")
Back to Journals » Drug, Healthcare and Patient Safety » Volume 11
Safety and tolerability of macitentan in the management of pulmonary arterial hypertension: an update
Authors Wong AK, Channick RN
Received 30 January 2019
Accepted for publication 12 July 2019
Published 3 September 2019 Volume 2019:11 Pages 71—85
DOI https://doi.org/10.2147/DHPS.S173050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rajender R Aparasu
Alexandra K Wong,1 Richard N Channick2
1Division of Pulmonary and Critical Care, Massachusetts General Hospital, Boston, MA 02114, USA; 2Division of Pulmonary and Critical Care, University of California Los Angeles Medical Center, Los Angeles, CA 90095, USA
Correspondence: Alexandra K Wong
Division of Pulmonary and Critical Care, Massachusetts General Hospital, 55 Fruit Street, Boston, MA 02114, USA
Tel +1 617 724 9674
Fax +1 617 726 6878
Email [email protected]
Abstract: Macitentan is a medication in the endothelin receptor antagonist class, approved for treatment of pulmonary arterial hypertension in 2013 based on the results of the pivotal SERAPHIN Trial (Study with an Endothelin Receptor Antagonist in Pulmonary arterial Hypertension to Improve cliNical outcome). Macitentan was shown in initial trials to reduce the likelihood of a morbidity/mortality event. Real-world use of this medication additionally reveals a reduced risk of hospitalizations related to pulmonary arterial hypertension, improved health-related quality of life scores, potential clinical utility in other conditions (such as chronic thromboembolic pulmonary hypertension and pulmonary hypertension related to congenital heart disease), and has a similar safety profile as demonstrated in initial trials.
Keywords: macitentan, pulmonary arterial hypertension, safety, tolerability, efficacy
Introduction
Pulmonary hypertension (PH) is defined as a mean pulmonary arterial pressure of greater than 25 mmHg, as measured during right heart catheterization, and encompasses a broad range of causes.1 Pulmonary arterial hypertension (PAH), or group 1 pulmonary hypertension according to the World Health Organization classification, is defined as PH without elevation in pulmonary capillary occlusion pressure and with increased pulmonary vascular resistance, in the absence of lung disease, chronic thromboembolic pulmonary hypertension (CTEPH), or other rare causes. PAH can be idiopathic in nature, related to drug or toxin exposure, associated with other conditions such as connective tissue disease or congenital heart disease, due to pulmonary veno-occlusive disease, or less commonly heritable in nature.2
The estimates of the prevalence of PAH are variable, but likely in the range of 15–60 cases per million population.3 The prognosis for this condition remains guarded, with mortality 1 year after diagnosis as high as 21% in patients classified as highest risk.4 Modifying this risk via various methods, including pharmacotherapy, has important implications for prognosis and survival in these patients.
Pharmacologic therapy for PAH has evolved dramatically over the last two decades, and there are several classes of medications available for treatment. Macitentan (OPSUMIT) was approved by the Food and Drug Administration (FDA) in 2013 after the Study with an Endothelin Receptor Antagonist in Pulmonary arterial Hypertension to Improve cliNical outcome (SERAPHIN) trial was published.5 Macitentan was the fourth medication in the class of endothelin receptor antagonists (ERAs) approved for the treatment of PAH; however, only three remain approved and available currently due to the withdrawal of the drug sitaxsentan from the market in 2010 due to hepatic toxicity. FDA approval encompasses only group 1 PAH as this was the study population in the large, randomized controlled trial SERAPHIN. Its use in other groups of pulmonary hypertension is also under investigation, as will be outlined later. Other classes of medications include phosphodiesterase-5 inhibitors (PDE-5i) and prostacyclin pathway agents. This review will focus specifically on the use and safety of macitentan over the past 5 years since approval in the United States and Europe.
Background of the drug/family
Increased tone in the pulmonary vascular bed increases pulmonary vascular resistance and is a key mechanism for the development of PAH. The endothelins are an important class of molecules that regulate vascular smooth muscle, and their upregulation is one of the key pathways in the pathogenesis of this disease. Endothelin-1 (ET-1) was the first peptide discovered in this class in 1988. Since then, two additional endothelins – endothelin 2 (ET-2) and endothelin 3 (ET-3) – have been identified which share a similar molecular structure, but have heterogeneous distribution in various tissues.6 There are two receptors for these molecules: endothelin receptor A (ETA), expressed on pulmonary vascular smooth muscle cells, and endothelin receptor B (ETB), expressed on both pulmonary vascular smooth muscle cells and endothelial cells. These receptors bind circulating endothelin peptides with slightly different affinities and appear to function somewhat differently in a normal compared to a pathologic state.
Production of ET-1 can be rapidly up- or downregulated by vascular endothelial cells to tightly regulate vascular tone.7 When this peptide binds the ETA receptor in the pulmonary vascular smooth muscle cells, a potent vasoconstrictive effect occurs through a pathway that ultimately results in the release of intracellular calcium. The presence of calcium and the resultant vasoconstriction persist even after ET-1 is no longer bound to the ETA receptor.8 Under normal physiological circumstances, activation of the ETB receptor by ET-1 on endothelial cells predominantly results in vasodilation (via nitric oxide and prostacyclin pathways), negative feedback and downregulation of ET-1 production, and clearance of ET-1. Activation of the ETB receptor also results in a small amount of vasoconstriction given its presence on pulmonary vascular smooth muscle cells. Under certain circumstances, particularly in the presence of pulmonary hypertension, the vasoconstrictive effect of the ETB receptor may be enhanced/upregulated.9
Similarly, increased serum concentrations of ET-1 and increased presence of ET-1 in the pulmonary arterial smooth muscle cells have been demonstrated in several forms of PH.10,11 One study by Giaid et al assessed lung tissue from 15 controls, 11 patients with plexogenic pulmonary arteriopathy (7 of whom had a clinical diagnosis of group 1 PAH), and 17 patients with secondary causes of pulmonary hypertension.11 Immunohistochemical analysis was performed to evaluate the expression of endothelin-1 in the vascular tissue. Their evaluation revealed little endothelin-1-like reactivity in the control groups but showed increasing reactivity in the patients with secondary pulmonary hypertension, and the most in the patients with plexogenic pulmonary arteriopathy. This study revealed evidence of increased endothelin-1 in vascular tissue in patients with pulmonary hypertension. Subsequent studies demonstrated that ETA and ETB receptors contribute to the proliferation of pulmonary arterial smooth muscle cells in-vitro, which is likely related to vascular remodeling in the pathogenesis of pulmonary hypertension.12 ERAs thus became a clear pharmacologic target for the treatment of pulmonary hypertension.
Several animal models were developed to assess and prove the favorable effect of ERAs in the pathogenesis of pulmonary hypertension. One study compared the effect of an ETA antagonist to a nonselective ETA/B antagonist in a rat model of pulmonary hypertension, induced by the administration of monocrotaline.13 In this model, the ETA receptor antagonsist-treated rats had a two-fold increased survival compared to the untreated PH model. In those rats treated with the dual ERA, there was an additional 10% improvement in survival. Furthermore, these rats experienced a reduction in right ventricular hypertrophy, an effect not seen with the selective ETA antagonist. However, there was no difference in the right ventricular systolic pressure (measured directly in surviving rats) or in a measure of the resistive properties of the lungs between the selective and nonselective groups.14 Both agents appeared to be effective in the treatment of a rat model of pulmonary hypertension, spurring further study in humans.
The first medication for the management of pulmonary hypertension targeted to block vasoconstriction mediated by the endothelin pathway was bosentan (TRACLEER), approved by the FDA in 2001. Bosentan is a dual ERA, blocking the effect of ET-1 on both ETA and ETB. In several randomized controlled studies, bosentan revealed improvement in patient exercise capacity, pulmonary vascular resistance, and time to clinical worsening compared to placebo. Ambrisentan (LETAIRIS), approved by the FDA in 2007, has affinity predominantly to the ETA receptor and was shown in two concurrent randomized trials to also improve exercise capacity and increase the time to clinical worsening. As noted earlier, sitaxsentan, also a selective ETA receptor antagonist, was pending FDA evaluation but was withdrawn from marketing and clinical study in 2010 due to fatal liver damage. Finally, macitentan (OPSUMIT) is the second dual ERA and was approved in 2013.
Mechanism
As noted earlier, macitentan is the second dual ERA available for the treatment of pulmonary arterial hypertension (Figure 1).15 It has a stronger affinity for the lipid phase as compared to other ERAs, thus promoting tissue penetration. Macitentan is metabolized into its active form in the liver. Peak concentration is reached in 8 hrs, and the half-life is 16 hrs for the parent compound but 48 hrs for the active metabolite.16 It differs from its dual ERA predecessor, bosentan, by demonstrating prolonged and enhanced attachment to the ETA and ETB receptors, as well as increased penetration into the pulmonary arterial smooth muscle, suggesting the potential for improved clinical efficacy.17,18 Further, the pharmacokinetic profile of macitentan allows for once-daily dosing, as opposed to twice daily for bosentan.19 Due to the prolonged binding and slower dissociation of the macitentan molecule to its receptors (15 to 20 times longer receptor binding as compared to prior ERAs), it is thought that this medication maintains its effect even in the presence of very high ET-1 concentrations.16 In addition, in an animal model of pulmonary hypertension, treatment with macitentan did not result in increased plasma bile salt concentrations, thought to be the mechanism of liver function test abnormalities with bosentan.20 Lastly, pharmacokinetic studies suggest that bosentan decreases the plasma concentration of sildenafil when prescribed as a second agent for pulmonary hypertension, whereas no such effect has been seen with macitentan.21–23 With the earlier preliminary data supporting efficacy and safety of macitentan, two randomized placebo-controlled trials were performed.
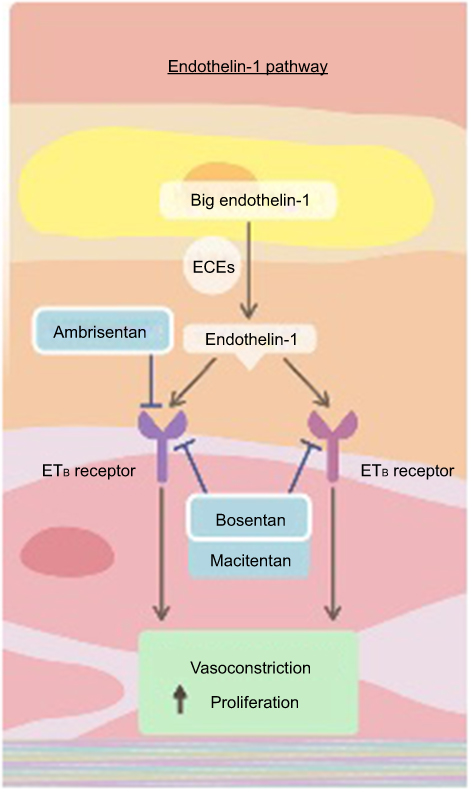 |
Figure 1 The endothelin pathway. Endothelin-1 and the endothelin receptor pathway. Dual endothelin receptor antagonists, such as macitentan, block the binding of endothelin-1 to both the ETA and ETB receptor and block the downstream vasoconstricting effects. Reproduced from Lai YC, Potoka KC, Champion HC, et al. Pulmonary arterial hypertension: the clinical syndrome, Circulation Research, 115, 1, 115–130, https://www.ahajournals.org/.15 Abbreviations: ECE, endothelin-converting enzymes; ET, endothelin. |
Evidence from clinical trials
The Macitentan USe in an Idiopathic Pulmonary Fibrosis Clinical (MUSIC) trial randomized patients from 2009 to 2011 with a relatively new diagnosis (<3 years) of idiopathic pulmonary fibrosis (IPF) to 10 mg daily of macitentan or placebo. The primary endpoint was a change in forced vital capacity (FVC) at 12 months; however, the safety and tolerability of macitentan over a prolonged period of time in that patient population were also of interest. While macitentan was not shown to have an effect on FVC in patients with IPF, the treatment group was maintained on macitentan for a median of 14.5 months, with 91 patients undergoing treatment with macitentan for greater than 12 months. Overall, the MUSIC trial suggested that macitentan was well tolerated at the 10 mg daily dosing for several months of treatment.24
The SERAPHIN trial, published in 2013, enrolled 742 patients in 39 countries and 151 centers to investigate whether long-term therapy with macitentan reduces morbidity and mortality in patients diagnosed with PAH. This trial randomized patients in a 1:1:1 fashion to placebo, 3 mg daily of macitentan, or 10 mg daily of macitentan, respectively. Kaplan–Meier curves for the composite primary endpoint in this study (first event related to PAH or death from any cause) revealed a significant treatment effect of macitentan at a once-daily dose of 10 mg compared to placebo, with a 45% reduction of morbidity and mortality events. PAH-related events were defined as worsening PAH, need for IV or subcutaneous prostacyclin medication, lung transplant, or the need for atrial septostomy. To meet criteria for “worsening PAH”, patients needed to demonstrate a decrease in 6-min walk test of 15% from baseline, worsening symptoms of PAH, and the need for additional treatment for PAH. As part of a secondary endpoint analysis, it was found that patients treated with 10 mg macitentan daily also experienced improvement in their World Health Organization (WHO) functional class and 6-min walk tests. In addition, the rate of a combined secondary endpoint of death or hospitalization due to PAH was reduced by 50% in the 10 mg macitentan group. Macitentan was well tolerated with few adverse events (see below for details).5 It is on the basis of these studies that FDA approval was obtained for macitentan for the treatment of pulmonary arterial hypertension. The randomized clinical trials that have been performed with the use of macitentan will be discussed and are summarized in Table 1.
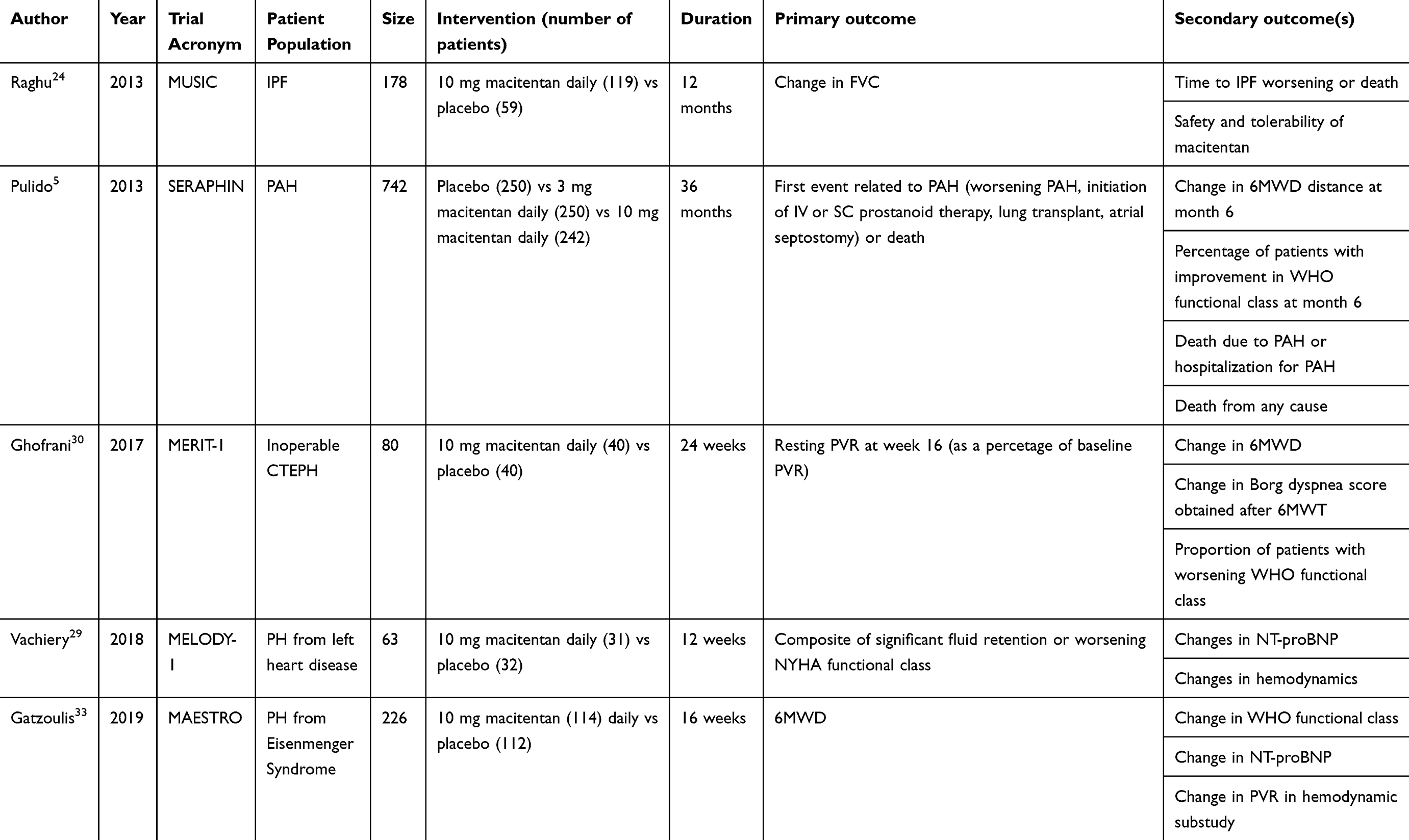 |
Table 1 Summary of clinical trials |
Effect on hospitalizations
Since the publication of the SERAPHIN Trial, additional analyses of that data have been reported. One such publication describes in detail the composite secondary endpoint of death or hospitalization due to PAH, as the reduction by 50% in this endpoint was driven mainly by a reduction in hospitalization.25 Throughout the duration of the trial, 117 patients in the placebo arm, 104 patients in the 3 mg macitentan arm, and 90 patients in the 10 mg macitentan arm were hospitalized for any reason. The risk of all-cause hospitalization was reduced by 32.3% in the 10 mg macitentan group as compared to placebo. In addition to a reduction in the rate of hospitalization, there was also a reduction in the average number of hospital days from 4.1 days in the placebo group to 2.8 days in the 10 mg macitentan group - a 31% reduction. There were 80 patients in the placebo arm, 53 in the 3 mg arm, and 46 in the 10 mg arm who were hospitalized due to PAH. The risk of PAH-related hospitalization was reduced by 49.8% in the 10 mg macitentan group compared to placebo (Figure 2). There was a 52.3% reduction in the average number of PAH-related hospital days in the 10 mg macitentan group as compared to placebo. Overall, macitentan decreased the rates of PAH-related and all-cause hospitalizations and did not increase the rates of non-PAH-related hospitalization.
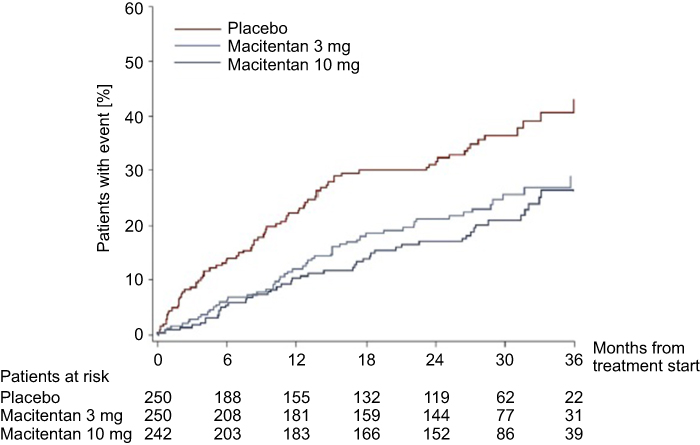 |
Figure 2 Effect of macitentan on PAH-related hospitalizations. The effect of macitentan 10 mg daily, 3 mg daily, and placebo on the rate of hospitalization due to pulmonary arterial hypertension (PAH). Macitentan at 10 mg daily showed a 49.8% reduction in PAH-related hospitalizations compared to placebo. Reprinted from JACC Heart Fail, 3, 1, Channick RN, Delcroix M, Ghofrani HA, et al, Effect of macitentan on hospitalizations, 1–8, Copyright (2015), with permission from Elsevier.25 |
Hemodynamic effects of macitentan
A total of 187 patients in SERAPHIN underwent right heart catheterization at baseline and at 6 months as part of a sub-study to evaluate the hemodynamic effects of macitentan and to investigate the association between hemodynamics and disease progression in the macitentan and placebo groups.26 Cardiac index (CI), mean pulmonary arterial pressure (mPAP), pulmonary vascular resistance (PVR), and NT-pro BNP were measured at baseline and at the 6-month time point. There was a significant improvement (reduction in mPAP, PVR, and BNP and increase in CI) in all parameters at 6 months in the 10 mg macitentan group compared to placebo. This study also determined thresholds of cardiac index, right atrial pressure, and NT-proBNP that predicted the risk of a primary endpoint event (ie, morbidity or mortality event). At 6 months, there was a 51% reduction in the primary endpoint in patients with a cardiac index greater than 2.5 L/min/m2. There was a 28% reduction in patients with a right atrial pressure less than 8 mmHg. Lastly, there was an impressive 78% reduction in the primary endpoint in patients with NT-proBNP less than 750 fmol/mL at 6 months. Patients were more likely to have achieved these favorable thresholds at 6 months if they were in the 10 mg macitentan group as compared to placebo.
Health-related quality of life
As part of the SERAPHIN Trial, the 36-item Short Form Survey (SF-36) was administered to patients aged 14 years or older at the time of enrollment, 6 months, 12 months, and at the end of study visit. It was found that patients treated with macitentan had an improvement in their health-related quality of life at 6 months and had a reduced risk of deterioration in their quality of life score over their enrollment period.27 The SF-36 categorizes final “domain” scores (physical functioning, limitations due to physical health, limitations due to emotional health, mental health, bodily pain, general health perceptions, vitality, social functioning) into a physical component summary (PCS) score and a mental component summary (MCS) score, which are then normalized to the US general population from a 1998 survey. Patients with PAH have a PCS score that is greater than 1 standard deviation below population norms, and more than one-quarter of PAH patients experience depression, anxiety, or stress.28 The absolute change between baseline and month 6 SF-36 scores was a pre-specified secondary endpoint within the SERAPHIN trial. Limited data were available at month 12 and the end of the trial, and the timing of the end-of-trial time point varied between patients making comparison to baseline potentially inaccurate. Instead, post-hoc analyses of trial data evaluated the time to a significant decline in SF-36 in macitentan treated patients vs placebo. A three-point decline in MCS or PCS was considered to be the minimally important difference. At 6 months, both the 3 mg and 10 mg macitentan groups experienced improvement in seven of the eight health domains as opposed to the placebo group, which experienced deterioration in both domain and physical and mental component scores. In addition, both the 10 mg macitentan dose and 3 mg macitentan dose significantly reduced the risk of a meaningful decline in the SF-36 scores until the end of treatment compared to placebo (Figure 3). The 10 mg dose of macitentan decreased the risk of significant PCS score deterioration by 40% and significant MCS score deterioration by 24%, which is paralleled by the drugs’ effect in delaying disease progression.
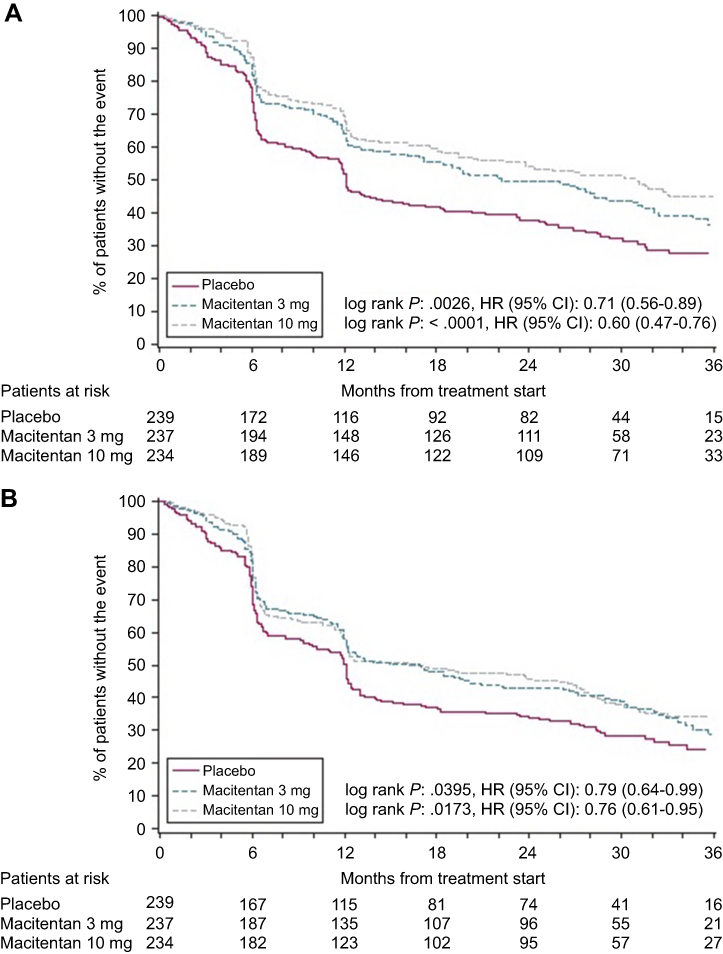 |
Figure 3 Time to significant decline in SF-36 score for macitentan (10 mg, 3 mg) vs placebo. Time to a significant decline in the SF-36 COMPONENT score. Overall, the 10 mg macitentan dose and 3 mg macitentan dose both significantly reduced the risk of a meaningful decline in the SF-36 scores until the end of treatment compared to placebo: Panel A displays the meaningful decline in the physical component score, Panel B depicts the meaningful decline in the mental component score. Reprinted from Chest, 151, 1, Mehta S, Sastry BKS, Souza R, et al, Macitentan improves health-related quality of life for patients with pulmonary arterial hypertension, 106–118, Copyright (2017), with permission from Elsevier.27 |
Use in group 2 pulmonary hypertension
The largest group of patients with pulmonary hypertension worldwide are those with pulmonary hypertension as a result of left heart disease or those people classified as WHO group 2. Thus, finding additional therapies that may benefit these patients is of utmost importance given their significant symptom burden and poor prognosis. In attempts to determine whether macitentan is effective in this population, the Macitentan in subjects with combined prE- and post-capiLlary pulmOnary hypertension due to left ventricular Dysfunction (MELODY-1) trial was performed.29 Patients with heart failure have elevated levels of circulating ET-1, making ERAs a possible therapeutic option. Previous studies in this population were not enriched for a population with a pre-capillary component, and thus had not shown benefit and suggested possible worsening of both peripheral edema and heart failure. This trial included patients with an elevated pulmonary capillary wedge pressure (PCWP, between 15 and 25 mmHg), elevated mean pulmonary artery pressure (mPAP, greater than 25 mmHg) as well as an increased pulmonary vascular resistance (PVR, greater than 3 Woods Units) and a diastolic pressure gradient (DPG) greater than 7 mmHg. A total of 63 patients were randomized to macitentan (10 mg daily) or placebo for a total of 12 weeks. The primary endpoint was a composite of significant fluid retention or worsening NYHA functional class at the end of treatment. Changes in NT-pro BNP and hemodynamics at the end of the trial were included as exploratory endpoints. Within the first month of treatment, patients in the macitentan arm had significantly more fluid retention than those in the placebo arm. There appeared to be a significant reduction in 6-min walk distance(6MWD) in patients in the macitentan arm. There was no difference at 12 weeks in PVR, mean right atrial pressure, PCWP, NT-proBNP, or cardiac index. More patients in the macitentan arm experienced serious adverse events, including hospitalization for heart failure, drug discontinuation, and anemia. Despite its small study population, lack of formal power calculations, and short study duration, MELODY-1 suggests that macitentan is not effective in patients with group 2 pulmonary hypertension and its use results in increased adverse events.
Use in chronic thromboembolic pulmonary hypertension
After the benefits of macitentan in group I pulmonary hypertension were elucidated, interest naturally shifted to its use in other types, namely in PH due to chronic thromboembolic disease (CTEPH). The Macitentan in thE tReatment of Inoperable chronic Thromboembolic pulmonary hypertension (MERIT-1) trial sought to evaluate the efficacy and safety of using macitentan in patients with inoperable CTEPH, including in those patients already on background therapy for pulmonary hypertension.30 A total of 80 patients who were deemed to have inoperable CTEPH were randomized to receive 10 mg macitentan daily versus placebo. Patients were required to have WHO functional class II–IV at the time of enrollment, 6MWT between 150 and 450 m, mPAP≥25 mmHg, pulmonary capillary wedge pressure≤15 mmHg, and PVR≥400 dyn*s/cm5. Those patients with WHO functional class III or IV were allowed to be on background therapy with PDE-5i or inhaled or oral prostanoid therapy. L-arginine or riociguat was not allowed as background therapies for the purposes of enrollment. Patients were monitored for 24 weeks, with RHC performed at baseline and at week 16. The majority of enrolled patients were in WHO functional class III, and thus on background therapy, mostly consisting of PDE-5i therapy. After 16 weeks of therapy, patients receiving 10 mg macitentan had a 27% reduction in their PVR compared to baseline pre-treatment values, indicating a significant treatment effect compared to the placebo arm. Data obtained at this time point also revealed a significant improvement in cardiac output and cardiac index in the treatment arm. After 24 weeks of therapy, there was a significant increase in the 6MWD as well as a significant decrease in NT-proBNP levels. Adverse events and side effects were similar in this population as in larger prior studies. Peripheral edema and anemia were more common in the treatment group, and 2 patients in the placebo arm discontinued the drug due to adverse events. There was no evidence of hypotension as an adverse event in the treatment arm, which is a known side effect of riociguat, the approved therapy for medical management of CTEPH.31
Congenital heart disease
Medical therapy for the treatment of PAH related to congenital heart disease is approached similarly to treatment of PAH due to other causes. However, data supporting the use of PDE-5i, ERAs, and prostacyclins are limited. Prior to macitentan, the only randomized, double-blind, placebo-controlled study in this population was the Bosentan Randomized Trial of Endothelin Antagonist THErapy-5 (BREATHE-5), which evaluated the effect of bosentan on patients with Eisenmenger syndrome who were treatment naïve and WHO functional class III with simple cardiac defects.32 This study suggested that bosentan improved hemodynamics and exercise capacity and was well tolerated in this population. In a similar vein, the MAcitentan in Eisenmenger Syndrome To RestOre exercise capacity (MAESTRO) study was recently published evaluating the safety and efficacy of macitentan in patients with PAH due to Eisenmenger syndrome.33 Unlike BREATHE-5, MAESTRO included patients with both simple and complex congenital heart disease, functional class II or III, on background therapy with PDE-5i, as well as patients with Down syndrome, as this demographic makes up 20% of the patients with Eisenmenger syndrome.
In MAESTRO, patients were randomized to macitentan or placebo and followed for a total of 16 weeks. The primary outcome was a change in 6MWD at the end of the study period. Secondary endpoints included a change in WHO functional class and dyspnea. Other parameters evaluated as exploratory endpoints included NT-proBNP and SpO2 changes. Some patients were included in a hemodynamic substudy, where pulmonary vascular resistance, mPAP, mean right atrial pressure, the ratio of pulmonary to systemic blood flow (Qp/Qs), and systemic vascular resistance were assessed after 16 weeks of treatment. In total, 226 patients were randomized, 40 of whom were also in the hemodynamic substudy. There was no difference in the primary endpoint between the two groups at the end of the study, nor was there a difference in WHO functional class, Borg Dyspnea scores, or oxygen saturation at the end of the trial. However, the improvement in 6MWD in patients with Eisenmenger’s syndrome was much less pronounced than the improvement seen with this group of patients in the BREATHE-5 trial. MAESTRO saw a significant improvement in 6MWD in patients in the placebo group; the reason for this is unclear and likely multifactorial, but certainly had an impact on the failure to show a significant difference in the primary endpoint. The macitentan group did have a significant reduction in NT-proBNP levels compared to the placebo group. In the hemodynamic substudy group, there was a decrease in PVR in the treatment group. Macitentan was well tolerated by this group of patients from a safety standpoint and had a similar adverse event profile as in prior studies. Namely, the rates of headache, upper respiratory tract infection, and bronchitis were increased. The rates of peripheral edema were similar between the two groups. There was a decrease in hemoglobin in patients receiving macitentan; however, as there was no change in oxygen saturation or Qp/Qs, it was not felt that this decline influenced the primary endpoint. Overall, this study suggests that macitentan is safe to use in a diverse group of patients with Eisenmenger syndrome, though failed to show an effect on 6MWD at a 14-week follow-up. Prior to the publication of this large trial, there were only some reports of the transition of patients with PAH related to congenital heart disease from bosentan (based on BREATHE-5) to macitentan (based on SERAPHIN), which was well tolerated (see below).
Comparison to other ERAs
No head-to-head comparison of the efficacy of one ERA compared to another has been performed. Each of the ERAs has been compared to placebo, but none of the approved medications in this class have been compared to one another. Thus, making an assessment of the benefits of one agent over another is challenging. An attempt to explore this question was made by Duo-Ji and colleagues in the performance of a network meta-analysis to compare the ERAs with the hopes of determining the most effective therapy for PAH in this class of drugs.34 As each included trial was designed differently, the authors used common outcome endpoints of 6MWD and clinical worsening as the primary efficacy indicators for comparison, and serious adverse events, death, and all-cause discontinuation as the acceptability outcomes for comparison. Traditional meta-analysis suggested that all ERAs improved 6MWD significantly compared to placebo, with bosentan suggesting the strongest effect. Only bosentan and ambrisentan revealed a reduced risk for clinical worsening in the comparison of odds ratios, and ambrisentan alone suggested a significantly reduced rate of all-cause discontinuation. Again, however, these comparisons were of each agent to placebo rather than a comparison of the agents to one another. Mixed treatment comparisons were performed to compare one agent to another. In this analysis, macitentan outperformed bosentan in a direct comparison of 6MWD. The other agents, when compared directly using Bayesian statistical methods, did not suggest superiority of one over another with regards to 6MWD or clinical worsening.
While potentially interesting, the above analysis does not replace direct comparison of the agents in a randomized controlled fashion. Thus, it is impossible to conclude that one ERA is superior to another. However, it is important to note that the SERAPHIN trial is the only study that assessed the effect of the drug for a long period of time (greater than 80 weeks) with clinically important endpoints of PAH-related morbidity and mortality. Other trials of ERAs have been short-term assessments on the order of 12 weeks and looked at endpoints such as 6MWD.
Safety and tolerability
Adverse events in clinical trials
As noted earlier, the MUSIC trial enrolled a total of 178 patients, 91 of whom completed more than 12 months of daily therapy with macitentan. Adverse events in this trial, which included IPF worsening, respiratory failure, pneumonia, and hypoxia, were similar between the macitentan and placebo group. In the SERAPHIN trial, similar numbers of patients in the three treatment groups (placebo, 3 mg macitentan, and 10 mg macitentan) discontinued the study drugs due to undesired adverse effects. The most frequently occurring adverse events across all treatment arms included worsening of PAH, upper respiratory infection, peripheral edema, nasopharyngitis, right ventricular failure, headache, anemia, dizziness, bronchitis, dyspnea, and cough. Rates of headache (8.8% placebo, 13.2% 3 mg, 13.6% 10 mg), nasopharyngitis (10.4% placebo, 14.8% 3 mg, 14% 10 mg), bronchitis (5.6% placebo, 8% 3 mg, 11.6% 10 mg), and anemia (3.2% placebo, 8.8% 3 mg, 13.2% 10 mg) were increased in the macitentan treatment groups and one patient in each group discontinued the drug due to anemia. The most common side effects and adverse reactions to the ERA class of medications are abnormalities in aminotransferase levels, peripheral edema, and anemia, and these are discussed in detail later (Figure 4).
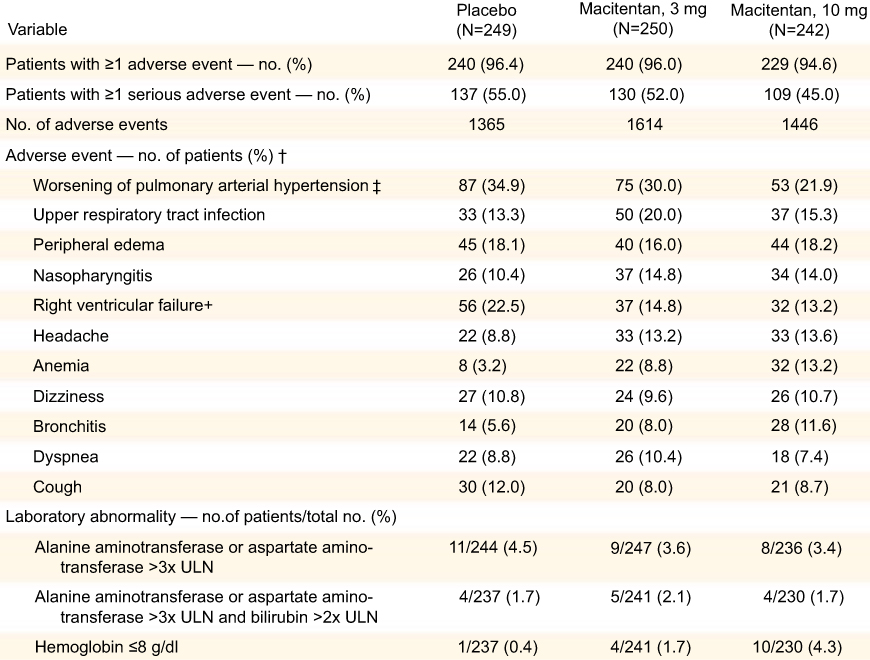 |
Figure 4 Adverse events, abnormalities in liver function tests and hemoglobin. Adverse events in the placebo, 3 mg, and 10 mg arms in the SERAPHIN trial, as well as rates of liver function abnormalities and anemia. From New England Journal of Medicine, Pulido T, Adzerikho I, Channick RN, et al, Macitentan and morbidity and mortality in pulmonary arterial hypertension, 369, 9, 809–818. Copyright © (2013) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.5 |
ALT/AST elevation
In the Phase II MUSIC Trial, the rates of aminotransferase elevations greater than three times the upper limit of normal were similar between groups. In SERAPHIN, such aminotransferase elevations were also similar between the three treatment groups.27 Interestingly, 4.5% of the patients in the placebo group, 4% in the 3 mg macitentan group, and 3.4% in the 10 mg macitentan group had these elevations of aminotransferases – rates decreasing as the dose of the drug increased, possibly highlighting a treatment effect. It has been hypothesized that this may be due to improved treatment of PAH and thus reduction in the rates of congestive hepatopathy with increasing doses of macitentan. The number of patients with an elevation of total bilirubin twice the upper limit of normal and aminotransferase elevation three times the upper limit of normal was the same across treatment groups. This pattern of elevation occurred in 1.7% of the patients treated with placebo, 2.1% of the patients in the 3 mg macitentan group, and 1.7% of the 10 mg macitentan group, suggesting that increasing doses of macitentan does not increase the risk for significant drug-induced liver injury. These rates of liver test abnormalities are similar to rates seen with the use of ambrisentan, and a significantly lower risk as compared to bosentan.35 The mechanism behind the development of hepatotoxicity is somewhat unclear. In in-vitro studies, it is suggested that the majority of the hepatotoxicity is a result of the ETB receptor antagonism, which results in severe cholestasis by an effect on several hepatobiliary transporters. There also appears to be enhanced portal sinusoid constriction, which further compounds the issue of cholestasis.36 The effect on the ETB receptor cannot be the sole mechanism for hepatotoxicity, however, as sitaxsentan was 6500 times more selective for the ETA receptor and was removed from the market for fatal hepatotoxicity. As noted earlier, the SERAPHIN trial did not reveal an increased rate of liver toxicity in patients treated with macitentan compared to placebo.
Peripheral edema
The MUSIC Trial revealed that the macitentan group had increased rates of peripheral edema compared to the placebo group, which is a previously known effect of the ERA class of drugs.24 In MUSIC, 11.8% of the treated patients versus 6.8% of the placebo-treated patients developed peripheral edema. Interestingly, in SERAPHIN, the rates of peripheral edema were similar among the three treatment groups, all approximately 20%. Peripheral edema was decreased in patients with WHO functional class III or IV disease treated with 10 mg daily of macitentan as compared to placebo, as well as in patients who were already receiving PAH treatment (with PDE-5i predominantly). Conversely, in patients age ≥65, the occurrence of peripheral edema was increased in those treated with macitentan as opposed to placebo. Nevertheless, these rates of peripheral edema are lower than seen with the other ERAs available in the United States.35 A large meta-analysis of 24 randomized trials looking at the clinical safety of ERAs was published in 2016 and suggested that the rates of peripheral edema are increased with the use of both bosentan and ambrisentan compared to placebo but that in real-world use this may not be the case with macitentan (RR 1.08).37 The exact mechanism for the development of peripheral edema is unknown. Peripheral edema appears to be more pronounced in ERAs selective to the ETA receptor.38 One possible mechanism of this appears to be activation of plasma vasopressin via stimulation of the uninhibited ETB receptors, resulting in increased plasma volume.39 Another proposed mechanism involves increased sodium retention in the renal collecting duct with blockade of the ETA and ETB receptors, resulting both in hypertension and fluid retention in knock-out mouse models.40 Of note, in a post-hoc analysis of patients who developed peripheral edema while taking ambrisentan, there was a significant reduction in brain natriuretic peptide (BNP) both in patients who developed edema and in those who did not, suggesting perhaps that the mechanism for edema is not related to cardiac dysfunction.41 It is as yet unknown if a similar effect on BNP is seen with macitentan use.
Anemia
The development of anemia has been an observed effect with prior ERAs, and this too was seen with the use of macitentan in both the MUSIC (10.9% vs 0% in the macitentan vs placebo groups) and SERAPHIN trials. The effect was dose-dependent in SERAPHIN, with worsening anemia with increasing doses of macitentan, hemoglobin stabilization by 12 weeks of therapy, and reversibility of anemia when the drug was discontinued.35,36 Anemia was an adverse event for 13.2% of the patients in the 10 mg group, and severe anemia was the cause for drug discontinuation for one patient in each of the treatment arms. Patients who experienced a significant decline in their hemoglobin to levels <10 g/dL often had a personal history of anemia or experienced this decline in the setting of clinically relevant events such as bleeding, vitamin/mineral deficiency, or renal failure. There is no bone marrow toxicity resulting in impaired red blood cell production, nor is there increased destruction via hemolysis to explain the anemia observed with macitentan or the other ERAs. Anemia is hypothesized to be mostly related to an increase in plasma volume.42
Post-marketing reported adverse events
Following initial “new drug application” approval of macitentan in the United States, the FDA required the creation of a registry of patients prescribed macitentan, to be followed for at least 1 year.43 Thus, the Opsumit Users (OPUS) registry was created in April 2014 to be a large (5000 patients) observational cohort study of patients newly prescribed macitentan in the real-world setting. The estimated completion date for this registry is in the summer of 2019, and thus final data are not yet available.44 However, several abstracts have been presented to begin to summarize some of the findings in this large registry.
One- and two-year follow-up data published in abstracts in 2015, 2016, and 2017, suggested that the rate of adverse events was similar to that seen in the clinical trial setting. The rate of liver-related adverse events in these reports was around 3%, and the other most common adverse events were dyspnea and peripheral edema. These adverse events were the most common reason for drug discontinuation, which was in the range of 15–20%. Of 718 patients, 15 patients experienced hepatic adverse events that were deemed to be of special interest; each of these events was evaluated by the Independent Liver Safety Data Review Board and were all felt to be unrelated to macitentan treatment (Figure 5).45 These reports also suggested that the majority of the use of macitentan was as combination therapy, most commonly with PDE-5i, and was well tolerated.46,47
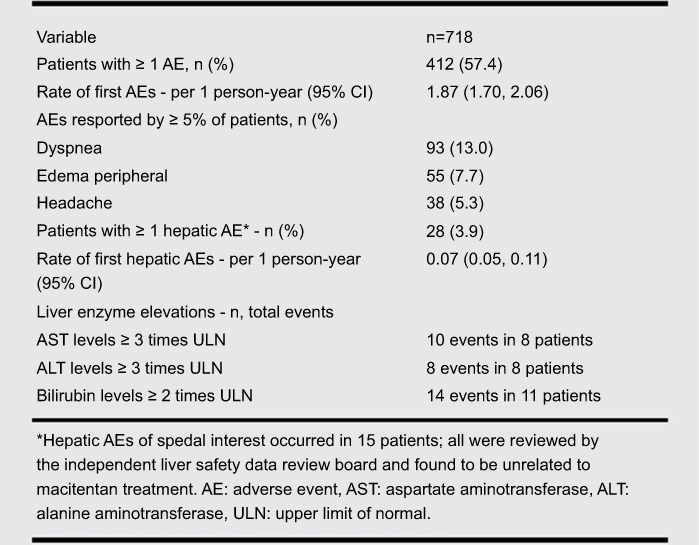 |
Figure 5 Rates of adverse events and liver function abnormalities with the use of macitentan in a real-world setting. Data from the OPUS registry evaluating adverse events and liver function test abnormalities with the use of 10 mg daily of macitentan in a real-world setting. Reprinted from The Journal of Heart and Lung Transplantation, 36, 4, Kim NH, Bergmark BA, Zelniker TA, et al, OPUS registry: safety and tolerability of macitentan in a real-world setting, S20-S21, Copyright (2017), with permission from Elsevier.45 |
A report of the subset of patients with PAH related to systemic sclerosis suggested that macitentan was also well tolerated in this subgroup. There was a lower rate of drug discontinuation in this subgroup, though the rates of adverse events of dyspnea and peripheral edema were slightly higher as compared to the PAH cohort as a whole. Patients with PAH related to systemic sclerosis had similar rates of hepatic dysfunction and PAH-related hospitalization compared to the overall cohort.48 A gender subgroup analysis suggested that survival is better in female patients.49
Pregnancy outcomes
The guidelines for administration of macitentan require that female patients of reproductive potential have a negative pregnancy test prior to initiation of macitentan and then monthly while undergoing treatment. It is required that women utilize a reliable form of contraception during and for 1 month following cessation of treatment. There is also some evidence in animal models that macitentan may cause abnormalities in spermatogenesis which could be transmitted to a fetus.50 At all doses tested, macitentan caused cardiovascular abnormalities and mandibular arch fusion abnormalities in rabbit and rat fetuses. At 5 times the human exposure dose, macitentan reduced fetal survival and offspring reproductive potential when administered to female rats. At 218 times the human exposure dose, macitentan caused fetal death in rabbits.51
Tolerability
Switch to macitentan from other ERA
With available data suggesting decreased side effects, once daily dosing, reduction in lab monitoring, and fewer drug–drug interactions with macitentan as opposed to bosentan, one study describing the patient tolerability of a direct switch from one agent to the other was published.52 This was a retrospective assessment of 24 patients who switched from bosentan to macitentan over a two-year period. Patients were instructed to take their first macitentan dose 12 to 24 hrs after their last bosentan dose. In this group of patients, 6 had idiopathic PAH, 7 related to underlying connective tissue disease, 5 related to congenital heart disease, 2 had PAH related to anorexigen use, 2 had PH following pulmonary thromboendarterectomy for CTEPH, and 2 had portopulmonary hypertension. Data were obtained just prior to the switch and at a 6-month follow-up visit, and included 6MWD, assessment of functional class, hemodynamics as assessed by echocardiography, laboratory monitoring (BNP, aminotransferase levels) as well as monitoring of side effects. 6MWD was unchanged, as was the Borg dyspnea score and oxygen saturation. WHO functional class remained unchanged except in two patients, one of whom had improvement in functional class and the other with deterioration. BNP and liver function testing remained stable. The presence of lower extremity edema remained unchanged. Hemodynamic parameters of right atrial pressure, cardiac output, cardiac index, right ventricular systolic pressure, or the presence of a pericardial effusion were similarly not influenced by the transition from bosentan to macitentan. The stability of these findings was preserved in patients on background PDE-5i therapy. Two patients in this cohort had to be switched back to bosentan. One was due to elevation of aminotransferase levels to greater than 5 times the upper limit of normal in a patient with portopulmonary hypertension. The second was due to tachyarrhythmias that developed after the initial switch in a patient with scleroderma. This report supports that a rapid transition between ERAs was well tolerated even in patients on background therapy with PDE-5i.
As noted earlier, bosentan was the only medication studied for the treatment of PAH related to congenital heart disease. In 2017, the results of a series of 43 patients, 75% of whom had Eisenmenger syndrome, who were switched from bosentan to macitentan were published.53 These patients were previously on bosentan for a median of 7.3 years and were prospectively observed after the decision to switch to macitentan. After 6 months of therapy, outcomes including heart failure, syncope, WHO functional class, 6MWD, oxygen saturation, NT-proBNP, and echocardiographic parameters. At baseline, approximately half of the patients were in WHO functional class II, the rest in class III. The average 6MWD was 394 m. After 6 months of therapy with macitentan, there were fewer patients in WHO functional class III (now in class II), NT-proBNP decreased significantly from 723 to 488 ng/L and a significant improvement in tricuspid annular plane systolic excursion (TAPSE). There was no difference in hospitalization or 6MWD. The switch was well tolerated. Two patients had a drop in hemoglobin without clinical consequences, and one patient had transient elevation of transaminases that resolved spontaneously and also had no clinical consequences. A previous trial evaluating the efficacy and safety of macitentan in patients with PAH due to congenital heart disease included 15 total patients, 9 of whom were also switched from bosentan to macitentan.54 Similar to the trial by Blok et al earlier, this switch was well tolerated with few adverse events.
Macitentan as part of combination therapy
There has been increasing attention given to the use of combination therapy in PAH to improve outcomes in patients with more severe disease. As part of the SERAPHIN trial, patients receiving background therapy were identified as a pre-specified subgroup. The majority of patients were in this group, as 63.7% of were on an additional agent for the treatment of PAH at the time of enrollment. The overwhelming majority of patients, 97.4%, in both the macitentan and placebo groups were receiving PDE-5 inhibitors as their background therapy. Similar to the overall trial, there was a statistically significant reduction in morbidity/mortality of 38% in the subgroup receiving background therapy plus macitentan as compared to placebo. This subgroup also had a reduction in the risk of PAH-related hospitalization by 37.4%, improvement in 6MWT and functional class, and similar effects on quality of life as the overall SERAPHIN population. Importantly, adverse events reported in patients receiving combination therapy were similar to patients receiving background therapy (ie, PDE-5i) and placebo. Rates of worsening PAH and RV failure were increased in patients on monotherapy (PDE-5i and placebo) compared to combination therapy. Anemia was increased in the combination therapy group (PDE-5i and macitentan), as was the rate of bronchitis. The rate of study drug discontinuation was similar between the two groups as well. Overall, macitentan was well tolerated as part of combination therapy with PDE-5i for PAH.55 Given increasing realization of the importance and potential significant clinical improvements in patients treated up front with combination therapy, macitentan is currently being investigated as an agent in initial triple therapy, in the ongoing Efficacy and Safety of Initial Triple Versus Initial Dual Oral Combination Therapy in Patients With Newly Diagnosed Pulmonary Arterial Hypertension (TRITON) study.56
Conclusion
Since approval, macitentan has proven to be a well-tolerated and effective medication for the treatment of pulmonary arterial hypertension. In addition to decreasing PAH-related morbidity and mortality, data support its beneficial effect in reducing PAH-related hospitalizations, improving hemodynamics, improving health-related quality of life, and potential efficacy in chronic thromboembolic pulmonary hypertension. Macitentan is generally well tolerated – adverse events which include liver function abnormalities, peripheral edema, and anemia occur at similar rates as was seen in the clinical trial setting. The transition to macitentan from a different ERA is also well tolerated. The majority of the use of macitentan is as part of combination therapy, typically with a PDE-5i. Real-world and trial data suggest that this strategy is effective and safe.
Disclosure
RNC has served as a consultant for and received research grants from Actelion, Bayer, United Therapeutics and Arena. The authors report no other conflicts of interest in this work.
References
1. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi:10.1016/j.jacc.2013.10.029
2. McGoon MD, Benza RL, Escribano-Subias P, et al. Pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D51–D59. doi:10.1016/j.jacc.2013.10.023
3. Galiè N, Humbert M, Vachiery, JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119.
4. Hoeper MM, Wedzicha JA, Iversen M, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50:1700740. doi:10.1183/13993003.00711-2017
5. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. doi:10.1056/NEJMoa1213917
6. Rubin LJ. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sci. 2012;91:517–521. doi:10.1016/j.lfs.2012.07.033
7. Yanagisawa M, Inoue A, Takuwa Y, Mitsui Y, Kobayashi M, Masaki T. The human preproendothelin-1 gene: possible regulation by endothelial phosphoinositide turnover signaling. J Cardiovasc Pharmacol. 1989;13:S13–S17.
8. Clarke J, Benjamin NI, Larkin SW, Webb DJ, Davies GJ, Maseri A. Endothelin is a potent long-lasting vasoconstrictor in men. Am J Physiol. 1989;257:H2033–H2035. doi:10.1152/ajpheart.1989.257.6.H2033
9. Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S62–S67. doi:10.1016/j.jacc.2004.02.042
10. Stewart D, Levy R, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertesnion: marker or mediator of disease. Ann Intern Med. 1991;114:464–469. doi:10.7326/0003-4819-114-6-464
11. Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–1739. doi:10.1056/NEJM199306173282402
12. Davie N, Haleen SJ, Upton PD. et al. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. 2002;165:398–405. doi:10.1164/ajrccm.165.8.2106104
13. Wilson DW, Segall HJ, Pan LC, Lamé MW, Estep JE, Morin D. Mechanisms and pathology of monocrotaline pulmonary toxicity. Crit Rev Toxicol. 1992;22:307–325. doi:10.3109/10408449209146311
14. Jasmin J-F, Lucas M, Cernacek P, Dupuis J. Effectiveness of a nonselective ET A/B and a selective ET A antagonist in rats with monocrotaline-induced pulmonary hypertension. Circulation. 2001;103:314–318. doi:10.1161/01.CIR.103.2.314
15. Lai Y-C, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–130. doi:10.1161/CIRCRESAHA.115.301146
16. Selej M, Romero AJ, Channick RN, Clozel M. Development of macitentan for the treatment of pulmonary arterial hypertension: macitentan for the treatment of pulmonary arterial hypertension. Ann N Y Acad Sci. 2015;1358:68–81. doi:10.1111/nyas.12856
17. Hong IS. Macitentan for the treatment of pulmonary arterial hypertension. Ann Pharmacother. 2014;48:538–547. doi:10.1177/1060028013518900
18. Iglarz M, Bossu A, Wanner D, et al. Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension. Life Sci. 2014;118:333–339. doi:10.1016/j.lfs.2014.02.018
19. Sidharta PN, van Giersbergen PLM, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67:977–984. doi:10.1007/s00228-011-1043-2
20. Sidharta PN, van Giersbergen PLM, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects: the journal of clinical pharmacology. J Clin Pharmacol. 2013; n/a-n/a. doi:10.1002/jcph.152
21. Paul GA, Gibbs JSR, Boobis AR, Abbas A, Wilkins MR. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br J Clin Pharmacol. 2005;60:107–112. doi:10.1111/j.1365-2125.2005.02383.x
22. Burgess G, Hoogkamer H, Collings L, Dingemanse J. Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur J Clin Pharmacol. 2008;64:43–50. doi:10.1007/s00228-007-0408-z
23. Sidharta PN, van Giersbergen PLM, Wolzt M, Dingemanse J. Investigation of mutual pharmacokinetic interactions between macitentan, a novel endothelin receptor antagonist, and sildenafil in healthy subjects: pharmacokinetic interactions between macitentan and sildenafil. Br J Clin Pharmacol. 2014;78:1035–1042. doi:10.1111/bcp.12447
24. Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42:1622–1632. doi:10.1183/09031936.00104612
25. Channick RN, Delcroix M, Ghofrani HA, et al. Effect of macitentan on hospitalizations. JACC Heart Fail. 2015;3:1–8. doi:10.1016/j.jchf.2014.07.013
26. Galiè N, Jansa P, Pulido T, et al. SERAPHIN haemodynamic substudy: the effect of the dual endothelin receptor antagonist macitentan on haemodynamic parameters and NT-proBNP levels and their association with disease progression in patients with pulmonary arterial hypertension. Eur Heart J. 2017;38:1147–1155. doi:10.1093/eurheartj/ehx025
27. Mehta S, Sastry BKS, Souza R, et al. Macitentan improves health-related quality of life for patients with pulmonary arterial hypertension. Chest. 2017;151:106–118. doi:10.1016/j.chest.2016.08.1473
28. Vanhoof JMM, Page RLEA, Ambardekar AV, Brieke A, Lindenfeld J. Emotional symptoms and quality of life in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2014;33:800–808. doi:10.1016/j.healun.2014.08.022
29. Vachiéry J-L, Delcroix M, Al-Hiti H, et al. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J. 2018;51:1701886. doi:10.1183/13993003.01886-2017
30. Ghofrani H-A, Casarin A, Ely EW, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med. 2017;5:785–794. doi:10.1016/S2213-2600(17)30234-5
31. Ghofrani H-A, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–329. doi:10.1056/NEJMoa1209657
32. Galiè N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114:48–54. doi:10.1161/CIRCULATIONAHA.106.630715
33. Gatzoulis MA, Landzberg M, Beghetti M, et al. Evaluation of macitentan in patients with Eisenmenger syndrome. Circulation. 2019;139:51–63. doi:10.1161/CIRCULATIONAHA.118.033575
34. Duo-Ji -M-M, Long Z-W. Comparative efficacy and acceptability of endothelin receptor antagonists for pulmonary arterial hypertension: a network meta-analysis. Int J Cardiol. 2017;234:90–98. doi:10.1016/j.ijcard.2016.12.092
35. Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13:391–405. doi:10.1517/14740338.2014.859674
36. Davila C, Monaco T. Safety, efficacy, and clinical utility of macitentan in the treatment of pulmonary arterial hypertension. Drug Des Devel Ther. 2016;1675. doi:10.2147/DDDT.S88612
37. Wei A, Gu Z, Li J, et al. Clinical adverse effects of endothelin receptor antagonists: insights from the meta‐analysis of 4894 patients from 24 randomized double‐blind placebo‐controlled clinical trials. J Am Heart Assoc. 2016;5. doi:10.1161/JAHA.116.003896
38. Trow T, Fares W, Kholdani C. Macitentan for the treatment of pulmonary arterial hypertension. Vasc Health Risk Manag. 2014;665. doi:10.2147/VHRM.S33904
39. Vercauteren M, Strasser D, Vezzali E. et al. Vasopressin in involved in endothelin receptor antagonist-induced fluid retention in rat. Differential effect of selective ETA and dual ETA/ETB receptor antagonists. Eur Respir J. 2012;40:716s. doi:10.1183/09031936.00213711
40. Ge Y, Bagnall A, Stricklett PK, Webb D, Kotelevtsev Y, Kohan DE. Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol- Ren Physiol. 2008;295:F1635–F1640. doi:10.1152/ajprenal.90279.2008
41. Shapiro S, Chun L, Patton KK, Matsushita MM, Page RL, Raskind WH. Frequency of edema in patients with pulmonary arterial hypertension receiving ambrisentan. Am J Cardiol. 2012;110:1373–1377. doi:10.1016/j.amjcard.2012.07.033
42. Gabbay E, Fraser J, McNeil K. Review of bosentan in the management of pulmonary arterial hypertension. Vasc Health Risk Manag. 2007;3:887–900.
43. Center for Drug Evaluation And Research; Approval Package for Macitentan. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204410Orig1s000Approv.pdf. Accessed December 2, 2018.
44. OPsumit users registry - full text view - ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02126943.
45. Kim NH, Bergmark BA, Zelniker TA, et al. OPUS registry: safety and tolerability of macitentan in a real-world setting. J Heart Lung Transplant. 2017;36:S20–S21. doi:10.1016/j.healun.2017.06.015
46. Kim NH, Chin KM, Muros-Le Rouzic E. et al. OPsumit® USers Registry (OPUS): insights into the safety and tolerability of Opsumit® | D54. CLINICAL PULMONARY HYPERTENSION. Am J Respir Crit Care Med. 2016;A7396.
47. Chin KM, Channick RN, Kim NH, et al. OPUS registry: treatment patterns with macitentan in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2017;195:A2299.
48. Chin KM, Kim N, Channick R, Muros-Le Rouzic E, Selej M, McLaughlin V. OPUS registry: treatment patterns and safety of macitentan in patients with Pulmonary Arterial Hypertension Associated with Systemic Sclerosis (PAH-SSc). Chest. 2017;152:A999–A1000. doi:10.1016/j.chest.2017.08.1036
49. McLaughlin VV, Channick R, Chin KM, et al. The OPSUMIT Users Registry: a Gender Subgroup Analysis of Patient Characetistics, Treatment Patterns and Survival in Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2018;71(Supplement). doi:10.1016/S0735-1097(18)32493-8
50. FDA Opsumit Package Insert. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204410s000lbl.pdf.
51. Monograph AO. Available from: https://www1.actelion.com/documents/ca/ca-opsumit-procuct-monograph-en.pdf.
52. Safdar Z, Thakur A, Frost A. Tolerability of switch to macitentan from bosentan in pulmonary arterial hypertension. South Med J. 2017;110:223–228. doi:10.14423/SMJ.0000000000000738
53. Blok I, van Riel AC, van Dijk AP, Mulder BJ, Bouma BJ. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: further improvement? Int J Cardiol. 2017;227:51–52. doi:10.1016/j.ijcard.2016.11.211
54. Herbert S, Gin-Sing W, Howard L, Tulloh RMR. Early experience of macitentan for pulmonary arterial hypertension in adult congenital heart disease. Heart Lung Circ. 2017;26:1113–1116. doi:10.1016/j.hlc.2016.12.011
55. Jansa P, Pulido T. Macitentan in pulmonary arterial hypertension: a focus on combination therapy in the SERAPHIN trial. Am J Cardiovasc Drugs. 2018;18:1–11. doi:10.1007/s40256-017-0255-y
56. The efficacy and safety of initial triple versus initial dual oral combination therapy in patients with newly diagnosed pulmonary arterial hypertension - full text view - ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02558231.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.