")
Back to Journals » Medical Devices: Evidence and Research » Volume 14
Safety of Medical Devices in Poland – Analysis of Withdrawn and Suspended Certificates of Compliance
Authors Patryn R, Zagaja A, Drozd M
Received 22 April 2021
Accepted for publication 22 June 2021
Published 26 July 2021 Volume 2021:14 Pages 239—247
DOI https://doi.org/10.2147/MDER.S316473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Rafał Patryn, Anna Zagaja, Mariola Drozd
Chair and Department of Humanities and Social Medicine, Medical University of Lublin, Lublin, Poland
Correspondence: Anna Zagaja
Chair and Department of Humanities and Social Medicine, Medical University of Lublin,, Poland ul. Codźki, Lublin, 720-093, Poland
Tel +48 81448 6850
Email [email protected]
Introduction: In the European Union, the process of controlling currently used medical devices is carried out and determined by legal provisions. The law stipulates that each entity responsible for using medical equipment must confirm its safety with an appropriate certificate issued by a notifying body. In Poland, the entity responsible for keeping records as to the withdrawn and suspended certificates, is the Office for Registration of Medicinal Products, Medical Devices, and Biocidal Products (URPL). Certification is required for all medical devices prior to their introduction onto the market and during their use.
Purpose: The article presents data concerning the number of medical devices that failed to meet the certification criteria.
Methods: The research method is an analysis of available subject literature and a report on withdrawn and suspended certificates of medical devices in Poland.
Results: In the years 2014– 2020, the notified bodies withdrew and suspended 13,354 certificates for medical devices, of which 9792 certificates were withdrawn, 2852 suspended and one falsified.
Conclusion: The suspension or withdrawal of a certificate for medical devices due to the inefficiency, obsolescence, imprecision, or safety of the devices is an action that improves the safety of patients. Such action reduces the number of medical damages and the obligation to pay compensation to those injured.
Keywords: safety, medical equipment, certification, law, damage, patient
Introduction
In the European, and by extension also Polish, law, medical devices are products intended for medical use. A medical device is any instrument, tool, machine, appliance, implant, in vitro reagent, software, material or other similar or related device, intended for use for one or more specific medical purposes such as diagnosis, prevention, monitoring, treatment, alleviation of ailments, examination, replacement, modification or support of an anatomical or physiological process or disinfection.1 Medical devices must undergo a conformity assessment to demonstrate that they meet legal requirements, to ensure that they are safe and perform as intended. Conformity assessment is conducted by notifying bodies (designated by Member States), which must fulfill necessary requirements as established in the decision no 768/2008/EC of the European Parliament and of the Council of 9 July 2008 on a common framework for the marketing of products, and repealing Council Decision 93/465/EEC. Such a body is assigned a unique identification number and is authorized to conduct conformity assessment and issue certificates of compliance. In practice, a notified body is authorized to test the product for compliance with relevant standards, and if those standards are not met, refrain from placing the product on the market, suspend or withdraw it. Manufacturers are free to choose any notified body that has been legally designated to carry out the conformity assessment procedure.
Conformity assessment usually includes an audit of the manufacturer’s quality system and, depending on the type of product, a review of the manufacturer’s technical documentation regarding the product’s performance and safety. After completing this process, the manufacturer receives a declaration of conformity, and the product is officially Conformité Européenne (CE) marked. Manufacturers may only affix the CE mark to a medical device after it has passed the conformity assessment. The CE marking confirms that the device meets all legal requirements established in Europe, including safety standards.
Prior to launching a medical device on the marketplace, European legislation requires proof of its effectiveness and clinical safety.2 The high quality, high functionality, and safety of medical products is, in fact, currently a necessity. However, these regulations vary from market to market, as there is no global harmonization.
In Europe, the In Vitro Diagnostic Regulation (IVDR) requires all IVDR devices to have new certification or be recertified. In parallel with the IVDR regulation, the International Organization for Standardization (ISO) and the European Committee for Standardization (CEN) published several standards for the processing of pre-analytical samples, which will be relevant for re-certification under this regulation.3 Re-certification of a medical device is carried out in order to confirm its continued compliance and effectiveness with the required standards. Medical devices undergo re-certification no later than 6 months before certificate expiry date. Re-certification must be performed within the validity period of the existing certificate. This is to ensure the continuity of certification and thus the possibility of using a specific medical device in practice. Entities applying for marketing authorization for a new medical device should assure the regulatory authorities of its production quality, and it must be emphasized, of its safety and effectiveness.4 The safety requirement is intended to protect the safety of patients, the operating staff, and other people, even if they come into contact with the device incidentally. Thus, a notified body is included in the system to protect all end users of such medical devices.
The USA has slightly different requirements. An entity wishing to place a medical device on the market must obtain the required approval for medical devices from the US Food and Drug Administration (FDA).5 The FDA issues a Marketing Authorization, which is available mainly in two versions: The Pre-Market Notification Procedure and the Pre-Market Approval (PMA). An application for a PMA must be supported by valid scientific evidence, which typically includes the collection of extensive technical, preclinical, clinical, and manufacturing data. In 2016, the 21st Century Cures Act established FDA oversight of software used in the medical field.5 Additionally, since 2007, a 243% increase in international and a 46% increase in domestic device inspections conducted by FDA was observed. Even though, in the USA, the number of unit recalls is alarming, as they show an increasing trend. Between 2017 and 2018, percent growth reached 50 million units quarterly, representing a 9.8% yearly growth. Nevertheless, the problems with the quality of medical devices have resulted in the death of 83,000 patients.6
Polish law introduces the rules of supervision over the production, introduction, and, in addition, control over medical devices already placed on the market and in use.1 Therapeutic devices, and in-vitro and in-vivo diagnostic devices, regardless of their classification, must be certified prior introduction on the market.7 Prior to introducing the product on the market and before clinical trials, the manufacturer is obliged to perform a conformity assessment. The assessment period varies (depending on product characteristics) and may be extended at the request of the manufacturer or an authorized representative, submitted within the period agreed in the contract, for subsequent periods not longer than 5 years.8 It is a procedure that ensures proper adherence to requirements in terms of safety, design, production, packing, and labeling of medical devices. This procedure is assessed and validated (through a certification process) by an independent entity (ie a notified body).9 Such an assessment may be conducted by the manufacturer (currently for one type of products only); however, it is most often done by an outside notified body. A notified body is an institution such as a laboratory, a research institute, or a specialist group, independent of both the manufacturer and the consumer. Its’ assessment is completed with the issuance of a Certificate of Conformity,1,10 a document assuring that the product, in its design, type, manufacturing, sterilization, inspection, and final testing process, complies with the stipulated requirements, rules, and parameters.
Certification by a notified body, according to Council Directive 90/385/EEC of June 20th 1990 stipulates that the conformity assessment procedure that is conducted, complies with essential requirements relating to a medical device. The notified body is authorized to issue certificates of conformity, and what is more, can change or impose limitations, supplement, suspend, restore validity and withdraw certificates of conformity that have already been issued when a medical device does not meet set requirements, or if a certificate of conformity has been issued contrary to the provisions, the notified body has the power to suspend, withdraw or impose restrictions on the issued certificate of conformity, unless the manufacturer implements corrective and remedial actions. In practice, such a product cannot be used, and legal provisions clearly state that it is forbidden to place products on the market if their certificates of conformity have expired or were withdrawn or suspended.1,10 Such medical devices should be withdrawn from use; hence, the notifying body conducts periodic inspections and evaluations in order to ensure that the manufacturer applies the approved quality systems and provides assessment reports.1,11 The frequency of inspections varies and depends on the type of devices, and the requirements set by the manufacturer (found in the devices technical description). Periodic examinations should take place at most every 36 months. For certain groups of devices, inspections should be carried out between 6 and 12 months. For example, incubators should be inspected every 12 months. The manufacturer or operator may carry out such a test at any time. In Poland, the top frequency of technical inspections is regulated by the Act on Medical Devices, which provides for technical inspections at least once a year.12
Information from certificates of conformity is placed in the devices’ IT database (Eudamed). Those authorized to place medical devices on the market include the manufacturer, an authorized representative, importer, distributor, or other appropriate institutions, such as the hospital. After carrying out tests that confirm requirement compliance, the product is CE marked. The notification must contain the number of notified bodies participating in the conformity assessment to register a medical product. The notification then must also contain copies of the certificates of compliance issued by the notifying bodies and a list of laboratories engaged in evaluating the performance of the product reported for assessment.1,7 So far, as many as 200 thousand various medical devices were introduced on the territory of the Polish Republic. This number is an estimate, because URPL does not possess precise data, which results from the fact that according to article 58 of the Act on Medical Devices, registration concerns a series of products and not individual products. The annual reports of the President of URLP also encompass individual reports regarding medical devices manufactured or marketed in Poland, which may be submitted by manufacturers, importers and distributors. Additionally, reporting may be done by companies’ legal representatives and entities, which deal with compiling medical devices into systems and procedure packs, as well as sterilizing such systems, packs and/or medical devices. Table 1 presents the number of cases reported to the URPL, including applications, notifications and changes to data found therein between 2014 and 2019 in terms of formal defects, product labeling, instructions for use and accompanying promotional materials (Table 1).
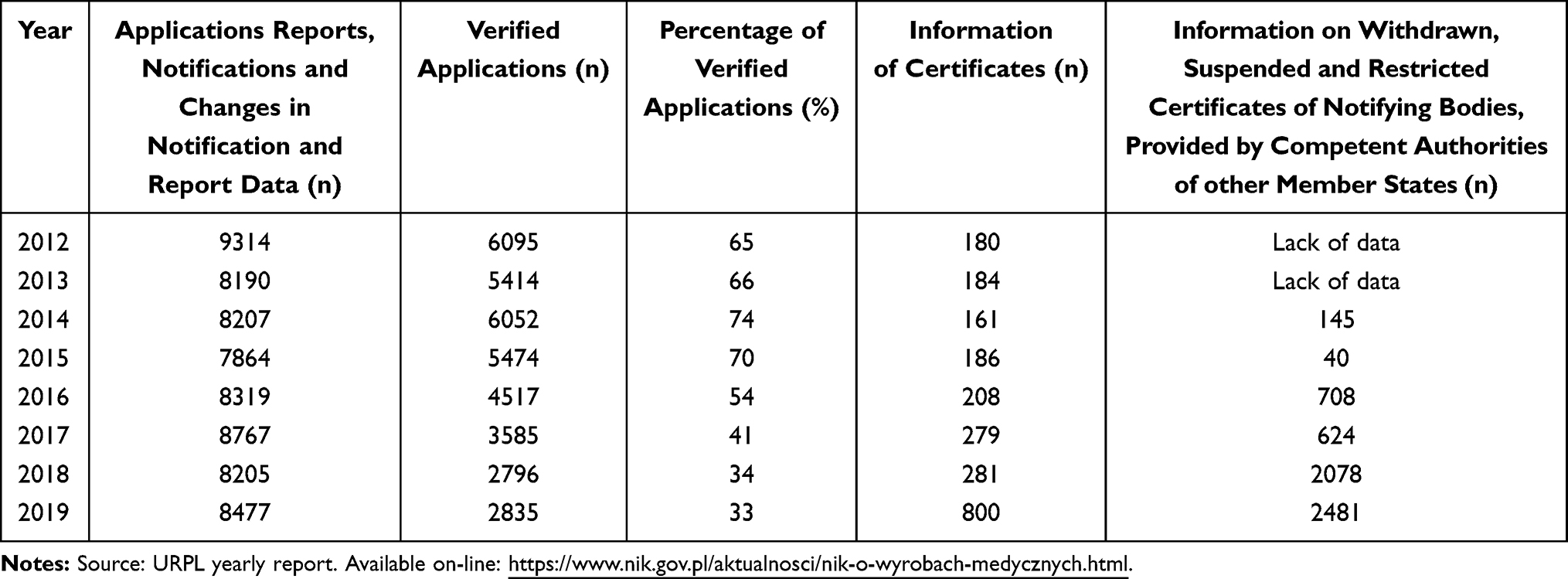 |
Table 1 Applications and Certificates of Medical Devices Received by URPL Between 2012 and 2019 |
The notifying body is obliged to immediately inform the Minister of Health and the President of URPL about issuing changes, imposing restrictions, supplementing, suspending, restoring the validity and withdrawing certificates of conformity, which it has issued and about refusing to issue a certificate of conformity.7,14 Data presented by the government agency’s report (the Supreme Audit Office) proves that the withdrawal or suspension of certificates by an authorized body is related to non-compliance with its use requirements and safety parameters. It is an action that is ultimately intended to serve patients’ safety.15 The study was designed to present the number of withdrawn and suspended medical equipment and, ultimately, to turn heads towards faulty devices, leading to an improvement of patient safety. The conducted analysis focuses on mandatory solutions in Europe and presents the extent to which medical devices have been withdrawn and suspended in Poland.
Purpose
The aim of the manuscript is to turn heads towards the problem of faulty medical devices.
Materials and Methods
Materials
Gathered data are a synthesis of available Polish and European legal acts and subjects literature. The main focus of the article is a Polish report drawn up by URPL, which is a document presenting, amongst others, the name of the manufacturer (the name of an authorized representative), the name of the product, the name of the notified body and the conclusion drawn from the quality check, ie, the withdrawal or suspension/restriction of use.15 The report contains 13,354 records (from 2014 to 2020) of faulty series of devices altogether.
Methods
In addition to literature review, the authors analyzed a publicly available report of the Supreme Audit Office. Included in the analysis were withdrawn and suspended medical devices and excluded were the positions that lacked the name of the device (Figure 1).
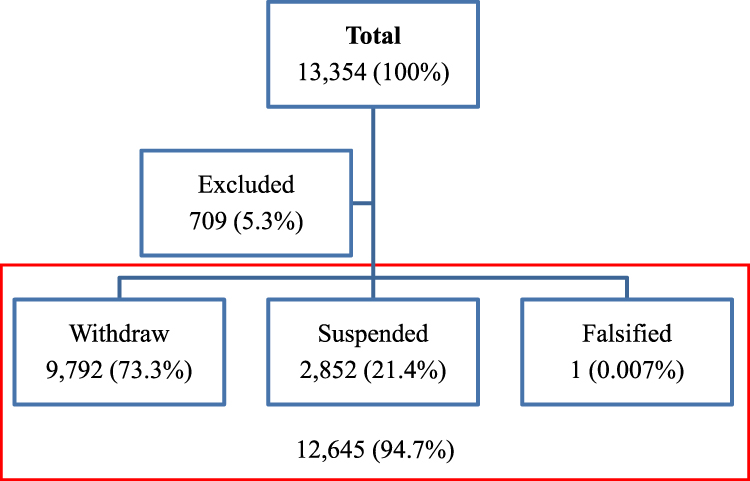 |
Figure 1 The figure shows the total number of analyzed certificates, certificates excluded from the analysis and in the red box, the ones that were included. |
Results
Results indicate that out of the 13,354 recalled certificates of medical devices within the 6-year period (from 2014 to 2020), 9792 were withdrawn (73.3%), 2852 (21.4%) suspended and one falsified. A total of 709 (5.3%) were excluded from the analysis due to missing data (name of the device) (Figure 1), (Table 2). The paste of the withdrawal of the medical equipment is stable and ranges (since 2016) between 69.1% and 79.1%, Me=1881 (Table 3).
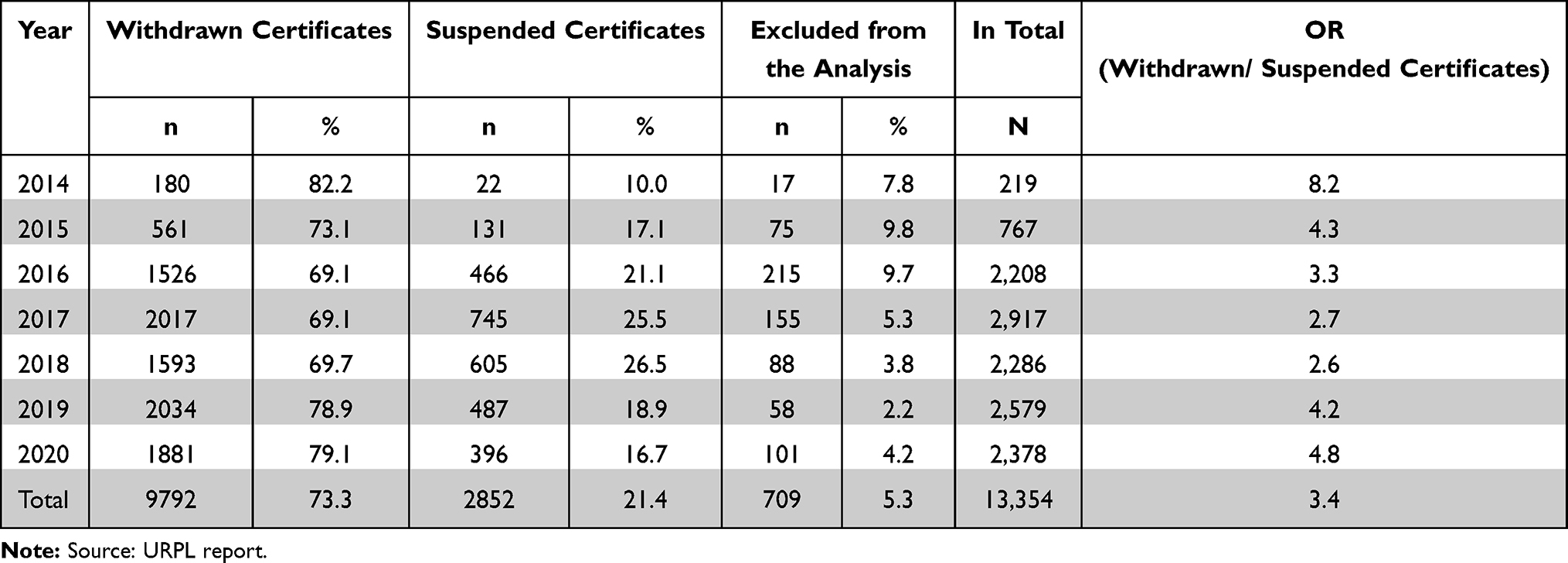 |
Table 2 Information on Withdrawn and Suspended Certificates of Medical Devices Between 2014 and 2020 |
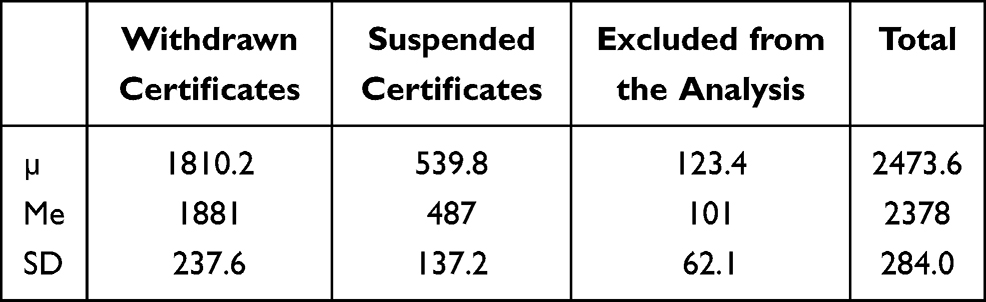 |
Table 3 Data Analysis (2016–2020) |
All mentioned devices (both withdrawn and suspended) were officially in use prior to their being listed in the report.
Discussion
The medical devices found on the market vary in shapes, sizes and application, all of them, however, may cause harm to the patient and further litigation. Therefore, the process of certification and re-certification plays a pivotal role in assuring patient safety and the safety of those who use the specific device type. What is more, the certification process enables the use of faulty series of devices, which in turn also ensures manufacturer’s safety as to placing faulty devices on the market.
Results indicate that there was as many as 13,354 recalled certificates of medical devices within the 6-year period (from 2014 to 2020). Certificates include a whole series of medical devices and it is impossible to determine the actual amount of specific products that were refrained from use within that time period. Because this information is unavailable, it is impossible to determine the ratio of overall medical devices in use to medical devices as well as the number of withdrawn to issued certificates. What is more, the report provides no specific reasoning for the withdrawal of individual medical devices. However, if the provisions concerning devices’ conformity assessment are analyzed, safety is key. A withdrawn or suspended device could endanger user safety, be imprecise, outdated, or unfit for use. Therefore, the certificate’s withdrawal or suspension could be dictated by the premises related to the use of devices that is (or has become) dangerous to use. The source of knowledge on the recalled medical devices includes manufacturers themselves, complaints made by patients or medical professionals as well as reported medical incidences. It should be emphasized that if such incidences occur, the withdrawal is not limited only to patients treated in Poland, but also include patients hospitalized in medical facilities in countries where the given medical devices were used. Based on safety notes found on the URLP, it is possible to elaborate on the type of withdrawn medical devices and the reasoning for their withdrawal. For example, resection electrodes for endoscopic diagnostic procedures in urology and endoscopy were withdrawn because the manufacturer received numerous complaints caused by breakage of the loop wire at the distal end of these electrodes. Upon investigation, it turned out that such a situation may occur even while using the tool as intended. Another example of a withdrawn device could be disposable tools for blunt dissection, where the cotton tip of the device separated from the device. This is in turn, potentially increased the risk of prolonging the time spent in the operating room or generated the need for inadvertent exposure to X-rays in order to detect a foreign body – interestingly, reports indicated that in four cases it was necessary to perform an intraoperative X-ray to detect a separate tip.17,19 These are only some out of the many cases of medical devices withdrawal. However, broken, outdated, or worn-out devices threatens patients’ safety and the safety of the medical personnel who use it.
The legal liability of the hospital, in which the harm occurred, due to faulty devices (or its inefficient use), is determined as an organizational fault. Under Polish law, it is the medical devices user, ie, the hospital, that must pay financial compensation to the patient who has suffered such injury. Cases concerning injury resulting from the use of malfunctioning medical devices are taken to court. In one of the cases, due to a hot water bottle leakage caused by an unexpected temperature increase, a patient suffered severe burns to her feet. The court granted her appropriate compensation.19 In another case, during a lung operation, a patient suffered first- and third-degree hand and foot burns because of faulty equipment (in this incident, an electric knife). The court determined permanent disability at 15% and granted compensation to the patient.19
In Poland, medical devices are subjected to regular inspections specified by technical requirements (Act on Medical Devices; of May 20, 2010, consolidated text: Journal of Laws of 2015, item 876, as amended). For example, pressure devices (sterilizers, autoclaves) are subject to technical inspection, with the frequency specified in the ordinance of the Ministry of Economy, Labor and Social Policy on technical conditions of technical inspection in the field of pressure equipment.
The audit of the used specialist medical devices in Poland 2006–2008, published in January 2010, carried out by a governmental institution, the Supreme Audit Office (polish: NIK), revealed that medical devices used in public hospitals pose a threat to both the patients’ and medical staffs’ health. The research revealed that hospitals provided medical services largely by using technically outdated diagnostic devices. Almost half of them were manufactured before 2000, most of them before 1997. The oldest medical devices were over 40 years old. Almost two-thirds (61.5%) of which the entities looked at did not comply with the statutory obligation of conducting tests using the devices checked in terms of safe use, ie, certificated.18 A study from 2015 in which 3410 questionnaires were filled out by physicians (heads of departments) and nursing personnel also drew attention to this phenomenon’s size. The analysis shows that 86.5% of the surveyed medical personnel had participated in adverse events. Every fifth respondent (20.2%) participated in an event related to pharmacotherapy, every sixth with diagnosis (16.2%) or with an infection (15.7%), and every seventh with medical devices failure (14.2%) or surgery (14.1%).19
Incidents related to the malfunctioning of medical devices are noted everywhere and threaten patients’ and medical staff’s safety and compromise the manufacturers reputation. What is more, they may cause medical errors and ultimately lead to legal consequences.20–22 These consequences consist of establishing, which entity is formally responsible for medical devices malfunctioning. In case of a breakdown, malfunction, or other technical difficulties, which lead to or contributed to placing the patient at risk, the entity responsible for the device should pay compensation. It may be its manufacturer, importer, or, most frequently, the hospital where the incident occurred. Therefore, procedures have been introduced to ensure the maximum safety level for medical device used during treatment.23
The process of introducing the medical device to everyday use pertains to mandatory procedures for its producer or importer (those responsible for placing the medical device on the market). The EU regulation on medical devices is designed to emphasize more on patient safety.24 The law clearly states that compliance with strict obligations to carry out conformity assessments is mainly carried out by notified bodies.25 In the analyzed report, each of the notified bodies was independent, as were those involved from outside Poland, which guarantees assessment objectivity. These entities are responsible for verifying and controlling whether the medical device presented for assessment meets the specified safety, design, manufacturing, packaging, and labelling requirements. The manufacturer or an authorized representative entrusts a notified body with the conformity assessment of a medical device. Once certified, the formal road is over. There is no requirement to assess the effectiveness of a certificate issued by a notified body. Conformity assessment is performed to verify the safety and the declared performance as stated by the manufacturer.26–28
Depending on the type of device, it is the manufacturers’ legal obligation to obtain a certificate from a Notifying Body (entity chosen by individual Member States and listed on the URPL’s website). The certificate is obtained for a series of devices and not individual equipment. It also allows to determine the level of acceptability of the benefit–risk ratio for the use of the assessed product (by assessing side effects and potential hazards that may occur in connection with the use of the product).
Moreover, various initiatives and activities are developed to evaluate medical devices’ performance and safety once their use has been approved.29,30, There are various solutions to this problem worldwide, including through the hospital’s internal activities, ie, the hospital IT network, which also performs the function of controlling the used medical devices.31 There are also special organizations like the British Medicines and Healthcare products Regulatory Agency (MHRA) that monitor medical devices’ safety and performance.32 Above all, however, entities that use medical equipment should implement activities related to its safety and those connected with improving existing procedures.33
It is worth to note that the certification mechanism itself is also not free of defects. The judgment of the Court of Justice of the European Union of Feb 16, 2017 was concerned with the possibility of incurring legal liability by a notified body towards the end-user of a medical device. The case concerned breast implants manufactured by the French company Poly Implant Prothès (the company used industrial implants that did not comply with the directive, instead of medical ones).34
The prepared and presented report35 precisely defines the number of certificates that suspended or withdrew medical devices from use in hospitals. The numbers presented indicate the variable scale of this phenomenon (eg, 2017–2020)
Conclusion
In Poland, and as a rule in other European countries, proceedings related to medical devices are introduced through legal means. Therefore, those proceedings are mandatory, and the failure to obey them could be interpreted as non-compliance with the law.
Legal provisions specify compliance with strict obligations to conduct conformity assessments, which are to be carried out by notified bodies. The degree of complexity of the procedures for assessing the conformity of medical devices established in Polish law is determined depending on the level of risk associated with the use of medical equipment. Therefore, an obligation has been introduced to certify each medical device that carries a specific level of risk of its use. The assessment is also intended to control the medical devices introduced into use.
Notified bodies are responsible for checking and controlling whether the medical devices presented for assessment meets the specified requirements regarding its safety, design, manufacture, packaging, and labelling. Notified bodies also have the power to assess products already placed on the market, that is, the supervision of products in use.
The new EU law on medical devices was to apply from May 2020. However, the European Commission considered that this timeframe had to be postponed. The powers of notified bodies are unified in the EU countries (where the same safety standards are to apply). A central database of medical devices in the EU territory is also to be established. First module on Actor registration operates since December 1, 2020. The following two modules ie on Unique Device Identification UDI/device registration and the module on Certificates and Notified Bodies will become available by May 2021.32 Currently, medical devices subject to certification may be marketed based on the existing documentation until the certificate expires, but no later than May 27, 2024.
The study was conducted as a quantitative descriptive exploratory study using a government audit agency’s report and documents and articles regarding the safety of the medical devices in use. The problem seems universal.
The requirement of authorization (in the USA) or certification (in Europe) for medical devices is a mechanism intended to ensure patients’ safety during tests or procedures. However, many medical entities try to save money on medical devices inspection and maintenance and neglect the use of mandatory protective procedures. As a result, certificates are lost or are withdrawn by the notifying bodies in Europe. Consequently, patients diagnosed with specialized medical devices may not be assured of the obtained results’ safety and reliability.
Studies Limitations
Main limitation concerns the impossibility of determining how many certificates were issued and this ultimately hampered the possibility of determining the scale of the withdrawn and suspended certificates.
We contacted notifying bodies in Poland, to ask for the overall number of issued certificates; however, we have not received an answer and even if such an answer would be provided, we are uncertain whether manufacturers from different countries would not apply for such certification, with medical devices that would not be used in Poland later.
What is more, we are not able to state, for how long medical devices were on the market prior to their recall.
Disclosure
Authors declare no conflict of interest.
References
1. Medical devices. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices.
2. Manita D, Aakash V, Rana AC, Sharma PC. Regulation and Clinical Investigation of Medical Device in the European Union. Appl Clin Res Clin Trials Regulatory Affairs. 2019;6(3):163–181. doi:10.2174/2213476X06666190821095407
3. Dagher G, Becker KF, Bonin S, et al. Pre-analytical processes in medical diagnostics:New regulatory requirements and standards. N Biotechnol. 2019;52:121–125. doi:10.1016/j.nbt.2019.05.002
4. Aronson JK, Heneghan C, Ferner RE. Medical Devices: definition, Classification and Regulatory Implications. Drug Saf. 2020;43:83–93. doi:10.1007/s40264-019-00878-3
5. Vila Wagner M, Schanze T. Comparison of approval procedures for medical devices in Europe and the USA. Current Directions Biomed Eng. 2019;5(1):605–608. doi:10.1515/cdbme-2019-0152
6. Fenton R. 5 Staggering Medical Device Recall Statistics. That Should Concern Everyone. Available from: https://www.qualio.com/blog/medical-device-recall-statistics. Accessed 08 July, 2021.
7. Act of 20, May 2010 about medical devices. Available from: https://isap.sejm.gov.pl/isap.nsf/download.xsp/WDU20101070679/U/D20100679Lj.pdf.
8. Office for Registration of Medicinal Products, Medical Devices and Biocidal Products. Obligation to notify. Available from: http://www.urpl.gov.pl/pl/wyroby-medyczne/pytania-i-odpowiedzi-faq.
9. Stelengowska M, Młynarczyk T. Zasady wprowadzania wyrobów medycznych do obrotu i dystrybucji na terenie Rzeczypospolitej Polskiej w świetle nowej ustawy o wyrobach medycznych. Acta Bio-Optica Et Informatica Medica. 2011;17(2):111–115.
10. Council Directive 90/385/EEC of June 20 1990 on the approximation of the laws of the Member States relating to active implantable medical devices. Available from::https://eur-lex.europa.eu/legal-content/PL/TXT/?uri=CELEX%3A31990L0385.
11. Maresova P, Hajek L, Krejcar O, Storek M, Kuca K. New Regulations on Medical Devices in Europe: are They an Opportunity for Growth? Adm Sci. 2020;10(1):16. doi:10.3390/admsci10010016
12. Częstotliwość przeglądów technicznych sprzętu medycznego [The frequency of technical inspections of medical equipment]. Available from: http://ppcito.home.pl/ztm/przeglady-techniczne-sprzetu-medycznego/. Accessed 15 July, 2021.
13. Regulation of the Minister of Health of Feb 17 2016 on assessment procedures of active implantable medical devices. Available from: https://www.infor.pl/akt-prawny/DZU.2016.034.0000211,rozporzadzenie-ministra-zdrowia-w-sprawie-wymagan-zasadniczych-oraz-procedur-oceny-zgodnosci-wyrobow-medycznych.html.
14. Najwyższa Izba Kontroli. Dostępność refundowanych wyrobów medycznych wydawanych na zlecenie i bezpieczeństwo ich stosowania. Informacja o wynikach kontroli 18.04.2019. KZD.430.006.2018. Available from: https://www.nik.gov.pl/kontrole/P/18/056/.
15. Office for Registration of Medicinal Products, Medical Devices and Biocidal Products. Available from: http://www.urpl.gov.pl/pl/wyroby-medyczne/wykaz-wycofanych-i-zawieszonych-certyfikat%C3%B3w-zgodno%C5%9Bci.
16. Ruling of the District Court in Lublin on Apr 04 2002. File reference number I C 656/99. Available from: https://fjkancelaria.pl/placowka-medyczna-a-odpowiedzialnosc-uzycie-wadliwych-urzadzen-medycznych/.
17. Ruling of the Supreme Court of May 11 1983. File reference number IV CR 118/83. Available from: https://sip.lex.pl/orzeczenia-i-pisma-urzedowe/orzeczenia-sadow/iv-cr-118-83-wyrok-sadu-najwyzszego-520096595.
18. Question n° 14530 to the Minister of Health regarding the defectiveness of medical equipment in Polish hospitals. Available from: http://orka2.sejm.gov.pl/IZ6.nsf/main/12B706FB.
19. Survey of medical and nursing staff’s opinion on reporting adverse events and the requirements that should be met by reporting systems in healthcare; 2015. Available from: https://www.cmj.org.pl/raporty.php?&drukuj=.
20. Badnjević A, Pokvić LG, Lemana SL. Chapter 74 Inspection of medical devices. In: Clinical Engineering Handbook.
21. Aruna M, Gunasilan U, Naeem S. Efficient Equipment Management for Biomedical Engineering Department in the Hospital. Int Rev Manage Marketing; Mersin. 2018;8(3):69–74.
22. Jang D, Kim S. Performance Evaluation of Diagnostic X-Ray Equipment Regarding the Hospital Size in the Republic of Korea. Iranian J Med Phy. 2019;16(3):195–199.
23. Gurbeta L, Izetbegović S, Badnjević-čengić A. Inspection and Testing of Pediatric and Neonate Incubators. In: Badnjević A, Cifrek M, Magjarević R, Džemić Z, editors. Inspection of Medical Devices. Series in Biomedical Engineering. Singapore: Springer; 2018.
24. Thienpont E, Quaglio G, Karapiperis T, Andersen K. Guest Editorial: new Medical Device Regulation in Europe: a Collaborative Effort of Stakeholders to Improve Patient Safety. Clin Orthop Relat Res. 2020;478(5):928–930. doi:10.1097/CORR.0000000000001154
25. Groennvold WC. New Regulations on Notified Bodies and Conformity Assessment of High- Risk Medical Devices in Europe: impact on Clinical Investigation from an Industry Perspective. Int J Clin Res Trials. 2017;2:13. doi:10.15344/2456-8007/2017/115
26. Rising J, Moscovitch B. The Food and Drug Administration’s Unique Device Identification System: better Postmarket Data on the Safety and Effectiveness of Medical Devices. JAMA Intern Med. 2014;174(11):1719–1720. doi:10.1001/jamainternmed.2014.4195
27. Yaeger KA, Martini M, Yaniv G, Oermann EK, Costa AB. United States regulatory approval of medical devices and software applications enhanced by artificial intelligence. Health Policy Technol. 2019;8(2):192–197. doi:10.1016/j.hlpt.2019.05.006
28. Gurbeta L, Badnjević A, Kurta E. eVerlab: software Tool for Medical Device Safety and Performance Inspection Management. In: Badnjevic A, Škrbić R, Gurbeta pokvić L, editors. CMBEBIH 2019. CMBEBIH 2019. IFMBE PROCEEDINGS. Vol. 73. Cham: Springer; 2020.
29. Kramer DB, Tan YT, Sato C, Kesselheim AS. Ensuring medical device effectiveness and safety: a cross--national comparison of approaches to regulation. Food Drug Law J. 2014;69(1):1–i.
30. Badnjević A, Gurbeta L, Bošković D, Džemić Z. Medical devices inlegal metrology, 2015 4th Mediterranean Conference on Embedded Computing (MECO). Budva. 2015;365–367. Available from: https://pl.booksc.eu/book/53170866/54bca8. Accessed 01 June, 2021.
31. Zhang M, Zheng K, Shen Y, Lin Z, Li Z. [Hospital Networked Medical Equipment Safety Management Chinese]. Zhongguo Yi Liao Qi Xie Za Zhi. 2018;30;42(4):303–304. Chinese. doi:10.3969/j.issn.1671-7104.2018.04.021. PMID: 30112902.
32. Wilkinson J, Crosbie A. A UK medical devices regulator’s perspective on registries. Biomed Eng Biomedizinische Technik. 2016;61(2):233–237.
33. Kedwani M, Schröttner J, Baumgartner C. Analysis of regulatory requirements of medical devices and in-vitro diagnostics worldwide for the development of an efficient procedure of registration for manufacturers of medical products. Current Directions Biomed Eng. 2019;5(1):609–612. doi:10.1515/cdbme-2019-0153
34. Rytel M Jednostki notyfikowane odpowiedzialne za wyroby medyczne trafiające do użytkowników. Available from: http://www.wyrobmedyczny.info/jednostki-notyfikowane-odpowiedzialne-wyroby-medyczne-trafiajace-uzytkownikow/.
35. Annual Reports (issued between 2012–2019) Of The President Of The Office For Registration Of Medicinal Products, Medical Devices And Biocidal Products. Available from: http://urpl.gov.pl/pl/urz%C4%85d/raport-roczny.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.