")
Back to Journals » International Medical Case Reports Journal » Volume 8
Sacrococcygeal teratoma in a female newborn with clinical features of trisomy 13: a case report from Central Africa
Authors Lubala T, Mukuku O , Shongo M, Mutombo A, Lubala N, Luboya O, Lukusa-Tshilobo P
Received 5 April 2015
Accepted for publication 4 August 2015
Published 14 December 2015 Volume 2015:8 Pages 333—336
DOI https://doi.org/10.2147/IMCRJ.S86098
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ronald Prineas
Toni Kasole Lubala,1,2 Olivier Mukuku,1 Mick Pongombo Shongo,1,2 Augustin Mulangu Mutombo,1 Nina Lubala,1 Oscar Numbi Luboya,1 Prosper Lukusa-Tshilobo3
1Department of Paediatrics, Faculty of Medicine, 2Center for Human Genetics, Faculty of Medicine, University of Lubumbashi, Lubumbashi, 3Department of Paediatrics and Centre for Human Genetics, University Hospital, University of Kinshasa, Kinshasa, Democratic Republic of Congo
Introduction: The objective of this report is to describe the first patient presenting clinical features of trisomy 13 in association with a sacrococcygeal teratoma.
Case presentation: We present the case of a Congolese female infant born with bilateral cleft lip and palate, hypotelorism, microcephaly, and capillary hemangioma on her face. She presented with a large sacrococcygeal mass (15.0 cm ×12.0 cm ×5.0 cm) with a cystic consistency and a positive transillumination.
Conclusion: This observation suggests that overexpression of certain genes on chromosome 13 may lead to tumor formation from remnant cells of Hensen’s node.
Keywords: Patau syndrome, Hensens’s Node, sacrococcygeal, teratoma
Introduction
Sacrococcygeal teratoma is a rare tumor with a birth prevalence of 1 in 21,700 births.1 In a relatively large proportion (20%), this tumor can have a malignant development, most commonly a papillary adenocarcinoma.2 Previously, teratome of the liver, umbilical cord, and oral region have been described in three patients with trisomy 13 syndrome (Patau syndrome), but thus far, no sacrococcygeal teratoma has been observed in this syndrome. Trisomy 13 has an incidence of 1 in 10,000–20,000 with variable expression and is characterized by the clinical triad of cleft lip and palate, microphthalmia/anophthalmia, and postaxial polydactyly.3 However, other anomalies are frequently associated.4,5 The objective of this report is to describe a Congolese newborn presenting with a sacrococcygeal teratoma and the clinical features of trisomy 13.
Case presentation
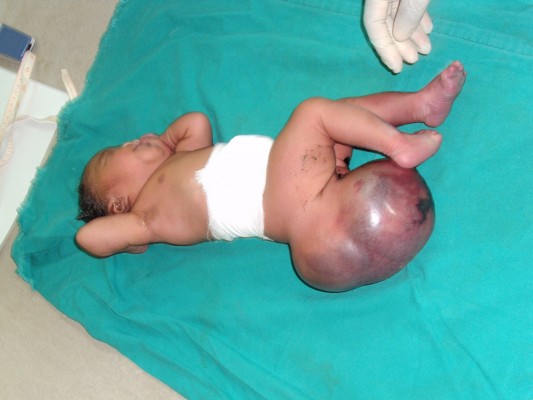
A female patient was born in Fungurume (mining area in southern Katanga Province, Democratic Republic of Congo) at 40 weeks of gestation via lower segment cesarean section delivery with a birth weight of 4,580 g (+2.48 standard deviation [SD], World Health Organization [WHO] growth charts 20066) and a length of 51 cm (0.9 SD, WHO growth charts 20066). Her mother was 40 years old and her father 51 years old, both were healthy and unrelated. The mother did not use medications during pregnancy. No prenatal ultrasound scan was performed. She was referred on day 2. On clinical examination, we noted a bilateral cleft lip and palate, hypotelorism, microcephaly (28 cm, −5.06 SD, WHO growth charts6), and capillary hemangioma on her face. No scalp defect has been observed. The infant had low-set ears and a short neck (Figure 1). No organized ocular tissues could be seen or palpated. The infant had five normal fingers in both hands and feet. She had a large sacrococcygeal mass (15.0 cm ×12.0 cm ×5.0 cm) with a cystic consistency and a positive transillumination (Figure 2). However, abdominal and pelvic urtrasonography could not be performed, and thus, the extent of the sacrococcygeal teratoma could not be graded according to the American Academy of Pediatrics classification. The genitalia or rectum was not involved. The anal sphincter and limb tone were normal. Since genetic diagnosis was not available in this part of the world when the infant was born, we were not able to assess genetic confirmation of the suspected diagnosis of trisomy 13. The child was hypertonic, had experienced feeding difficulties, and died on day 8 from severe sepsis and acute anemia.
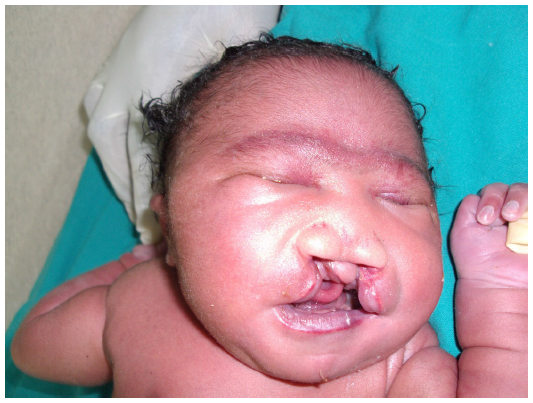 | Figure 1 Craniofacial abnormalities observed in our patient. |
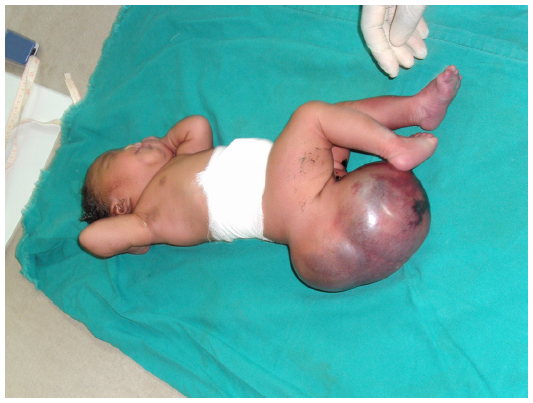 | Figure 2 Large sacrococcygeal teratoma. |
Discussion
Dysmorphology is a major challenge in low-income countries, where imaging and genetic technologies are not available. Since comparative genomic hybridization array and karyotyping are not available in this part of the world, the diagnosis of trisomy 13 in this patient relied on clinical grounds. Two signs of the characteristic clinical triad were present in our patient (cleft lip and palate and microphthalmia/anophthalmia), but no postaxial polydactyly has been observed.3 Table 1 shows clinical features in trisomy 13 with molecular confirmation versus our patient.7–11 As we can see in Table 1 as well as in Caba et al’s paper, full classical triad of trisomy 13 is not constant, which is an indirect evidence supporting the argument that our patient likely had trisomy 13.3
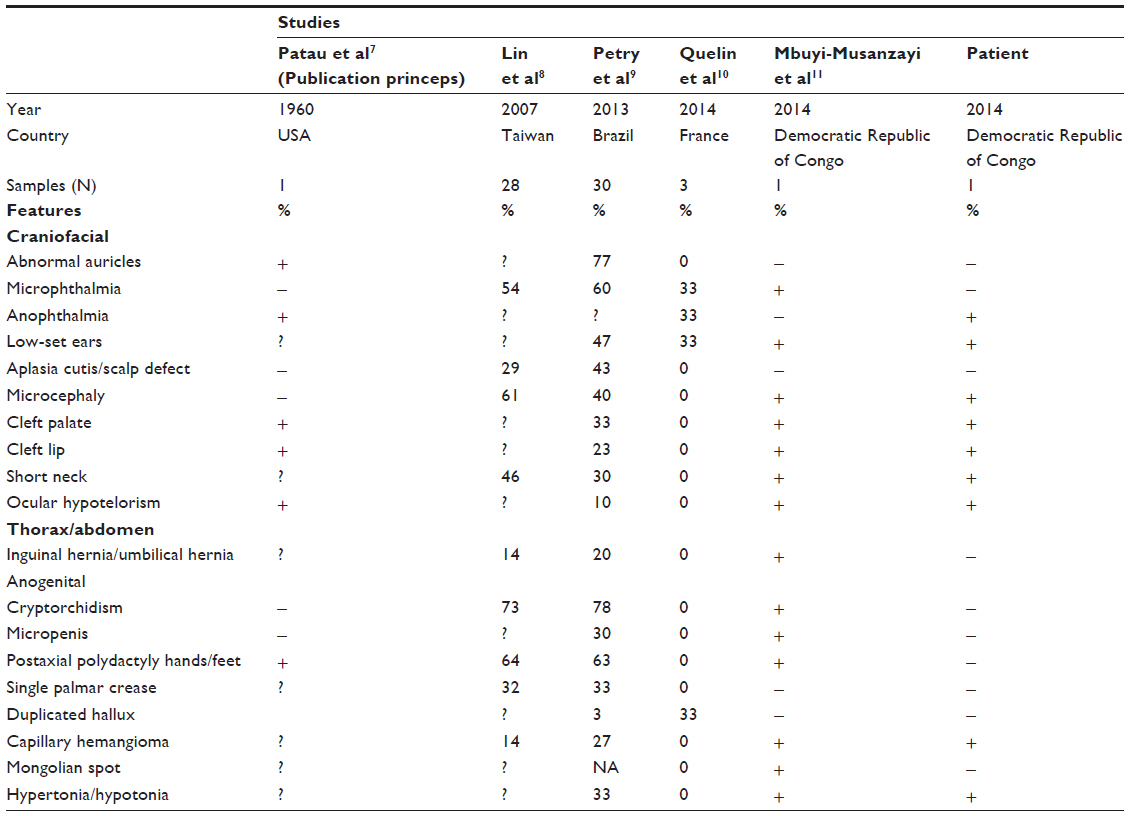 | Table 1 Clinical features in trisomy 13 with molecular confirmation versus our patient |
Whereas in most industrialized countries, trisomy 13 is diagnosed prenatally,12 ~24% of pregnant women have no access to prenatal care services, and the vast majority of them currently do not have access to prenatal ultrasound follow-up in the Democratic Republic of Congo.11
The identification of cell-free DNA in maternal blood and the recent development of polymerase chain reaction-based noninvasive DNA hold enormous promise for diagnosis of fetal trisomies 21, 18, and 13 in general pregnant populations. However, the lack of teaching, and thus interest and knowledge, in human genetics might be barriers to this testing in Democratic Republic of Congo.
Upon reviewing the literature, the relationship of aneuploidy and chromosomal instability to neoplasia has been discussed over the past decades. Three patients with trisomy 13 disorders have been reported with teratoma. In Dische and Gardner’s paper, a fetus with trisomy 13 syndrome and teratomas of liver and neck has been described.13 Yapar et al have presented a case of epignathus (oral cavity teratoma) detected prenatally in a fetus with trisomy 13 syndrome.14 More recently, an unusual solid and cystic mass was diagnosed at 17 weeks fetus with trisomy 13. Fetopathological examination revealed an umbilical cord teratoma.15 These three cases suggest the association between trisomy 13 and teratoma. However, the mechanism of origin of these tumors is probably not the same. Extragonadal teratomas are thought to arise from germ cells that migrate aberrantly during embryogenesis, whereas sacrococcygeal teratoma is believed to arise from totipotent cells derived from the remnant of Hensen’s node located in the coccyx region, the caudal eminence.16
As we can see in Table 2, sacrococcygeal teratoma has already been described in some chromosomal disorders syndrome such as partial trisomy 1q, partial trisomy 3q, and mosaic r (21),13–15,17–24 and genomic gain in 1q and 12p has been observed in sacrococcygeal teratoma. Furthermore, it has been shown that some genes in individuals with trisomy 21 may inhibit the generation and growth of tumors originating from the remnant of Hensen’s node.16
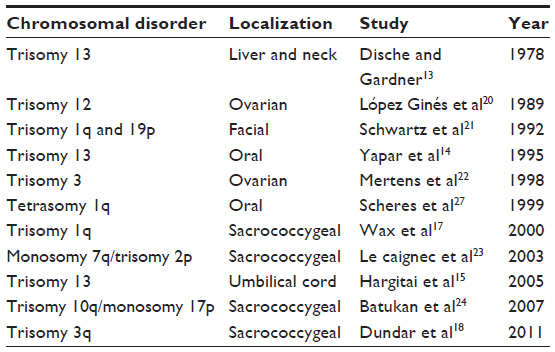 | Table 2 Overview of reported teratoma in patients with chromosomal disorder syndromes |
Trisomies as a gain-of-function mechanism by selection of a gain-of-function mutation or by gene dosage have been also implicated in hematologic malignancies as well as solid tumors through duplication and amplification of mutated alleles (RUNX1 or MET oncogene).25
It is not excluded that this represents a chance of association of two pathogenetically unrelated conditions, and the lack of genetic testing is a weakness of this report. Alternatively, given the previous reports of sacral appendage26 and teratomas with other localizations (Table 2) in other patient with trisomy 13, it is not excluded that overexpression of certain genes on chromosome 13 may lead to tumor formation from remnant cells of Hensen’s node.
Consent
Written informed consent was obtained from the patient’s legal guardians for publication of this case report and any accompanying images. The Hôpital Général De Référence de Kamalondo Ethics Committee advised ethical approval was not required.
Acknowledgment
The authors are grateful to the parents of the reported infant for their kind collaboration.
Author contributions
All authors were responsible for the study conception, data retrieval, and drafting the manuscript. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Gucciardo L, Uyttebroek A, De Wever I, et al. Prenatal assessment and management of sacrococcygeal teratoma. Prenat Diagn. 2011;31:678–688. | |
Lister J. Sacro-coccygeal teratoma. Arch Dis Child. 1955;30:66–69. | |
Caba L, Rusu C, Butnariu L, et al. Phenotypic variability in Patau syndrome. Rev Med Chir Soc Med Nat Iasi. 2013;117:321–327. | |
Duarte AC, Menezes AI, Devens ES, Roth JM, Garcias GL, Martino-Roth MG. Patau syndrome with a long survival. A case report. Genet Mol Res. 2004;3:288–292. | |
Carey JC. Perspectives on the care and management of infants with trisomy 18 and trisomy 13: striving for balance. Curr Opin Pediatr. 2012;24:672–678. | |
WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards: Length/height-for-age, Weight-for- age, Weight-for-length, Weight-for-height and Body mass index-for-age: Methods and Development. Geneva: World Health Organization, 2006. Available from: http://www.who.int/childgrowth/standards/technical_report/en/. Accessed September 10, 2015. | |
Patau K, Smith DW, Therman E, Inhorn S, Wagner H. Multiple congenital anomaly caused by an extra autosome. Lancet. 1960;275:790–793. | |
Lin HY, Lin SP, Chen YJ, et al. Clinical characteristics and survival of trisomy 18 in a medical center in Taipei, 1988–2004. Am J Med Genet. 2006;140A:945–951. | |
Petry P, Polli JB, Mattos VF, et al. Clinical features and prognosis of a sample of patients with trisomy 13 (Patau syndrome) from Brazil. Am J Med Genet. 2013;161A:1278–1283. | |
Quelin C, Spaggiari E, Khung-Savatovsky S, Dupont C, Pasquier L, Loeuillet L. Inversion duplication deletions involving the long arm of chromosome 13: phenotypic description of additional three fetuses and genotype-phenotype correlation. Am J Med Genet. 2014;164A:2504–2509. | |
Mbuyi-Musanzayi S, Lumaka AZ, Yogolelo HB, et al. Preaxial polydactyly of the foot: variable expression of trisomy 13 in a case from Central Africa. Case Rep Genet. 2014;2014:365031. | |
Kroes I, Janssens S, Defoort P. Ultasound features in trisomy 13 (Patau syndrome) and trisomy 18 (Edwards syndrome) in a consecutive series of 47 cases. Facts Views Vis Obgyn. 2014;6:245–249. | |
Dische MR, Gardner HA. Mixed teratoid tumors of the liver and neck in trisomy 13. Am J Clin Pathol. 1978;69:631–637. | |
Yapar EG, Ekici E, Gökmen O. Sonographic diagnosis of epignathus (oral teratoma), prosencephaly, meromelia and oligohydramnios in a fetus with trisomy 13. Clin Dysmorphol. 1995;4:266–271. | |
Hargitai B, Csabai L, Bán Z, et al. Rare case of exomphalos complicated with umbilical cord teratoma in a fetus with trisomy 13. Fetal Diagn Ther. 2005;20:528–533. | |
Kobayashi T, Sakemi Y, Yamashita H. Increased incidence of retroperitoneal teratomas and decreased incidence of sacrococcygeal teratomas in infants with Down syndrome. Pediatr Blood Cancer. 2014;61:363–365. | |
Wax JR, Benn P, Steinfeld JD, Ingardia CJ. Prenatally diagnosed sacrococcygeal teratoma. A Unique Expression of Trisomy 1q. Cancer Genet Cytogenet. 2000;117:84–86. | |
Dundar M, Uzak A, Erdogan M, et al. Partial trisomy 3q in a child with sacrococcygeal teratoma and Cornelia de Lange syndrome phenotype. Genet Couns. 2011;22:199–205. | |
Chen CP, Chang SD, Lee YX, et al. Ring chromosome 21 presenting with sacrococcygeal teratoma: prenatal diagnosis, molecular cytogenetic characterization and literature review. Gene. 2013;522:111–116. | |
López Ginés C, Gil R, Pellin A, Martorell M, Vilar F, Llombart-Bosch A. Trisomy 12 and translocation (7;9) in an ovarian immature teratoma. Int J Gynecol Pathol. 1989;8:277–285. | |
Schwartz S, Raffel LJ, Sun CC, Waters E. An unusual mosaic karyotype detected through prenatal diagnosis with duplication of 1q and 19p and associated teratoma development. Teratology. 1992;46:399–404. | |
Mertens F, Kullendorff CM, Hjorth L, Alumets J, Mandahl N. Trisomy 3 as the sole karyotypic change in a pediatric immature teratoma. Cancer Genet Cytogenet. 1998;102:83–85. | |
Le Caignec C, Winer N, Boceno M, et al. Prenatal diagnosis of sacrococcygeal teratoma with constitutional partial monosomy 7q/trisomy 2p. Prenat Diagn. 2003;23:981–984. | |
Batukan C, Ozgun MT, Basbug M, Caglayan O, Dundar M, Murat N. Sacrococcygeal teratoma in a fetus with prenatally diagnosed partial trisomy 10q (10q24. 3→qter) and partial monosomy 17p (p13. 3→ pter). Prenat Diagn. 2007;27:365–368. | |
Dicker F, Haferlach C, Kern W, Haferlach T, Schnittger S. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007;110(4):1308–1316. | |
Pachajoa H, Escobar LEM. Mosaic trisomy 13 and a sacral appendage. BMJ Case Rep. 2013;2013:bcr2012008150. | |
Scheres JMJC, De Pater JM, Stoutenbeek P, Wijmenga C, Rosenberg C, Pearson PL. Isochromosome 1q as the sole chromosomal abnormality in two fetal teratomas: possible trisomic or tetrasomic zygote rescue in fetal teratoma with an additional isochromosome 1q. Cancer genetics and cytogenetics 1999;115(1):1–10. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.