")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 12
Safety And Efficacy Of Princess® FILLER Lidocaine In The Correction Of Nasolabial Folds
Authors Grablowitz D , Sulovsky M , Höller S, Ivezic-Schoenfeld Z , Chang-Rodriguez S, Prinz M
Received 8 April 2019
Accepted for publication 6 October 2019
Published 26 November 2019 Volume 2019:12 Pages 857—864
DOI https://doi.org/10.2147/CCID.S211544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Doris Grablowitz,1 Monika Sulovsky,2 Sonja Höller,3 Zrinka Ivezic-Schoenfeld,3 Souyet Chang-Rodriguez,3 Martin Prinz3
1Medizinisch Ästhetisches Zentrum Wien, Wien 1010, Austria; 2YUVELL® Fine Aesthetics, Wien 1010, Austria; 3Croma-Pharma GmbH, Leobendorf 2100, Austria
Correspondence: Martin Prinz
Croma-Pharma GmbH, Industriezeile 6, Leobendorf 2100, Austria
Tel +43 2262 684 68-0
Fax +43 2262 684 68-165
Email [email protected]
Purpose: Nasolabial folds (NLFs) are one of the most noticeable signs of facial aging. NLFs negatively affect self-confidence and social acceptance often leading to a person’s desire to improve their appearance using dermal fillers. The hyaluronic acid injectable gel implant Princess® FILLER Lidocaine (PFL) is a minimally invasive easy to administer the product. In this investigation, we assessed the safety and efficacy of PFL to correct moderate to severe NLFs over a 36-week period.
Methods: Adult women and men with moderate to severe NLFs received one injection of PFL to both NLFs. After 2 weeks, a touch-up treatment could be performed, if deemed necessary by the investigator. The change in NLF severity was assessed using the Nasolabial Fold Severity Rating Scale (NFL-SRS) developed by Croma-Pharma and the Global Aesthetic Improvement Scale (GAIS).
Results: Out of 60 analyzed subjects, 59 (98.3%) had improved their NLF severity by at least 1 grade on the NFL-SRS at week 4, 58 subjects (96.7%) at weeks 24 and 36. All subjects showed aesthetic improvement (GAIS), at weeks 4 and 24. The investigator judged the aesthetics as very much improved (score of 1) in 45 (75.0%) at week 4, 48 (80.0%) at week 24, and in 39 of 60 subjects, respectively (65.0%) at week 36. Thirty-six weeks post-initial treatment, 56 of 60 subjects (93.3%) were very satisfied or satisfied with the treatment. Adverse device effects (ADEs) were mild or moderate and resolved at latest 25 days post-onset. The most commonly reported ADEs were injection site hematoma and injection site pain.
Conclusion: PFL was safe and effective in reducing the severity of NLFs. Most subjects were (very) satisfied with the treatment outcome throughout a 36 weeks investigation period.
Keywords: hyaluronic acid, dermal filler, lidocaine hydrochloride, mid to deep dermis, facial wrinkles, injection
Introduction
Nasolabial folds (NLFs) are one of the typical clinical manifestations of facial aging, which further include flaccidity of the skin and subcutaneous tissue, wrinkle expression in the upper third of the face, tear through, drop of the angle of the mouth, loss of definition in the mandibular border, cervicofacial platysmal bands, changes in skin pigmentation, and evident veins.1 The perception of age and health is a critical aspect in the common judgment of attractiveness. Younger faces are generally perceived as more attractive than older-looking faces, and estimated age is negatively correlated with perceived attractiveness.2,3 Attractiveness influences both the self-perception and the social behavior toward others and is related to traits such as self-confidence and social acceptance. Thus, it is not surprising that aesthetic interventions can improve the psychological well-being and quality of life in people who chose to undergo such rejuvenation procedures.4 Positive changes in individuals undergoing aesthetic interventions include increased satisfaction with their self-appearance, reduced incidences of depression or anxiety, improved emotional wellbeing, and increased self-confidence.5 Dermal fillers are widely used for facial rejuvenation and the correction of deep wrinkles, including NLFs. Treatments with dermal fillers provide desirable aesthetic outcomes with minimal invasiveness and without the downtime associated with surgery. Dermal fillers also stimulate the cell turn-over in the dermis to produce elastin and collagen, which also fills the depressed facial regions.6 Hyaluronic acid (HA) dermal fillers are the most popular, with about 2.1 million injections performed in the US in 2017.7 HA dermal fillers are easy to administer, have predictable effectiveness, a good safety profile, and quick recovery.8–11 Pain was the most commonly reported patient complaint with dermal fillers. Consequently, a local anesthetic (lidocaine hydrochloride) was included in their formulation to reduce procedural pain thus bypassing the need for additional anesthesia.12
Princess® FILLER Lidocaine (PFL) is a soft tissue filler manufactured by Croma-Pharma GmbH containing HA and lidocaine hydrochloride as a supplemental anesthetic. HA is a natural component of human skin; hence, PFL is naturally absorbed, with the lifetime of the device anticipated to be 6–9 months. Lidocaine hydrochloride is rapidly released from the device after injection and quickly eliminated from the body due to a short half-life of approximately 90 mins.
The device is designed to be injected into the mid to deep dermis.
Rheological measurements are performed to evaluate the physical characteristics of HA fillers. The storage modulus Gʹ is a suitable parameter for determination of the stiffness of HA-based, cross-linked products like PFL, where the elasticity is more pronounced than the viscosity. The Gʹ specification of the PFL is 45,000–195,000 mPa.
PFL is approved for the use in adults to correct moderate to severe facial wrinkles and folds, to increase lip volume, and for medical reconstructive purposes in the treatment of facial lipoatrophy, debilitating scars, or morphological asymmetry of the face. It received the CE mark in 2016.
The present clinical investigation was undertaken to assess the safety and efficacy of PFL in the correction of moderate to severe NLFs.
Materials And Methods
Materials
Commercially available PFL (Croma-Pharma GmbH, Industriezeile 6, 2100 Leobendorf, Austria) was used for this investigation.
Subjects And Clinical Investigation
This prospective, open-label, multicenter, post-market investigation was conducted between 20-Sep-2017 and 02-Jul-2018 at 2 centers in Vienna, Austria. The Ethics Committee approval was obtained from the Ethikkommision der Stadt Wien (Vienna, A). The study was registered at http://www.clinicaltrials.gov (NCT 03611491).
Subjects had to be male or female adults with 2 fully visible, approximately symmetrical NLFs, with each fold scoring 2 (moderate) to 3 (severe) according to the 5-grade Nasolabial Fold Severity Rating Scale (NLF-SRS; developed by Croma-Pharma GmbH, see Figure 1). Eligible subjects had healthy facial skin, were free of diseases that could have interfered in the cutaneous aging evaluation and were willing to abstain from any aesthetic or surgical procedures in the treatment area for the duration of the clinical investigation. Subjects were excluded from participation for any of the following reasons (among others): pregnancy, lactation, planned pregnancy or unwillingness to use contraception at any time during the investigation (for women of childbearing potential only); mental disorders or emotional instability; allergic reaction or hypersensitivity to HA, lidocaine, or any amide-based anesthetic; any corrective procedures performed or planned in the nasolabial region (eg, silicone implants, permanent fillers, absorbable and non-absorbable sutures, laser therapy, dermabrasion, botulinum toxin application, chemical peeling); infectious, inflammatory, or proliferative lesions in the nasolabial region; cutaneous lesions in the treatment area; human immune deficiency virus-positive; allergies against aesthetic filler; recurrent herpes simplex virus 1; tendency to hypertrophic scars, keloid formation, and/or pigmentation disorders; any autoimmune or connective tissue disease, or current treatment with immune therapy; diabetes mellitus or uncontrolled systemic diseases; and use of anticoagulant, antiplatelet or thrombolytic medication.
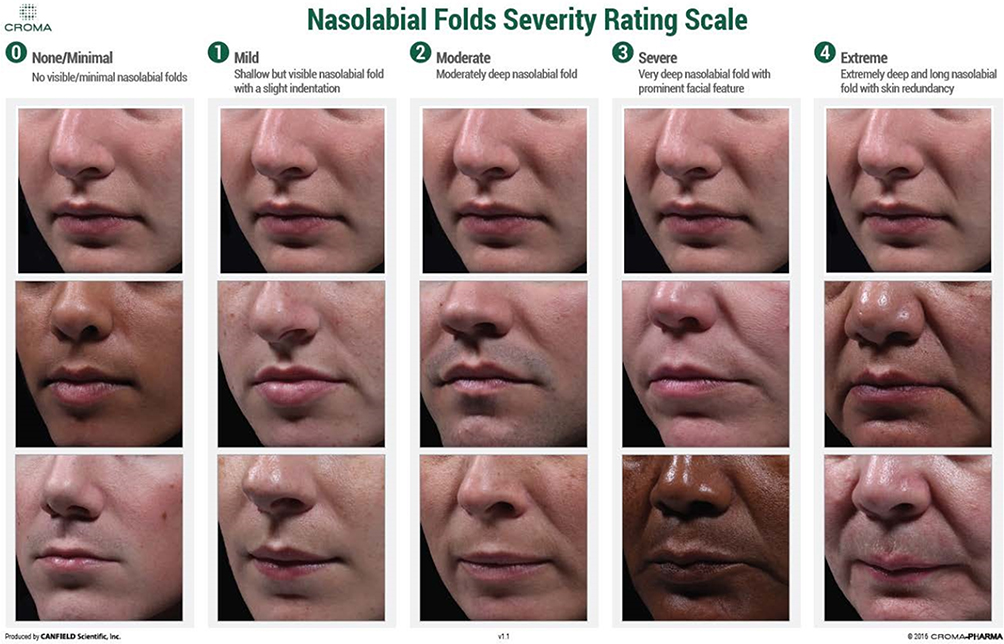 |
Figure 1 Croma-Pharma nasolabial folds severity rating scale. Notes: Grade 0 (none/minimal)=no visible or minimal NLFs, 1 (mild)=shallow but visible NLF with a slight indentation, 2 (moderate)=moderately deep NLF, 3 (severe)=very deep NLF with prominent facial feature, 4 (extreme)=extremely deep and long NLF with skin redundancy. Used with permission fromCroma-Pharma. Abbreviation: NLF, nasolabial fold. |
Before any investigation-related procedures or assessments were performed, subjects received the subject information and voluntarily signed and dated the informed consent form. At least 3 facial photographs (frontal view, left oblique and right oblique) including both NLFs were taken on day 0 (before treatment) and at all post-treatment visits following instructions provided in a photography manual.
The investigation was conducted in accordance with the International Standards Organization 14155:2011, the principles of the Declaration of Helsinki, and the applicable sections of the national medical device law. Ethics committee approval was obtained before any investigation-specific procedures were performed. Before receiving treatment, subjects were medically examined including the documentation of their medical history and current medication.
Treatments
PFL was injected into the mid to deep dermis using a prefilled syringe and a 27G½ʺ disposable needle. Sensitive skin could be pre-treated with a local anesthetic patch or cream. The injection technique (retrograde or fan) and the injected volume were chosen at the investigator’s discretion based on characteristics of the defect under correction. The maximum-recommended volume was 10 mL per treatment session and a total of 20 mL per subject per year.
Subjects received the initial treatment to both NLFs on day 0, with an optional touch-up treatment at week 2, if the investigator was not satisfied with the result of the first treatment. Subjects were followed up for 36 weeks.
Assessments
Immediately after injection and 15 mins thereafter on day 0 and, if applicable, at week 2, subjects were asked to quantify the pain associated with the procedure on a semi-quantitative numeric rating scale from 0 (no pain) to 10 (worst imaginable pain).
The severity of NLFs was graded on the NLF-SRS live by the investigator on day 0 (before treatment), and at week 4, week 24, and week 36. Additionally, photographs were taken at each visit and evaluated by an independent reviewer. The NLF-SRS ranges from none/minimal (Grade 0) to extreme (Grade 4) and uses sample guide photographs labeled with a differentiating description for each severity grade (Figure 1).
The aesthetic improvement of the subject’s appearance after NLF correction was assessed by the investigator at week 4, week 24, and week 36 comparing the result for each NLF with photographs of the pre-treatment appearance using the 5-point Global Aesthetic Improvement Scale (GAIS). The GAIS ranges from “very much improved” (score = 1) to “worse” (score = 5).
Aesthetic improvement based on the GAIS was achieved with scores <4.
At each visit and before the investigator’s aesthetic evaluation, subjects rated their satisfaction with the treatment on a 5-point scale ranging from “very unsatisfied” to “very satisfied”. The safety and tolerability of the treatment with PFL was assessed based on spontaneous reporting of adverse events (AEs) by the subjects, and through clinical examinations and asking non-leading verbal questions about the subjects’ general well-being by the investigator at each scheduled visit. The investigators also examined the treatment area for injection site reactions at each scheduled visit.
Measures And Endpoints
The primary efficacy endpoints were the average change versus baseline (day 0) in the NLF-SRS grade of NLFs at week 24 as evaluated by the investigator and the proportion of subjects with the NLF-SRS grade reduced by ≥1 point versus baseline at week 24. The secondary endpoints included the average change in NLF-SRS grade and the proportion of subjects with improvement at all visits as evaluated by investigators and independent reviewers of photographs, the improvement by using GAIS, subject satisfaction and pain rating.
Statistical Analysis
All primary and secondary endpoints were analyzed descriptively. For the change in the average grade of NLF severity based on the NLF-SRS from baseline to week 24, 95% confidence intervals of the mean were calculated. Responder analyses based on the proportion of subjects achieving a ≥1 grade or 2 grades improvement from baseline were performed. Sixty subjects received treatment and were included in the safety analyses. Fifty-nine subjects completed all visits until the end of the investigation at week 36 and were included in all efficacy analyses.
Results
Subjects
Sixty subjects were enrolled, and each received at least 1 injection with PFL to both NLFs. All subjects had NLF severity of 2 (moderate) to 3 (severe) according to the NLF-SRS at baseline. Fifty-nine women and 1 man were enrolled. All subjects were white and between 30 and 76 years old with a median age of 55.5 years.1 subject was lost to follow up after week 2, the remaining 59 subjects completed all visits until the end of the investigation at week 36.
Dosing And Administration
At baseline (day 0), 60 subjects were injected, of whom 29 subjects (48.3%) were treated with anesthetic cream before injection. The median-injected volume was 1.45 mL (range: 0.5–2.5 mL) PFL to the right and 1.50 mL (range: 0.7–2.6 mL) to the left NLF. The retrograde injection technique was used in 57% and the fan technique in 15% of subjects. In 28% of subjects another injection technique or the combination of techniques was used. At week 2, the investigator judged the initial treatment to be incomplete for 20 subjects, who then received touch-up treatment. For 7 subjects, anesthetic cream was applied before injection. The median volume of the touch-up treatment was 1.0 mL to both NLFs (range: 0–2 mL). Subjects were injected using the fan technique in 15% of subjects (3 of 20), the retrograde technique in 35% of subjects (7 of 20), and the combination of fan and retrograde injection technique in 50% of subjects (10 of 20).
Efficacy
NLF Severity Based On The NLF-SRS
The proportion of subjects with an improvement of at least 1 grade of the NLF-SRS as assessed by the investigator is shown in Table 1 and by the independent reviewer of photographs is shown in Figure 2A. Based on the investigator’s evaluations, all of the 59 evaluated subjects at week 4 (98.3%) and all but one subject (58 subjects, 96.7%) at week 24 and week 36 had improved their NLF severity by at least 1 grade. The independent reviewer of photographs assessed all subjects to be improved by at least 1 grade at all post-baseline visits. The investigator's rating of a ≥2-grade improvement was 88% of subjects at week 4, 68% at week 24 and 60% at week 36 (Figure 2B).
 |
Table 1 (%) Of Subjects With ≥1 Grade Reduction Of NLF Severity By At Least 1 Grade On The NLF-SRS As Assessed By The Investigator |
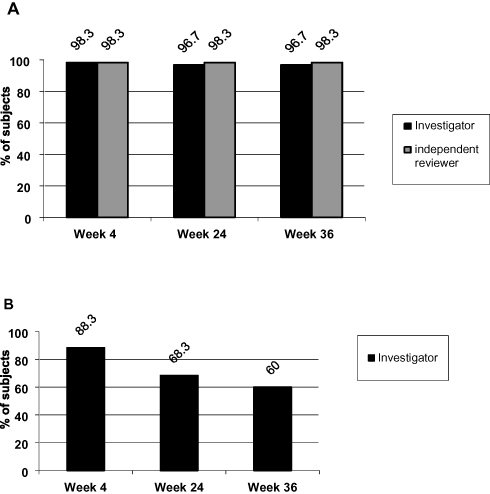 |
Figure 2 (A) Proportion of subjects with ≥1 grade improvement in NLF severity based on the NLF-SRS as assessed by the investigator and the independent reviewer of photographs (“reviewer”). (B) Proportion of subjects with ≥2 grades improvement in NLF severity based on the NLF-SRS as assessed by the investigator. Notes: Percentages are based on the number of subjects in the analysis set (N=60). The evaluation of 1 subject was missing at all post-baseline visits. Abbreviations: N, number of subjects; NLF, nasolabial fold; NLF-SRS, nasolabial fold-severity rating scale. |
Aesthetic Improvement Assessed By GAIS
All 59 evaluated subjects (98.3%) had improved aesthetics (below score 4 of the GAIS) at week 4 and week 24. Most subjects had a “very much improved” aesthetic appearance (corresponding to a score of 1) at all post-baseline visits. At week 4, 76.3% had GAIS Score 1 (very improved), 20.3% had GAIS Score 2 (much improved), and 3.4% had GAIS Score 3 (improved). At week 24 81.4% had GAIS Score 1, 15.3% had GAIS Score 2 and 3.4% had GAIS Score 3. At week 36 this trend continued with the exception of one subject (96.7%). At week 36 66.1% had GAIS Score 1, 27.1% had GAIS Score 2, 5.1% had GAIS Score 3 and 1.7% had GAIS Score 4 (no change) (Figure 3).
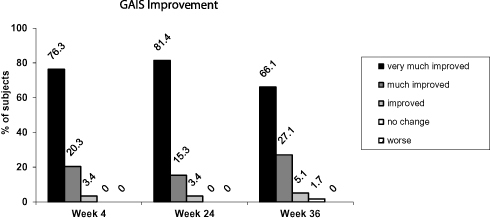 |
Figure 3 Proportion of subjects with improved aesthetics as assessed by the investigator using the GAIS. Notes: The GAIS score was averaged over both nasolabial folds. Percentages are based on the number of subjects in the analysis set (N=60). The evaluation of 1 subject was missing at all postbaseline visits. Abbreviations: GAIS, Global Aesthetic Improvement Scale; N, number of subjects. |
Subject Satisfaction
At week 36, 56 subjects (93.3%) were “very satisfied” or “satisfied” with the treatment, while no subject was “very unsatisfied“ (Figure 4). This high degree of satisfaction with the treatment outcome was noted at week 4 and consistently maintained until the end of the investigation at week 36.
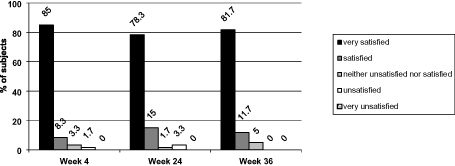 |
Figure 4 Subject satisfaction ratings. Notes: Percentages are based on the number of subjects in the analysis set (N=60). The evaluation of 1 subject was missing at all post-baseline visits. Abbreviation: N, number of subjects. |
Pain Intensity
Immediately after the injection 36.7% of subjects had pain score 0, 18.3% had pain score 1, 18.3% had pain score 2, 18.3% had pain score 3, 5% had pain score 4 and 3.3% had pain score 5, no patient reported pain scores 6–10 (Figure 5). Twenty subjects received a touch-up treatment 8.3% had pain score 0, 6.7% had pain score 1, 8.3% had pain score 2, 6.7% had pain score 4, 1.7% had pain score 5 and 1.7% had pain score 6, no patient reported pain scores 6−10 (Figure 5). After both treatments, on day 0 and at week 2, subjects reported no pain 15 mins after the first pain intensity assessment.
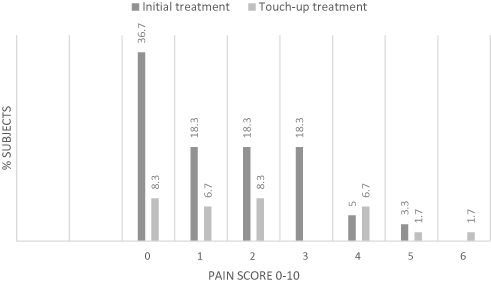 |
Figure 5 Pain score immediately after injection. Notes: Percentages of subjects with their Pain Score immediately after injection at Initial treatment and at Touch-up treatment. Percentages are based on the number of subjects in the analysis set (N=60) for the initial treatment and N=20 for touch-up treatment. The evaluation of 1 subject was missing at all post-baseline visits. Abbreviation: N, number of subjects. |
Safety
The treatment was generally safe and well tolerated. In total 38 AEs were reported by 28 subjects (46.7%). Most of these AEs (31 of 38, 81.6%) were related to the procedure and classified as adverse device effects (ADEs), with no event being related to the investigational medical device.
All ADEs were mild or moderate temporary effects that were localized to the injection site (Table 2). Nearly all ADEs resolved within 14 days post-onset, with one ADE (injection site hematoma) resolving 25 days post-onset. Two subjects experienced serious AEs, one moderate sub-mammary hematoma infection and one severe papillary thyroid cancer, both of which the investigator considered as not related to the investigational medical device or the procedure.
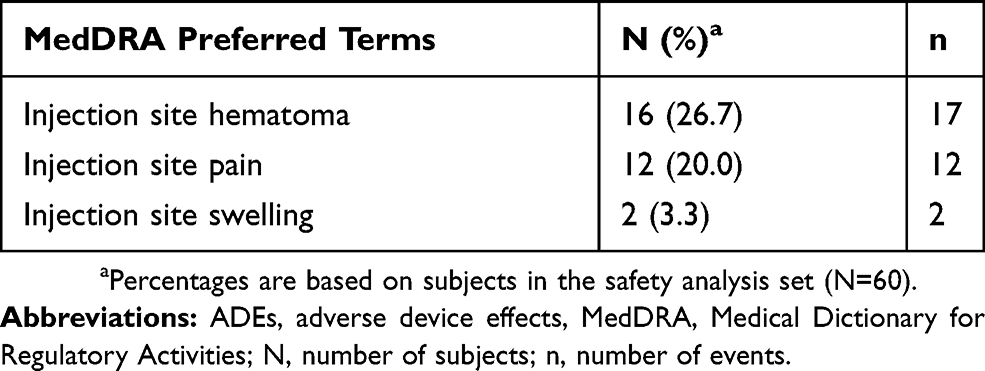 |
Table 2 Incidence Of ADEs Reported During The Investigation |
Discussion
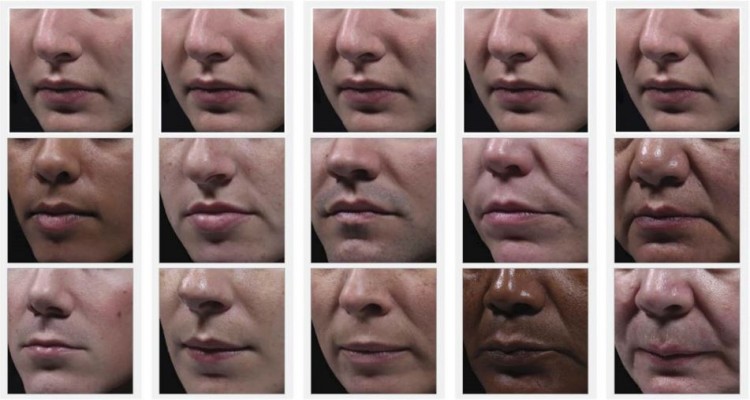
In this investigation, 60 white subjects (female; N=59, male; N=1), with a median age of 55.5 years were enrolled. Of these, 59 subjects were evaluated at week 4, week 24, and week 36; while 1 subject was lost to follow-up after week 2. The objectives of this investigation were to evaluate the efficacy of PFL in the correction of moderate to severe NLFs and to evaluate the safety of PFL when used to correct moderate to severe NLFs. After 24 weeks in all but one evaluated subjects (96.7%), the severity of NLFs improved by at least 1 NLF-SRS grade, as assessed by the investigator, and in all evaluated subjects (98.3%), as assessed by the independent reviewer of photographs (Figure 6).
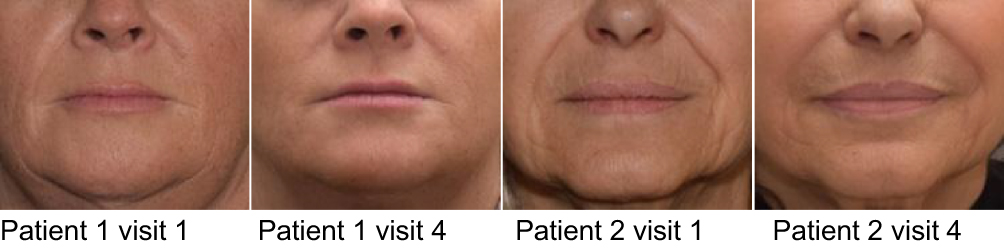 |
Figure 6 Clinical photos of treated patients. |
The evaluation of the efficacy of the treatment of nasolabial folds in the improvement of at least 1 point, as per scales defining severity of nasolabial folds, and using other comparable devices were already assessed by investigators in several clinical studies at 6 months (24 weeks) time-point: the Nasolabial Fold Severity Score (NLFSS) responder rates for Juvéderm Ultra Plus were 90.8%, compared to 89.9% for Restylane.19 In another investigation, Lupo et al reported that 96% of subjects treated with Juvéderm Ultra Plus maintained clinically significant improvement.20 In a study performed by Goodman et al Juvéderm Ultra Plus showed an improvement of 90% whereas Perlane® showed 65%.21 Juvedérm Vollure XC (VYC-17.5L) revealed a NLFSS responder rate of 93.2% as reported by Monheit et al.13 In all instances, the efficacy or clinically significant improvement was defined as at least 1 point improvement on a nasolabial folds severity scale.
Based on this data, it can be concluded that the efficacy results using PFL are similar to other benchmarked devices.
The NLF improvement was apparent at week 4 and was maintained throughout the investigation until week 36 confirming a tissue residency for PFL of at least 6 months after a maximum of 2 treatments. Thereby the median-injected volume was comparable or even slightly lower than the volume used with other HA fillers.13,14
Treatment with PFL also improved the subject’s aesthetic appearance as indicated by improved GAIS scores all evaluated subjects from week 4 (96.6% “very much improved”/”improved”) until week 24 (96.7% “very much improved”/”improved”). This improvement was also seen at week 36 (93.2% “very much improved”/”improved”) in all but one evaluated subjects. The subject satisfaction evaluation matched both the investigator’s and independent reviewer’s assessments, with over 90% of subjects being satisfied to very satisfied at the investigation completion. The subjective treatment outcome of PFL injections matched both the investigator’s and independent reviewer’s assessments with over 90% of subjects being satisfied to very satisfied at the investigation completion.
Moreover, the acceptance of pain was satisfactory throughout the study and for those subjects experiencing pain after injections, there was no pain sensation left at 15 mins post-treatment.
Taken together, these results were consistent with results of previous investigations using the HA Fillers, Princess® FILLER, the equivalent device but without lidocaine hydrochloride, and Princess® VOLUME, the equivalent device but with a higher dynamic viscosity. In these investigations, the use of the Princess® fillers was highly successful in the treatment of facial lipoatrophy, morphological asymmetry and debilitating scars (Princess® FILLER) and in the treatment of NLFs (Princess® VOLUME) over a 6-month period, as judged by investigators, subjects, and independent reviewers.15–17 All ADEs that were reported in this investigation (ie, injection site hematoma, pain, and swelling) were mild or moderate, transient, and are commonly reported following treatment with dermal fillers.18
Conclusion
Treatment with PFL is a safe, well tolerated and effective method to reduce the severity of NLFs; >96% of subjects showed an improvement in their NLF severity of at least 1 grade after 36 weeks compared to their baseline status, and this improvement was maintained for 6 months; 93.3% of the subjects were “very satisfied” or “satisfied” with the treatment outcome.
Data sharing statement
The authors do not intend to share individual’s de-identified participant data, nor other study-related documents.
Disclosure
DG and MS were principal investigators in this investigation. SH and MP are employees of Croma-Pharma GmbH. ZIS and SCR are former employees of Croma-Pharma GmbH. MP is partial owner of Croma-Pharma. The authors report no other conflicts of interest in this work.
References
1. Beer K, Beer J. Overview of facial aging. Facial Plast Surg. 2009;25(5):281–284. doi:10.1055/s-0029-1243075
2. Samson N, Fink B, Matts PJ, et al. Visible changes of female facial skin surface topography in relation to age and attractiveness perception. J Cosmet Dermatol. 2010;9:79–88. doi:10.1111/jcd.2010.9.issue-2
3. Porcheron A, Latreille J, Jdid R, et al. Influence of skin ageing features on Chinese women’s perception of facial age and attractiveness. Int J Cosmet Sci. 2014;36:312–320. doi:10.1111/ics.2014.36.issue-4
4. Waldman A, Maisel A, Weil A, et al. Patients believe cosmetic procedures affect their quality of life: an interview study of patient-reported motivations. J Am Acad Dermatol. 2019;
5. Sadick NS. The impact of cosmetic interventions on quality of life. Dermatol Online J. 2008;14:2.
6. Fallacara A, Manfredini S, Durini E, et al. Hyaluronic acid fillers in soft tissue regeneration. Facial Plast Surg. 2017;33:87–96. doi:10.1055/s-0036-1597685
7. American Society of Plastic Surgeons (ASPS) [Homepage online]. 2017 Complete plastic surgery statistics report. Available from: https://www.plasticsurgery.org/documents/News/Statistics/2017/plastic-surgery-statisticsfull-report-2017.pdf.
8. Bogdan Allemann I, Baumann L. Hyaluronic acid gel (Juvéderm) preparations in the treatment of facial wrinkles and folds. Clin Interv Aging. 2008;3(4):629–634. doi:10.2147/CIA.S3118
9. Fakhari A, Berkland C. Applications and emerging trends of hyaluronic acid in tissue engineering, as a dermal filler and in osteoarthritis treatment. Acta Biomater. 2013;9(7):7081–7092. doi:10.1016/j.actbio.2013.03.005
10. Beasley KL, Weiss MA, Weiss RA. Hyaluronic acid fillers: a comprehensive review. Facial Plast Surg. 2009;25(2):86–94. doi:10.1055/s-0029-1220647
11. Raspaldo H. Volumizing effect of a new hyaluronic acid sub-dermal facial filler: a retrospective analysis based on 102 cases. J Cosmet Laser Ther. 2008;10(3):134–142. doi:10.1080/14764170802154607
12. Smith L, Cockerham K. Hyaluronic acid dermal fillers: can adjunctive lidocaine improve patient satisfaction without decreasing efficacy or duration? Patient Prefer Adherence. 2011;5:133–139. doi:10.2147/PPA.S11251
13. Monheit G, Beer K, Hardas B, et al. Safety and effectiveness of the hyaluronic acid dermal filler VYC-17.5L for Nasolabial folds: results of a randomized, controlled. Study. Dermatol Surg. 2018;44(5):670–678. doi:10.1097/DSS.0000000000001529
14. Nast A, Reytan N, Hartmann V, et al. Efficacy and durability of two hyaluronic acid-based fillers in the correction of nasolabial folds: results of a prospective, randomized, doubleblind, actively controlled clinical pilot study. Dermatol Surg. 2011;37(6):768–775. doi:10.1111/j.1524-4725.2011.01993.x
15. Dai X, Li L, Peterson W, et al. Safety and effectiveness of hyaluronic acid dermal filler in correction of moderate-to-severe nasolabial folds in Chinese subjects. Clin Cosmet Investig Dermatol. 2019;12(12):57–62. doi:10.2147/CCID
16. Kopera D, Ivezic-Schoenfeld Z, Federspiel IG, et al. Treatment of facial lipoatrophy, morphological asymmetry, or debilitating scars with the hyaluronic acid dermal filler Princess® FILLER. Clin Cosmet Investig Dermatol. 2018;27(11):621–628. doi:10.2147/CCID.S181964
17. Kopera D, Palatin M, Bartsch R, et al. An open-label uncontrolled, multicenter study for the evaluation of the efficacy and safety of the dermal filler Princess VOLUME in the treatment of nasolabial folds. Biomed Res Int. 2015;2015:195328. doi:10.1155/2015/195328.
18. Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Plast Surg Nurs. 2015;35(1):13–32. doi:10.1097/PSN.0000000000000087
19. Dong L, Xie Y, Qin L, et al. Safety and effectiveness of Juvéderm ultra plus injectable gel in correcting severe nasolabial folds in Chinese subjects. Plast Reconstr Surg Glob Open. 2017;16:e1133. doi:10.1097/GOX.0000000000001133
20. Lupo M, Smith S, Thomas J, et al. Effectiveness of Juvéderm ultra plus dermal filler in the treatment of severe nasolabial folds. Plast Reconstr Surg. 2008;121(1):289–297. doi:10.1097/01.prs.0000294968.76862.83
21. Goodmann G, Phillip B, Rich M, et al. A comparison of the efficacy, safety, and longevity oft two different hyaluronic acid dermal fillers in the treatment of severe nasolabial folds: a multicenter, prospective, randomized, controlled, single-blind, within-subject study. Clin Cosmet Investig Dermatol. 2011;4:197–205. doi:10.2147/CCID.S26055
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.