")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Safety and pharmacokinetics of dicloxacillin in healthy Chinese volunteers following single and multiple oral doses
Authors Wu G, Zheng Y, Zhou H, Hu X, Liu J, Zhai Y, Zhu M, Wu L, Shentu J
Received 9 July 2015
Accepted for publication 6 September 2015
Published 16 October 2015 Volume 2015:9 Pages 5687—5695
DOI https://doi.org/10.2147/DDDT.S92117
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Guolan Wu, Yunliang Zheng, Huili Zhou, Xingjiang Hu, Jian Liu, You Zhai, Meixiang Zhu, Lihua Wu, Jianzhong Shentu
Research Center for Clinical Pharmacy, State Key Laboratory for Diagnosis and Treatment of Infectious Disease, First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
Background: Dicloxacillin, a semisynthetic isoxazolyl penicillin antibiotic, has antimicrobial activity against a wide variety of gram-positive bacteria including Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumonia, Streptococcus epidermidis, Streptococcus viridans, Streptococcus agalactiae, and Neisseria meningitidis. The objective of this study was to evaluate the safety and pharmacokinetic profile of dicloxacillin after single and multiple oral dose in healthy Chinese volunteers.
Methods: A single-center, open-label, randomized, two-phase study was conducted in 16 subjects. In the single-dose phase, subjects were randomly assigned to receive single doses of 0.25, 0.5, 1.0, and 2.0 g of dicloxacillin sodium capsule in a 4-way crossover design with a 5-day washout period between administrations. In the multiple-dose phase, subjects were assigned to receive 0.25 or 0.5 g every 6 hours for 3 days in a 2-way crossover design. Plasma and urine pharmacokinetic samples were assayed by a validated high-performance liquid chromatography-tandem mass spectrometry method. Pharmacokinetic parameters were calculated and analyzed statistically. Safety assessments were conducted throughout the study.
Results: Following a single oral dose of 0.25–2.0 g dicloxacillin sodium, the maximum plasma drug concentration (Cmax) and the corresponding values for the area under the concentration–time curve from 0 to 10 hours (AUC0–10 h) increased in a dose-proportional manner. The mean elimination half-life (t1/2) was in the range of 1.38–1.71 hours. Dicloxacillin was excreted in its unchanged form via the kidney, with no tendency of accumulation, and varied from 38.65% to 50.10%. No appreciable accumulation of drug occurred with multiple oral doses of dicloxacillin. No serious adverse events were reported. Adverse events were generally mild.
Conclusion: Dicloxacillin was safe and well tolerated in the volunteers and displayed linear increases in the Cmax and AUC0–10 h values.
Keywords: dicloxacillin, safety, tolerability, pharmacokinetics, healthy volunteers
Introduction
Dicloxacillin is a semisynthetic isoxazolyl penicillin antibiotic. It can be given by injection or by mouth as its sodium salt has significant activity against gram-positive β-lactamase-producing microorganisms.1 Compared with isoxazole penicillins in current clinical use, namely, oxacillin, cloxacillin, and flucloxacillin, dicloxacillin has been proved as active against Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumonia, Streptococcus epidermidis, Streptococcus viridans, Streptococcus agalactiae, and Neisseria meningitidis.2–4 It is currently used in the treatment of bacterial infections such as pneumonia and bone, ear, skin, and urinary tract infection.5–8
Dicloxacillin is characterized by time-dependent bactericidal activity, which means that drug concentration higher than the minimum inhibitory concentration (MIC) for an adequate percentage of time in a dosing interval (%T > MIC) must be maintained for efficacy. For these agents, increasing the concentration does not necessarily increase the rate or extent of killing; the best results are achieved by optimizing the duration of exposure to effective concentrations. Previous study indicated that a dosing regimen of 1 g dicloxacillin every 6 hours should be effective for S. aureus infections with a mean MIC value of 0.125 μg·mL−1 (achieving 67%T > MIC).9
T > MIC is dependent on the half-life (t1/2) of dicloxacillin and its serum concentration. Previous studies have proved that it is rapidly absorbed after oral administration, with the higher and more prolonged serum concentration than other isoxazole penicillins.10,11 A previous pharmacokinetic study reported that the mean absolute bioavailability of dicloxacillin was higher than cloxacillin (48.8% versus 36.9%) in healthy volunteers, with the higher cumulative urinary excretion in their unchanged form after oral administration of 2 g dicloxacillin or 2 g cloxacillin (74.1% versus 48.5%).12 The t1/2 was slower than cloxacillin (42 minutes versus 33 minutes). Furthermore, James et al reported that the higher protein binding of dicloxacillin did not prevent it having equal or superior antistaphylococcal activity, comparing with cloxacillin, oxacillin, and nafcillin, when given at equal doses.13–15
Several studies on its pharmacokinetic properties have been carried out in Western population, including healthy subjects16,17 of young and old age, children,18 and hemodialysis subjects.12 These studies revealed that its pharmacokinetics may vary between healthy subjects and patients, as well as between younger and older subjects, so further investigation of dicloxacillin pharmacokinetics was required for these different subgroups. Because the drug is not marketed in mainland China, its pharmacokinetics in Chinese population is poorly understood. Furthermore, race, age, body weight, hepatic, and renal functions or co-administered drugs, genetic polymorphism of drug metabolism enzymes, and drug transporters are important causes of pharmacokinetic variation, which may affect their safety, efficacy, and dosage. Therefore, the ability to extrapolate clinical efficacy and safety data from one region to another requires an understanding of ethnic similarities and differences in drug pharmacokinetics. The present study was conducted to determine the pharmacokinetics and tolerability of a generic formulation of dicloxacillin sodium in healthy Chinese adult subjects. The effect of sex on the pharmacokinetics of dicloxacillin was evaluated as a secondary objective. The study was conducted to meet China Food and Drug Administration requirements for marketing of the new generic formulation. These results will facilitate the understanding of pharmacological characteristics of dicloxacillin and provide a basis for better clinical use of the drug in Chinese population in the future.
Materials and methods
Subjects
Healthy male and female subjects, aged 18–45 years, with body mass index (BMI) between 18 and 25 kg·m−2, were eligible to participate in this study. Health status was determined by medical history, physical examination, and laboratory tests at screening. All female subjects were required to have a negative pregnancy test result at screening and expected to use appropriate contraceptives throughout the study. The subjects had not received any medications within 14 days prior to the current study and had not participated in any study within 3 months prior to the current study. Subjects who had a history of hypersensitivity, allergy, serious adverse drug reaction, especially to any β-lactam antibiotic, were excluded from the study. Subjects with positive tests for human immunodeficiency virus, hepatitis B virus surface antigen, or anti-hepatitis C virus antibody were also excluded.
Study design
This study has been registered at www.chictr.org.cn under registration No ChiCTR-TTRCC-13003696. This single-center, open-label, two-part (parts 1 and 2) study was conducted in healthy adult subjects. It was conducted from October 8, 2013 to January 25, 2014.
Part 1 was a randomized 4-way crossover, open-label, single-dose study in which 16 subjects received each of the following four treatments in a randomized sequence: 0.25–0.5–1.0–2.0 g, 0.5–1.0–2.0–0.25 g, 1.0–2.0–0.25–0.5 g, and 2.0–0.25–0.5–1.0 g according to a computer-generated randomization schedule (SAS 9.1, SAS Institute Inc., Cary, NC, USA). Treatments were separated by 5-day washout periods. All subjects fasted (ie, no food and liquid except water) for at least 10 hours prior to administration of study drug and continued fasting for 4 hours thereafter. Water was allowed as needed for up to 2 hours before drug intake and from 2 hours after intake.
In brief, 15 subjects (eight males and seven females) who completed the single-dose phase, and another female volunteer rescreening were enrolled in the multiple-dose phase (Part 2). They were randomly assigned to receive 0.25 or 0.5 g every 6 hours for 3 days in a 2-way crossover design. Subjects were confined to the study center, received standard meals daily (at 8 am, 11 am, and 5 pm) for two consecutive days. At approximately 7 am on day 3, subjects received the last dose of their assigned treatment in the fasted state. Standard meals were provided to all subjects at 4 and 10 hours after study drug administration.
For both parts of this study, dicloxacillin sodium capsules (0.25 g per capsule; lot 9130574) were supplied by Bright Future Pharmaceuticals Factory (Yuen Long, N.T., Hong Kong). Study treatment was administered orally with 240 mL of water, with mouth checks to ensure compliance. Carbonated drinks, alcohol, caffeine, tobacco (or nicotine-containing products), and grapefruit (or other citrus fruit) were not permitted during the study.
The protocol and written informed consent about the study was reviewed and approved by the Ethical Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (approval No: 2013-EC-58). The study was conducted in accordance with the principles of the current revision of the Declaration of Helsinki concerning medical research in humans,19 the International Conference on Harmonization Guideline for Good Clinical Practice,20 the Guideline for Good Clinical Practice, and the Guideline for Pharmacokinetics studies recommended by the China Food and Drug Administration.21,22 Each subject was informed about the nature and risk associated with the study, and has signed an informed consent form before participating.
Safety assessment
Safety evaluations included adverse event (AE) counts, monitoring for serious adverse events (SAEs) with the severity, and relationship to the study drug and regular monitoring of vital signs, physical condition, clinical laboratory tests (eg, hematology parameters, liver function tests [alanine transaminase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase], kidney function tests [blood urea nitrogen and serum creatinine], and urine analysis), and 12-lead electrocardiogram. The physicians were responsible for determining the clinical significance of the AEs. All information, including undesirable symptoms or medical conditions after dosing, were recorded on the case-report form by investigators regardless of the suspected relationship to the study drug.
Pharmacokinetic parameters and assessment
In Part 1 of the study (single dosing), blood samples (4 mL) were collected from an indwelling venous catheter into heparinized tubes before each dose (0 hours) and at 0.167, 0.333, 0.5, 0.75, 1, 1.25, 2, 2.5, 3, 4, 6, 8, and 10 hours after each dose. The blood samples were centrifuged at 3,000× g for 10 minutes at 4°C, and the plasma samples were stored at -70°C until analyzed. Urine samples were collected from 0 hours (pre-dose) and from 0 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 12, and 12 to 24 hours after each treatment. At the end of the interval, the urine bottles were shaken and the overall urinary excretion was measured and noted with accuracy. Approximately 15 mL of urine from each volunteer was collected and stored at -70°C for future experiments.
In Part 2 (0.25 or 0.5 g q6h for a total of nine doses), blood samples were collected at 0.167, 0.333, 0.5, 0.75, 1, 1.25, 2, 2.5, 3, 4, 6, 8, and 10 hours after the last dose. Additional blood samples were obtained before the seventh, eighth, and ninth dose administration to determine the plasma drug concentration at a steady state. Other experimental conditions were the same as in the single-dose phase.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was used to measure the concentrations of dicloxacillin in plasma and urine samples. The LC-MS/MS system included an Agilent 1200 high-performance liquid chromatography and Agilent 6410 triple quadrupole mass spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA). Chromatography was performed with an Agilent ZORBAX SB-Aq column (3.0 mm ×100 mm, 3.5 μm) and the mobile phase was water with 0.5% formic acid (A)–acetonitrile (B) at a flow rate of 0.5 mL·min−1. The gradient elution program: 0–4.5 minutes, 55%A/45%B; 4.6–6.0 minutes, 95%A/5%B; 6.1–8.0 minutes, 55%A/45%B.
Samples were analyzed by LC-MS/MS in multiple reaction monitoring mode using electrospray ionization as the ionization technique. The optimized instrumental parameters for mass spectral acquisition were as follows: capillary voltage was set to 4,000 V; nitrogen was used as a nebulizer gas and nebulizer pressure was set at 35 psi; desolvation gas (nitrogen) was heated to 300°C and delivered at a flow rate of 8 L·min−1. In order to precipitate proteins, plasma samples were pretreated with two volumes of acetonitrile. The resulting supernatant (10 μL) was injected into the LC-MS/MS system for analysis. Calibration curves were constructed at concentrations of 0.1808–72.32 μg·mL−1 for plasma. Plasma samples above the limit of quantitation were diluted and reanalyzed to yield results within the calibrated range. Stability tests showed that plasma sample decomposition was <15% for all conditions examined (6 hours at room temperature, 3 freeze–thaw cycles [-70°C], and 127 days after storage at or below 70°C). Separate sets of analytical quality control samples (for concentrations of 0.3619, 5.424, and 54.24 μg·mL−1) were used to assess assay precision and accuracy. The intra- and inter-day precisions (relative standard deviation, %RSD) were ≤3.77% and ≤6.52%, respectively. The intra- and inter-day recovery rates were 94.02%–108.13% and 95.86%–106.55%, respectively.
Urine samples were diluted using 1 mL of acetonitrile–water (40:60 [vol/vol]), and the supernatant (2 μL) was injected into the LC-MS/MS system for analysis. Two calibration curves were used for analyzing dicloxacillin in urine. The calibration curves showed the linear correlation (R2≥0.9991) within the range of 0.9288–928.8 μg·mL−1 and 461.1–7377.0 μg·mL−1. The intra- and inter-day batch precisions (%RSD) were lower than 10.83%, and the accuracy ranged from 89.18% to 106.40% in terms of percent accuracy.
The assays mentioned above were validated according to US Food and Drug Administration guidelines on bioanalytical method validation.23
The population for pharmacokinetic analysis consisted of all subjects who had received at least one dose of study medication and had an evaluable pharmacokinetic profile. Pharmacokinetic parameters were calculated by a non-compartment method using WinNonlin 6.3 software (Pharsight Corporation, Sunnyvale, CA, USA). The following pharmacokinetic parameters were obtained for Part 1: maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), elimination half-life (t1/2), area under the concentration–time curve from time zero to the last quantifiable concentration (AUC0–10 h), AUC from time zero extrapolated to infinity (AUC0–∞), apparent oral clearance (CL/F), apparent volume of distribution during the terminal elimination phase following extravascular administration (Vz/F). For multiple-dose study, the time to reach peak concentration (Tmax,ss) and maximum and minimum plasma concentration at the steady state (Cmax,ss and Cmin,ss) were determined directly from experimental observations. The accumulation ratio was calculated as the ratio of AUC0–τ,ss to AUC0–τ (single dose), and the fluctuation ratio was calculated by (Cmax,ss - Cmin,ss)/Cavg, where Cavg is the average steady-state drug concentration during multiple dosing, which is calculated as AUC0–τ,ss/τ, where τ is the dosing interval (6 hours). t1/2, CL/F, and Vss/F (volume of distribution at the steady state) were calculated using the same method as for single-dose period. In addition, the amount of drug excreted into the urine from time zero to time 24 hours (Ae0–24) and renal clearance were determined for each single-dose pharmacokinetic group from the urine drug concentration–time data.
Statistical methods
Pharmacokinetic parameters were expressed using arithmetic means and standard deviations, except for Tmax, for which median values and ranges are reported. For single- and multiple-dose studies, descriptive statistical analyses for pharmacokinetic parameters were performed using SPSS (Version 16.0, SPSS Inc., Chicago, IL, USA). To test dose proportionality in the single-dose range studied, log-transformed pharmacokinetic parameters AUC and Cmax were analyzed by using the power model, PK = A × (Dose)β, where PK is the pharmacokinetic parameter, A is the intercept, and β is the dose-proportionality coefficient. A 90% confidence interval (CI) for the dose-proportionality parameter β was then calculated. Note that a slope of 1 would correspond to perfect dose proportionality. A t-test was used to determine whether the multiple-dose pharmacokinetic parameters were consistent with those in the single-dose phase. The results for Tmax were evaluated using the Wilcoxon rank-sum analysis. Data were categorized by sex to determine statistical differences in PK parameters between males and females. P<0.05 was considered statistically significant.
Results
Study participants
In the single-dose study, a total of 16 healthy subjects (eight males and eight females) were enrolled in and completed the study. The mean age (range) of subjects was 22.2 (20–26) years, mean body weight (range) was 58.4 (45.0–69.0) kg, mean height (range) was 1.68 (1.55–1.80) m, and mean BMI (range) was 20.6 (18.7–22.8) kg·m−2. In the multiple-dose study, 16 healthy subjects (eight males and eight females) were aged 20–26 years and had a mean body weight of 58.9±6.0 kg and a mean BMI of 20.7±1.0 kg·m−2. Demographic data for the subjects receiving dicloxacillin sodium are shown in Table 1.
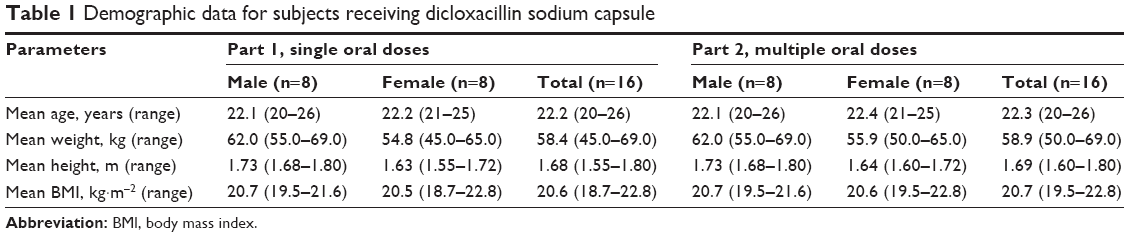 | Table 1 Demographic data for subjects receiving dicloxacillin sodium capsule |
Safety
All randomized subjects were included in the safety analysis. There were no deaths or SAEs. None of the subjects withdrew from the study or were discontinued by the investigators because of AEs.
Single oral dose of dicloxacillin sodium were well tolerated at all dose levels. No SAEs s occurred during the study, and all subjects were in good compliance with the protocol. No clinically significant laboratory results or trends were reported for any dose at any time point. No clinically meaningful changes in physical examinations, vital signs, or electrocardiogram (ECG) studies were apparent during the study.
Multiple oral-dose regimens of dicloxacillin sodium administered to healthy adult subjects for nine doses were well tolerated at the study dose levels. No subjects experienced SAEs, or were withdrawn from the study after dosing due to an AE. No clinically significant changes in physical exams, vital signs, or ECGs were observed at any dose level. Only one subject (1/16, 6.25%) developed leucopenia and neutropenia that were assessed as mild AE; both were suspected to be related to the study drug. The abnormal laboratory results rapidly returned to normal values within 7 days after dosing was completed. No other clinically meaningful shifts or trends were noted for laboratory parameters, including hematology, analyses of serum chemistry (liver function and kidney function), and urinalysis.
Pharmacokinetics
Single-dose administration in healthy volunteers (0.25–2.0 g, oral administration)
The mean plasma concentration–time profile of dicloxacillin following oral administration of a 0.25–2.0 g dose is shown in Figure 1. Pharmacokinetic parameters are presented in Table 2. The mean Cmax values for the 0.25, 0.5, 1.0, and 2.0 g group were 13.62 μg·mL−1, 24.28 μg·mL−1, 45.02 μg·mL−1, and 79.97 μg·mL−1, respectively. The AUC0–10 h values were 31.60 μg·h·mL−1, 61.40 μg·h·mL−1, 108.73 μg·h·mL−1, and 203.29 μg·h·mL−1, respectively. Plasma t1/2 was independent of the dose, averaging 1.51 hours (0.92–2.56 hours). The trend for an increasing t1/2 with increasing dose has been observed, and at the highest dose of 2.0 g, t1/2 was in the range of 1.13–2.56 hours. The apparent volume of distribution and apparent clearance ranged between 9.7 and 40.97 L and 5.33 and 16.89 L·h−1, respectively, over the entire dose range. The rates of drug excreted 24 hours after administration of the four doses were 50.10%, 49.00%, 38.65%, and 42.39%, respectively (Figure 2).
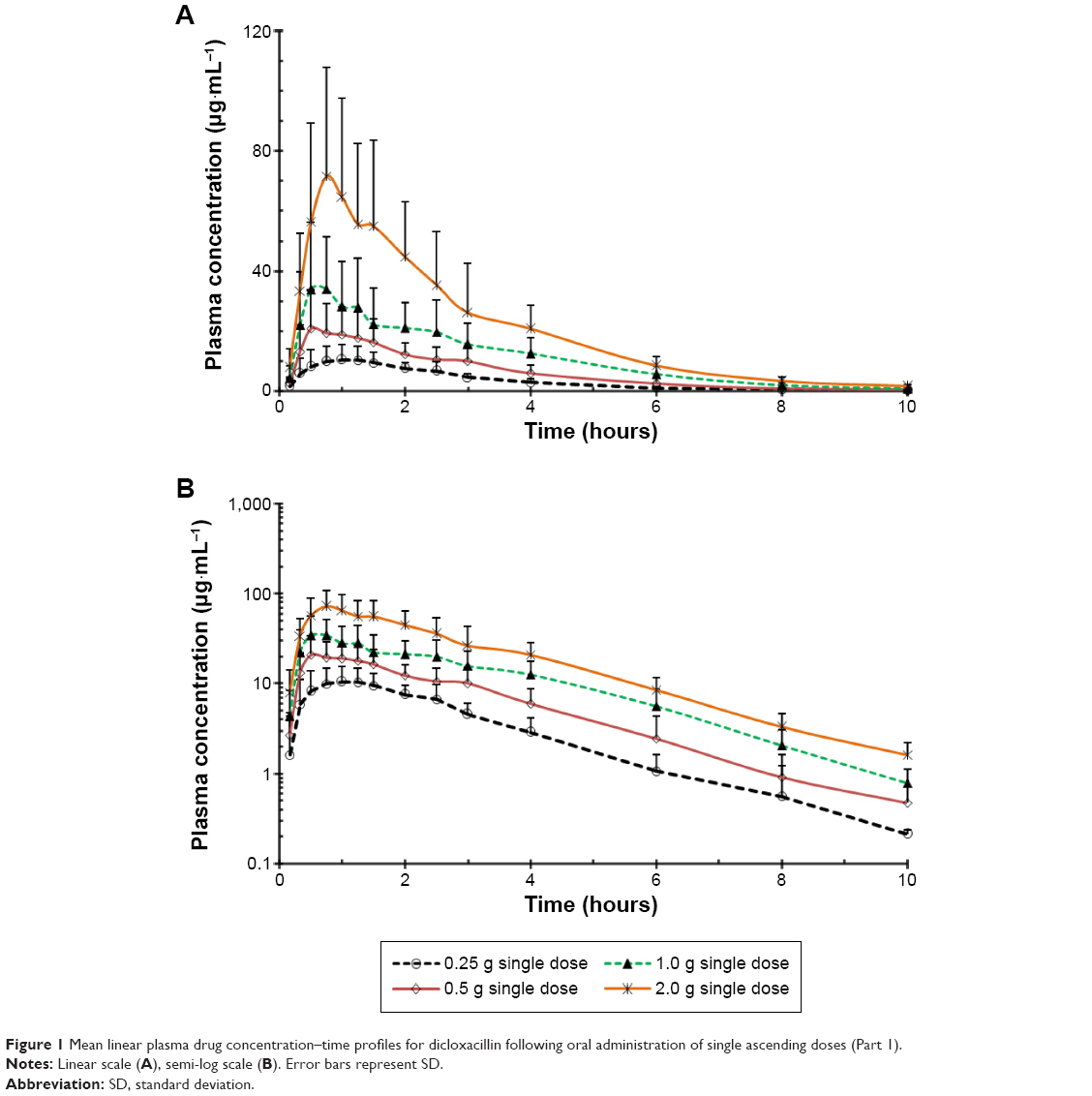 | Figure 1 Mean linear plasma drug concentration–time profiles for dicloxacillin following oral administration of single ascending doses (Part 1). |
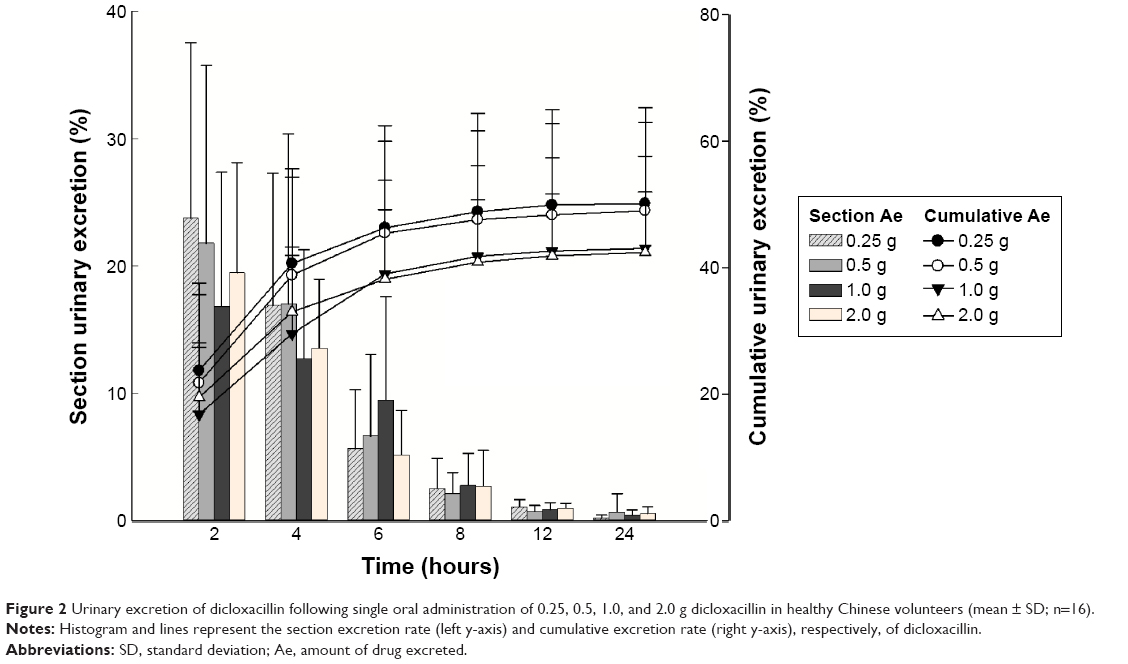 | Figure 2 Urinary excretion of dicloxacillin following single oral administration of 0.25, 0.5, 1.0, and 2.0 g dicloxacillin in healthy Chinese volunteers (mean ± SD; n=16). |
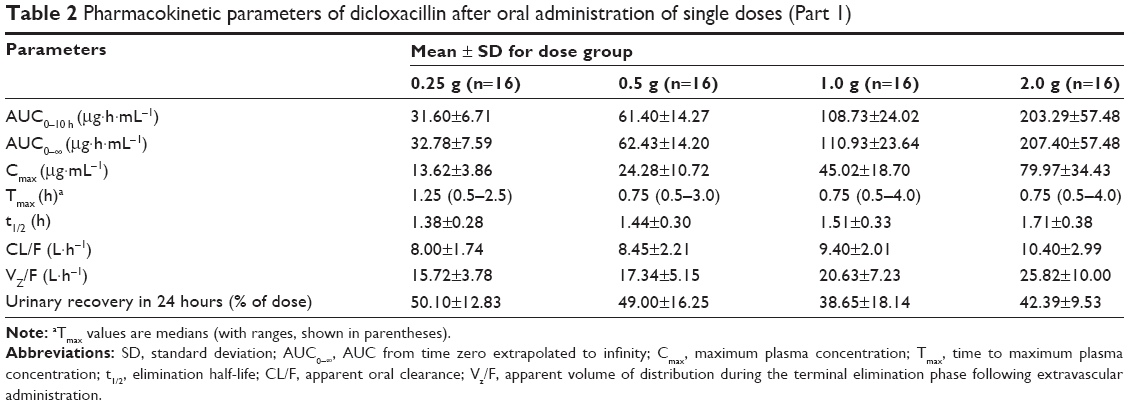 | Table 2 Pharmacokinetic parameters of dicloxacillin after oral administration of single doses (Part 1) |
For the relationship between non-compartmental parameters (AUC0–∞ and/or AUC0–10 h and Cmax) and dose, analysis results indicated that the estimated proportionality coefficients (β) for AUC0–∞, AUC0–10 h, and Cmax were 0.876 (90% CI: 0.812–0.940), 0.881 (90% CI: 0.816–0.945), and 0.828 (90% CI: 0.709–0.948), respectively. This indicated a slightly less than dose-proportional increase of AUC and Cmax in the dose range of 0.25–2.0 g, with a slope β of slightly less than 1.
PK parameters were analyzed by sex. No significant differences in Tmax, t1/2, MRT0–10 h, MRT0–∞, Cmax, CL/F, Vz/F, AUC0–10 h, AUC0–∞, or Ae were detected between male and female subjects who received 0.5, 1.0, or 2.0 g dicloxacillin sodium. In subjects who received 0.25 g dicloxacillin sodium, significant differences in AUC0–∞ (P=0.044) and CL/F (P=0.026) were observed between the two sex. In male volunteers, the mean drug excretion rate 24 hours after administration was 46.89%, 49.36%, 35.77%, and 41.01% in the 0.25 g, 0.5 g, 1.0 g, and 2.0 g groups, respectively. In female subjects, the proportions of drug excreted after 24 hours were 53.31%, 48.63%, 41.52%, and 43.77%. No significant differences in Ae0–24 h (P>0.05) were observed between the male and female subjects.
Multiple-dose administration in healthy volunteers (0.25 or 0.5 g q6h for nine doses, oral administration)
The mean plasma concentration–time profiles of dicloxacillin following multiple oral doses of dicloxacillin sodium are presented in Figure 3 and the pharmacokinetic parameters are summarized in Table 3. The steady state, based on plasma concentration of drug in pre-seventh, -eighth, and -ninth dose samples, was achieved after administration of dicloxacillin for six consecutive doses. Dicloxacillin at doses of 0.25 g q6h and 0.5 g q6h was rapidly absorbed, with Tmax,ss ranging from 0.33 to 3.0 hours after dose across subjects on day 3. The values for Cmax and AUC0–10 h after single- and multiple-dose administration were similar. The mean Cmax,ss values on day 3 were 14.76 μg·mL−1 and 26.54 μg·mL−1, respectively. The corresponding AUC0–10 h values on day 3 were 33.72 μg·h·mL−1 and 62.06 μg·h·mL−1. Plasma t1/2 was independent of the dose and dosing duration, averaging 1.46 hours (range, 0.97–2.35 hours). The accumulation ratio was to found to be in the range of 1.01 and 1.21, indicating that there was no accumulation after multiple doses of dicloxacillin sodium capsules. There were no statistically significant differences in multiple-dose pharmacokinetic parameters between female and male subjects (P>0.05).
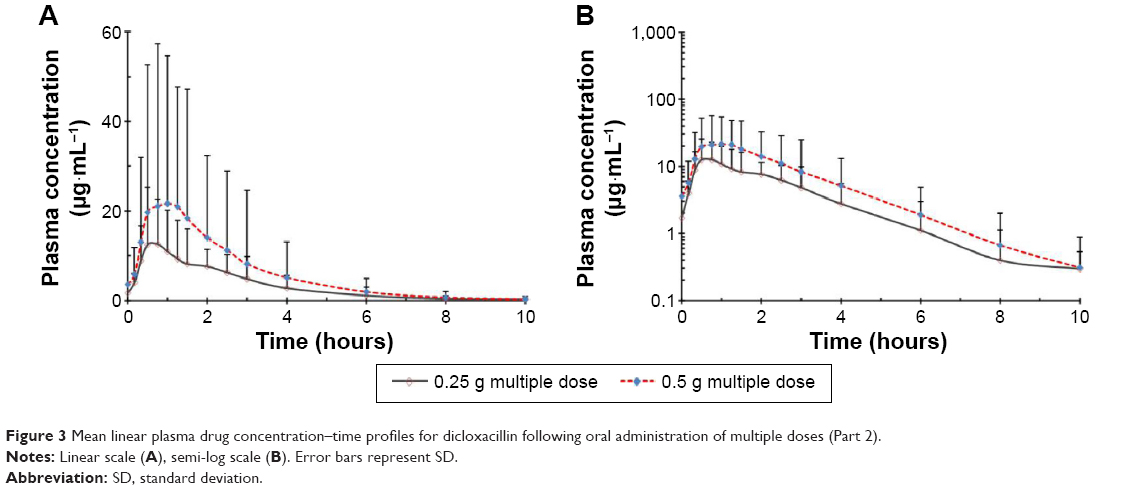 | Figure 3 Mean linear plasma drug concentration–time profiles for dicloxacillin following oral administration of multiple doses (Part 2). |
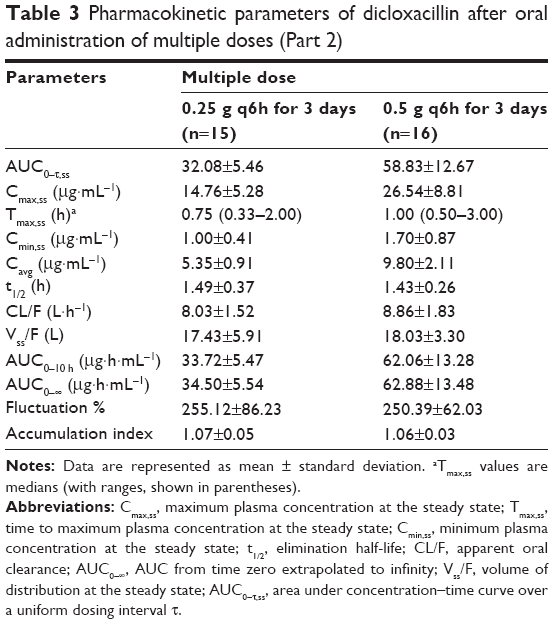 | Table 3 Pharmacokinetic parameters of dicloxacillin after oral administration of multiple doses (Part 2) |
Discussion
To our knowledge, this is the first published study to characterize the tolerability and pharmacokinetic profile of dicloxacillin after administration of single and multiple doses in healthy Chinese subjects. In our study, common AEs, such as fever, diarrhea, nausea, hypersensitivity symptoms, and increased ALT, AST, or γ-glutamyl transferase levels, associated with the treatment of dicloxacillin in patients with infections, were not found. Clinical significant shifts from a normal to below normal range were noted for white blood cell count and for neutrophil count in 6.25% (1/16) subjects. The reason for fewer AEs in our study lies in the small number of enrolled subjects; further evaluation in larger Chinese population will provide a more exact safety profile of dicloxacillin.
In single-dose study, our results demonstrated that dicloxacillin was rapidly absorbed after dosing, with median Tmax being attained within 0.75–1.25 hours, and the exposure of dicloxacillin increased in a dose-dependent manner. The reported pharmacokinetic parameters in a Mexican population after oral administration with 0.5 g of dicloxacillin17 showed a similar Tmax (0.75–1.0 hours) as our study. In our study, interindividual variability in Cmax values was considerable, with the coefficient of variation (CV) ranging from 28.3% to 44.2% for each treatment group. In the 0.25 g group, the CV of Cmax was lower than that in the other treatment groups (28.3% in the 0.25 g group, 44.2% in the 0.5 g group, 41.5% in the 1.0 g group, and 43.1% in the 2.0 g group). Of the 16 subjects (No 8) in the 2.0 g group, one showed higher clearance (16.89 L·h−1) than average (10.4 L·h−1), so Cmax and AUC0–∞ of the subject were lower (21.99 μg·mL−1 and 111.10 μg·h·mL−1, respectively) than average Cmax (79.97 μg·mL−1) and AUC0–∞ (207.40 μg·h·mL−1). Furthermore, the mean plasma Cmax and AUC0–∞ were approximately four times and five times higher in healthy Chinese subjects, respectively, compared with the corresponding PK parameters observed in a healthy Western population.17 There are several reasons for this difference. First, as dicloxacillin previously was reported to be a P-glycoprotein substrate,24 the duodenal expression and activity of P-glycoprotein could have been one of the reasons for the large interindividual variability in systemic exposure of dicloxacillin. Second, the lower body weight may result in greater dicloxacillin exposure compared with healthy Western population. Third, dosing in the fast or fed state may affect the extent of absorption. In addition, the difference in the bioavailability of the various dicloxacillin formulations is also an important reason for the difference. A longer t1/2 was obtained in the present study (0.92–2.21 hours) than that obtained in Mexican subjects (0.7–0.92 hours). These differences were unclear, but may be associated with differences in sample processing, analytical methods, age, and possible racial differences.
In the multiple-dose study, the median Tmax,ss of dicloxacillin on day 3 was 0.33–3.0 hours. The average half-life on day 3 was 0.97–2.35 hours. These parameters were similar to single-dose study. No appreciable accumulation occurred when study drug was administered at total daily doses of up to 8 g in the q6h regimen. This observation was consistent with the relatively short half-life.
This pharmacokinetic study had some limitations that should be considered. First, these results were obtained from healthy subjects, so the pharmacokinetics of dicloxacillin cannot be extrapolated to an older population or to patients with a disease. Second, despite the fact that a modest number of subjects per treatment group are usually acceptable for Phase I trial, the sample size of the present study was relatively small. Furthermore, the study was only conducted in the fasted state, the absorption rate at the fed state of dicloxacillin was unknown. Therefore, future studies are needed to investigate tolerability, efficacy, and pharmacokinetics including the effect of food in Chinese patients.
Acknowledgments
The authors deeply appreciated the contributions of the study team during the conduct of this study. This study was sponsored by Bright Future Pharmaceuticals Factory (Hong Kong). This research was funded by the Key Technologies R&D Program of 12th Five-year Plan (2012ZX09302-003-03), People’s Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Gravenkemper CF, Bennett JV, Brodie JL, Kirby WM. Dicloxacillin: in vitro and pharmacologic comparisons with oxacillin and cloxacillin. Arch Intern Med. 1965;116:340–345. | ||
Bergan T. Penicillins. Antibiot Chemother (1971). 1978;25:1–122. | ||
Reimer LG, Stratton CW, Reller LB. Minimum inhibitory and bactericidal concentrations of 44 antimicrobial agents against three standard control strains in broth with and without human serum. Antimicrob Agents Chemother. 1981;19(6):1050–1055. | ||
Dimitrova DJ, Pashov DA, Dimitrov DS. Dicloxacillin pharmacokinetics in dogs after intravenous, intramuscular and oral administration. J Vet Pharmacol Ther. 1998;21(5):414–417. | ||
Zamotaev IP, Sokolova VI, Konstantinova LA, Pozdniakova VP. [Effectiveness of capsules of the sodium salt of dicloxacillin in bronchopulmonary diseases]. Antibiotiki. 1976;21(3):266–269. Russian. | ||
Smith LG. Chronic staphylococcal infections of bone and soft tissue. Treatment with dicloxacillin. J Med Soc N J. 1970;67(5):219–222. | ||
Blaszczyk-Kostanecka M, Dobozy A, Dominguez-Soto L, et al. Comparison of two regimens of oral clindamycin versus dicloxacillin in the treatment of mild-to-moderate skin and soft-tissue infections. Curr Ther Res – Clin Exp. 1998;59(6):341–353. | ||
Stewart C, Shaver TS, Flournoy DJ, et al. Methicillin-resistant staphylococcal urinary tract infection treated with dicloxacillin. J Okla State Med Assoc. 1993;86(2):66–68. | ||
Nissen JL, Skov R, Knudsen JD, et al. Effectiveness of penicillin, dicloxacillin and cefuroxime for penicillin-susceptible Staphylococcus aureus bacteraemia: a retrospective, propensity-score-adjusted case-control and cohort analysis. J Antimicrob Chemother. 2013;68(8):1894–1900. | ||
Knott T, Lange A, Volkening R. [Dicloxacillin, a penicillinase-fast oral penicillin. Antibacterial efficacy and pharmacokinetics in humans]. Arzneimittelforschung. 1965;15(4):331–340. German. | ||
Bennett JV, Gravenkemper CF, Brodie JL, Kirby WM. Dicloxacillin, a new antibiotic: clinical studies and laboratory comparisons with oxacillin and cloxacillin. Antimicrob Agents Chemother (Bethesda). 1964;10:257–262. | ||
Nauta EH, Mattie H. Dicloxacillin and cloxacillin: pharmacokinetics in healthy and hemodialysis subjects. Clin Pharmacol Ther. 1976;20(1):98–108. | ||
Bass JW, Bruhn FW, Merritt WT, et al. Comparison of oral penicillinase-resistant penicillins: contrasts between agents and assays. South Med J. 1982;75(4):408–410. | ||
Sutherland R, Croydon EA, Rolinson GN. Flucloxacillin, a new isoxazolyl penicillin, compared with oxacillin, cloxacillin, and dicloxacillin. Br Med J. 1970;4(5733):455–460. | ||
Roder BL, Frimodt-Moller N, Espersen F, Rasmussen SN. Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection. 1995;23(2):107–112. | ||
Lofgren S, Bucht G, Hermansson B, et al. Single-dose pharmacokinetics of dicloxacillin in healthy subjects of young and old age. Scand J Infect Dis. 1986;18(4):365–369. | ||
Alderete O, Gonzalez-Esquivel DF, Del Rivero LM, Castro Torres N. Liquid chromatographic assay for dicloxacillin in plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805(2):353–356. | ||
Smith AL, Meeks CA, Koup JR, et al. Dicloxacillin absorption and elimination in children. Dev Pharmacol Ther. 1990;14(1):35–44. | ||
World Medical Association. Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended by the 59th WMA General Assembly, Seoul, Korea; 2008. | ||
European Medicines Agency (EMEA). Guidance on Good Clinical Practice. ICH topic E 6 (R1). In: CPMP. London, UK; 2002. | ||
China Food and Drug Administration. Guideline for Good Clinical Practice. 2003. Available from: http://www.sda.gov.cn/WS01/CL0053/24473.html. Accessed 24 September, 2015. | ||
China Food and Drug Administration. Guideline for pharmacokinetics studies of generic drug products. 2005. Available from: http://www.sfda.gov.cn/WS01/CL1616/83420.html. Accessed 24 September, 2015. | ||
U.S. Food and Drug Administration. Bioanalytical Method Validation. FDA Center for Drug Evaluation and Research, Rockville, MD; 2001. | ||
Putnam WS, Woo JM, Huang Y, Benet LZ. Effect of the MDR1 C3435T variant and P-glycoprotein induction on dicloxacillin pharmacokinetics. J Clin Pharmacol. 2005;45(4):411–421. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.