteristics]. 2023.
7. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–2046. doi:10.1182/blood-2015-02-629873
8. Peyvandi F, Mamaev A, Wang JD, et al. Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery. J Thromb Haemost. 2019;17(1):52–62. doi:10.1111/jth.14313
9. Leebeek FWG, Peyvandi F, Escobar M, et al. Recombinant von Willebrand factor prophylaxis in patients with severe von Willebrand disease: phase 3 study results. Blood. 2022;140(2):89–98. doi:10.1182/blood.2021014810
10. Smilowitz NR, Gupta N, Guo Y, Bangalore S, Berger JS. Perioperative bleeding and thrombotic risks in patients with von Willebrand disease. J Thromb Thrombolysis. 2017;44(1):67–70. doi:10.1007/s11239-017-1504-2
11. Desprez D, Drillaud N, Flaujac C, et al. Efficacy and safety of a recombinant von Willebrand factor treatment in patients with inherited von Willebrand disease requiring surgical procedures. Haemophilia. 2021;27(2):270–276. doi:10.1111/hae.14242
12. Rugeri L, Thomas W, Schirner K, Heyder L, Auerswald G. A systematic review of efficacy and safety of plasma-derived von Willebrand factor/factor VIII concentrate (Voncento) in von Willebrand disease. Thromb Haemost. 2024;124(9):828–841. doi:10.1055/a-2253-9701
 © 2025 The Takeda Pharmaceutical Company Limited. This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Takeda Pharmaceutical Company Limited. This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.