Back to Journals » OncoTargets and Therapy » Volume 13
Ruxolitinib Plus Decitabine Effectively Treats Myelodysplastic Syndrome/Myeloproliferative Neoplasm, Unclassifiable, by Decreasing the Variant Allele Frequency of KRAS
Authors Luo S, Xu X ![]() , Ye X, Zhu X
, Ye X, Zhu X ![]() , Wu C, Chen D, Jin J
, Wu C, Chen D, Jin J ![]() , Zheng Y, Zheng M, Huang J
, Zheng Y, Zheng M, Huang J
Received 15 July 2020
Accepted for publication 15 September 2020
Published 9 October 2020 Volume 2020:13 Pages 10143—10148
DOI https://doi.org/10.2147/OTT.S272207
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Shuna Luo,1 Xiaofei Xu,1 Xingnong Ye,1,2 Xiaoqiong Zhu,1 Cai Wu,1 Dan Chen,1 Jingxia Jin,1 Yan Zheng,1 Mengli Zheng,1 Jian Huang1,2
1Department of Hematology, The Fourth Affiliated Hospital of Zhejiang University School of Medicine, Yiwu, Zhejiang, People’s Republic of China; 2Department of Hematology, The First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Jian Huang
Department of Hematology, The Fourth Affiliated Hospital of Zhejiang University School of Medicine, N1 Shangcheng Road, Yiwu, Zhejiang 322000, People’s Republic of China
Tel +86 1 886 796 1032
Fax +86 5 798 993 5555
Email [email protected]
Abstract: Myelodysplastic syndrome/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U) is a subtype of MDS/MPN that exhibits a combination of the features of both MDS and MPN. To date, no curative treatment is available for MDS/MPN-U; however, previous studies have suggested a potential survival advantage for ruxolitinib and hypomethylating agents. We reported a case of a JAK2-negative but KRAS-positive MDS/MPN-U patient treated with ruxolitinib plus decitabine. After treatment, the patient’s clinical symptoms were moderated, and the size of the spleen and the peripheral blood cell counts were reduced. These effects might be due to the regimen’s ability to reduce STAT5 activation and upregulate microRNA-181c to downregulate the variant allele frequency (VAF) of KRAS.
Keywords: myelodysplastic syndrome, myeloproliferative neoplasm, decitabine, ruxolitinib, KRAS
Introduction
Myelodysplastic syndrome/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U) is a subtype of MDS/MPN that spans from MDS characterized by morphologic dysplasia and ineffective haematopoiesis to MPN with proliferative features.1 Poor prognosis and without standard treatment facilitated diverse therapeutic studies. Previous studies suggested that hypomethylating agents (HMAs) could improve overall survival (OS) among high-risk MDS/MPN-U.2 Moreover, a Phase II trial of ruxolitinib in combination with azacytidine in MDS/MPN demonstrated that the protocol was beneficial to MDS/MPN-U. Here, we found that the decitabine plus ruxolitinib regimen was effective for a JAK2-negative but KRAS-positive MDS/MPN-U patient.
Patient and Methods
Patient
An 80-year-old female who presented with a greater than 6-month history of dizziness along with fatigue and a 5-month history of leukopenia and anaemia was admitted to our hospital. Excluding penicillin allergy history and amaurosis, she denied a past medical history. Physical examination revealed anaemia appearance and petechial scatter on the palate. Before admission, routine blood tests showed that the white blood cell (WBC) count was 3.6×10^9/L, the haemoglobin (HB) level was 76 g/L, and the platelet (PLT) count was 661×10^9/L (Figure 1I). Ultrasonography showed that the spleen was 4.30 cm thick (Figure 1E). A peripheral blood smear revealed 8% blasts with teardrop-shaped red blood cells (Figure 1A). Moreover, the cell morphology of bone marrow (BM) suggested granulocyte hyperplasia along with pathological haematopoiesis (6%), and erythroid lineage hypoplasia revealed only 9% erythroblasts with teardrop-shaped red blood cells. In addition, the number of megakaryocytes decreased with hypersegmentation and multinucleation, whereas the function of megakaryocytes was fine (Figure 1B). Flow cytometry of BM demonstrated that the myeloid progenitor cell percentage was 2.95% and that CD45 expression was increased (Figure 1K). In addition, BM biopsy showed that BM haematopoietic tissue was hyperplastic with immature cell proliferation and morphologic dysplasia as well as slight reticular fibre hyperplasia (Figure 1C and D). Furthermore, KRAS p. G12D (34.7%), EZH2 p. E22Rfs*15 (35.6%), EZH2 p. N692S (40.8%), RUNX1 p. G165Afs*12 (34.6%), RUNX1 p. D332N (49.8%), STAG2 c.2097–2A>G (30%), PLCG1 p. A578D (47.6%), and CREBBP p. E1050k (37.3%) were detected by next-generation DNA sequencing (NGS), whereas JAK2, CALR and MPL genes were not (Figure 1J, Tables 1 and 2). Moreover, the chromosome karyotype was normal. The patient harboured cytopenia for 5 months with blasts (8% in peripheral blood smear) and morphologic dysplasia that met the characteristics of MDS. Otherwise, prominent thrombocytosis with BM fibrosis and splenomegaly implied MPN. Furthermore, neither a preceding history of MPN or MDS nor recent cytotoxic or growth factor therapy information was found. BCR-ABL, PDGFR and FGFR fusion in gene analysis or isolated del(5q), chr3 inversion in chromosome or features of mixed MDS MPN were not revealed. The patient cannot be assigned to MDS, MPN or MDS/MPN other categories; therefore, she was diagnosed with MDS/MPN-U according to the World Health Organization’s 2016 version of haematologic neoplasm classification.3
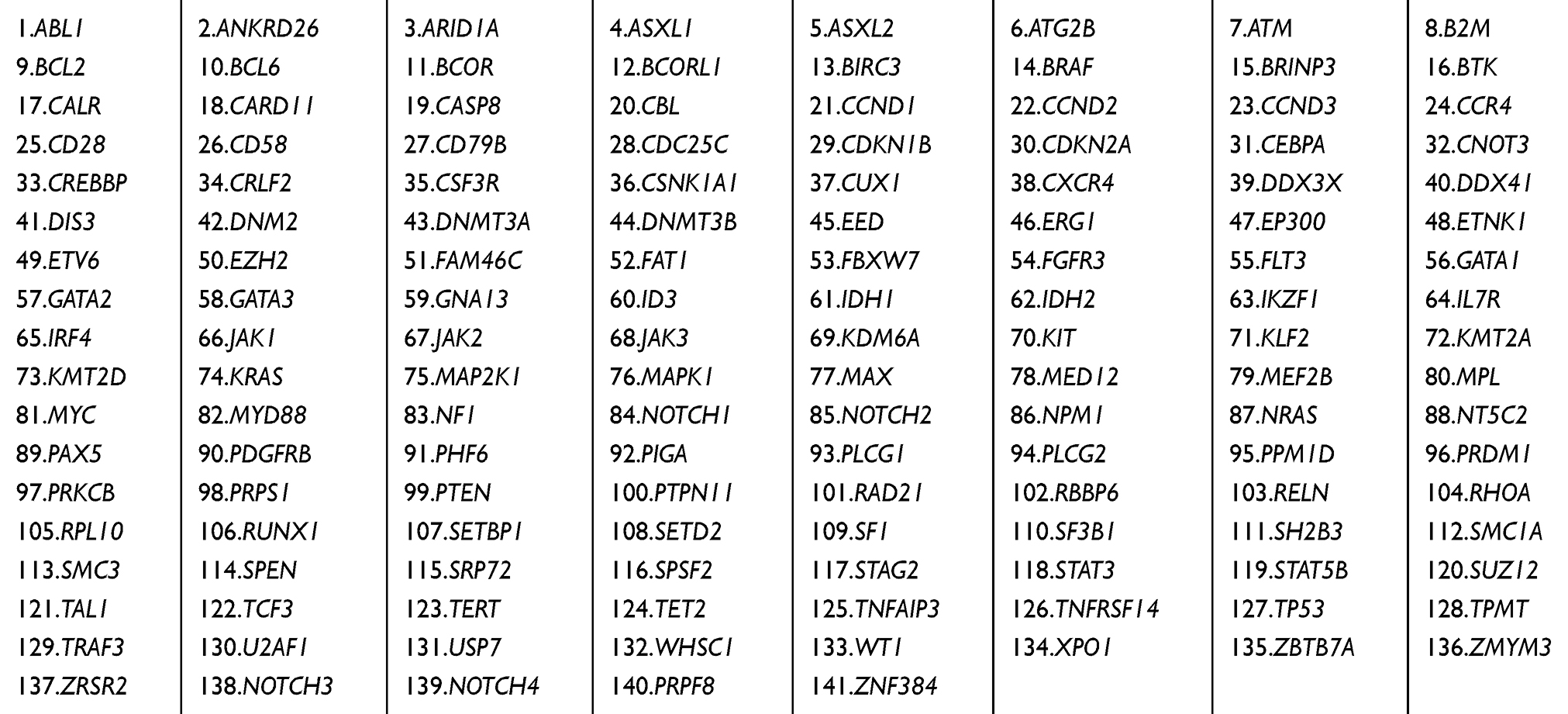 |
Table 1 Genes Assessed by Targeted Sequencing |
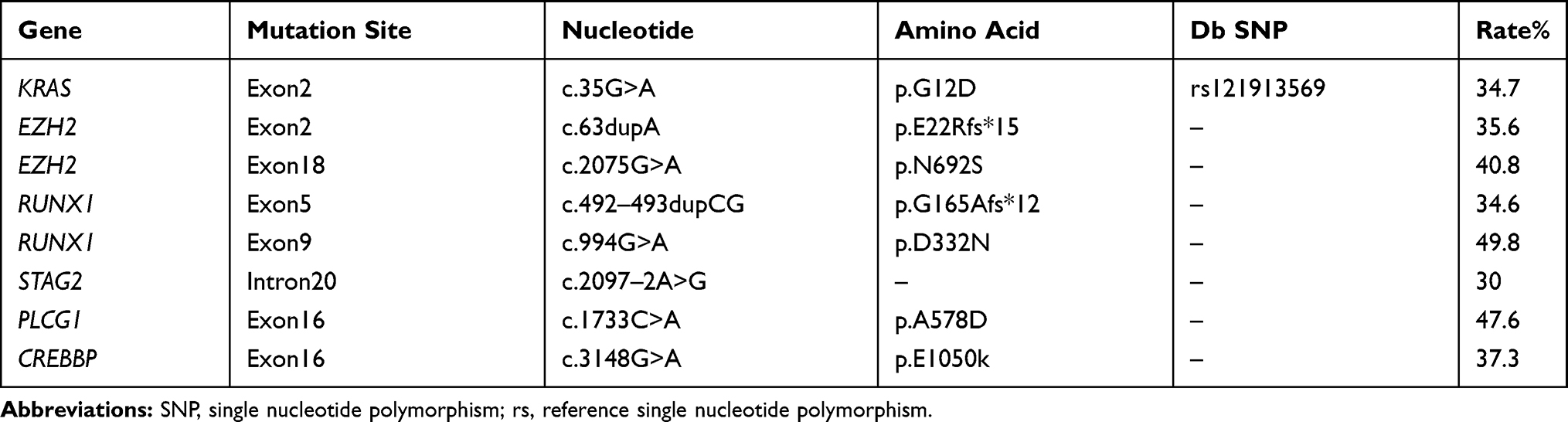 |
Table 2 Next-Generation DNA Sequencing of Bone Marrow at Diagnosis |
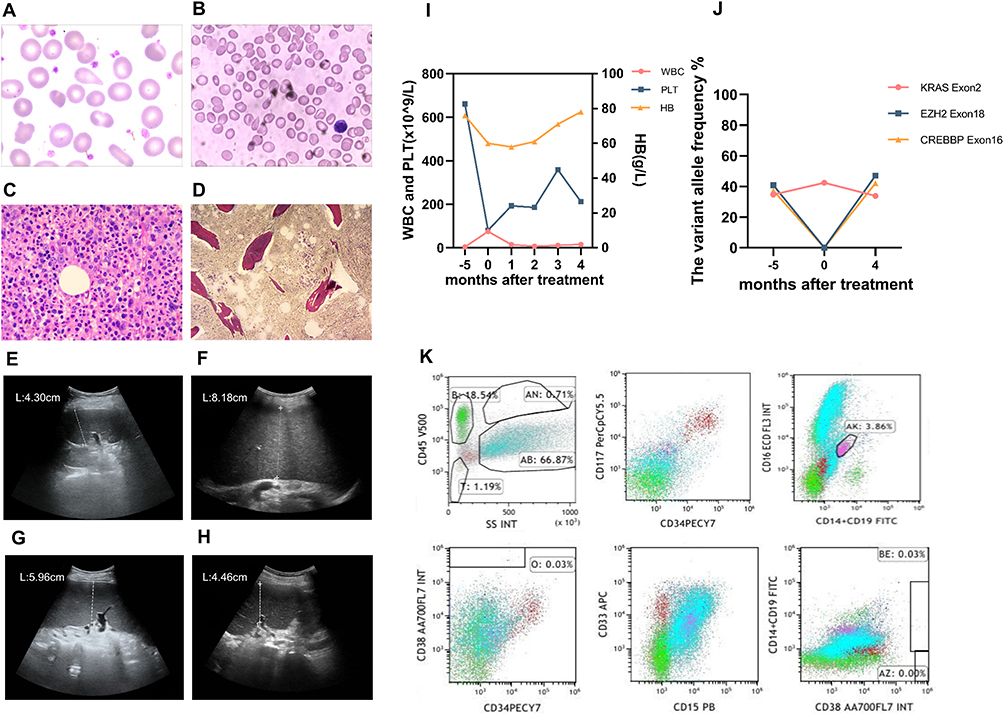 |
Figure 1 Laboratory and imaging results of patients. (A) Morphological assessment of peripheral blood (Wright’s staining, oil-immersion lens, × 1000) revealed teardrop-shaped red blood cells. (B) Morphological assessment of bone marrow (Wright’s staining, oil-immersion lens, × 1000) revealed erythroid lineage hypoplasia with teardrop-shaped red blood cells. (C) Bone marrow biopsy (haematoxylin-eosin staining, low power lens, × 400) revealed erythroid lineage hypoplasia. (D) Reticular fibre staining of bone marrow (reticular fibre staining, low power lens, × 100) revealed slight reticular fibre hyperplasia. (E) Splenic ultrasonography revealed that the thickness of the spleen at diagnosis was 4.30 cm. (F) The thickness of the spleen at disease progression was 8.18 cm. (G) The thickness of the spleen at day 15 of the first cycle was 5.96 cm. (H) The thickness of the spleen after 4 cycles of treatment was 4.46 cm. (I) The white blood cell (WBC) count, haemoglobin (HB) level and platelet (PLT) count at diagnosis (5 months before treatment), disease progression (treatment initiation) and after 1 to 4 cycles of treatment (1–4 months after treatment). (J) The variant allele frequency (VAF) of KRAS exon 2, EZH2 exon 18, and CREBBP exon 16 mutation at diagnosis (5 months before treatment), disease progression (treatment initiation) and after 4 cycles of treatment (4 months after treatment). (K) The immunophenotype of bone marrow cells revealed increased CD45 expression. |
After diagnosis, the patient rejected chemotherapy considering its side effects and was only treated with symptomatic methods. Approximately 5 months later, she complained of decreased appetite and returned to our department. Physical examination showed anaemia, hepatomegaly (one finger under the rib) and splenomegaly (two fingers under the rib and right edge over midline). The thickness of the spleen was 8.80 cm, whereas the previous thickness was 4.30 cm by ultrasonography (Figure 1F). Shockingly, blood examination revealed that the WBC count was 75.1×10^9/L, the HB level was 60 g/L, and the PLT count was 80×10^9/L (Figure 1I). BM cell morphology displayed extremely increased proliferation of the granulocyte lineage, whereas proliferation of other lineages was inhibited. The myeloid progenitor cell count was 3.60%, which was accompanied by CD117 overexpression and decreased CD45 expression as determined by flow cytometry of BM. BM biopsy illuminated that BM haematopoietic tissue exhibited increasing hyperplasia, and hyperplasia was most prominent in the granulocyte lineage. NGS showed that the variant allele fractions of KRAS, EZH2, RUNX1 STAG2, and PLCG2 mutations were increased (Figure 1J, Table 2).
These manifestations and examinations implied that the disease progressed, so we tried a new regimen that combined ruxolitinib and decitabine. She received 15 mg/m2 decitabine intravenously weekly four times for each 28-day cycle and 5 mg ruxolitinib orally twice a day continuously in cycles. The dose of ruxolitinib was adjusted according to the condition, and the maximum dose was 15 mg orally twice a day.
Methods
Genomic DNA was purified from bone marrow with Gentra Puregene Blood Kit (Qiagen, Hil- den, Germany) according to the protocol. High-throughput gene sequencing was performed using ultrahigh multiple PCR exon enrichment technology with an average sequencing depth of 800×. Mutation analysis was performed using the Ion Reporter System and Variant Reporter Software.
Ethics Statement
This study was approved by the Ethics Committee of the Fourth Affiliated Hospital of Zhejiang University School of Medicine. Before collecting clinical isolates from the patient, we informed her of our research purposes, and written informed consent for participation in the study was obtained. Written informed consent for publication of the case details and clinical images was obtained from the patient.
Results
On day 15 of the first cycle, hepatobiliary pancreaticosplenic ultrasonography was performed with a reduced spleen thickness of 5.96 cm (Figure 1G). The uncomfortable symptoms were alleviated, and the WBC count was 14.3×10^9/L after one cycle. After three cycles, the thickness of the spleen was reduced to 4.80 cm. After four cycles, she felt better. Blood examination revealed that the WBC count was 15.3×10^9/L, the HB level was 78 g/L, and the PLT count was 212×10^9/L (Figure 1I). In addition, the thickness of the spleen was decreased to 4.46 cm (Figure 1H). The variant allele frequency (VAF) of KRAS, EZH2, RUNX1, STAG2 and PLCG1 mutations was decreased (Figure 1J, Table 3). During the course of disease, no abnormal karyotypes were found. According to the MDS/MPN proposed response criteria for MDS/MPN published by the MDS/MPN International Working Group in 2015,4 clinical benefit was obtained.
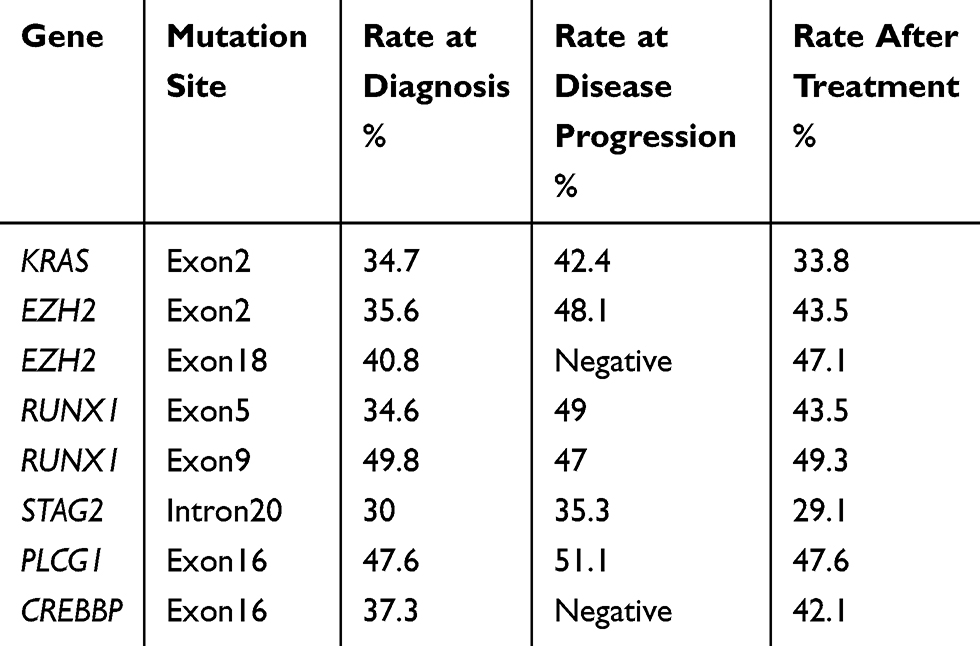 |
Table 3 The Variant Allele Fraction of Mutations Throughout the Disease Course |
Safety was assessed based on Common Terminology Criteria for Adverse Events version 4.0. During the treatment, neither haematological toxicity nor nonhaematological toxicity occurred, and all the doses were conducted as planned.
Discussion
MDS/MPN, an overlap disorder, may have mutational profiles that differentiate this condition from analogous disorders.5 This disorder is more likely to harbour gene mutations in epigenetic regulators or splicing factors that are related to morphologic dysplasia in conjunction with mutations associated with the activation of growth factor signalling pathways.5 Meggendorfer M demonstrated that MDS/MPN-U tended to be associated with mutations in epigenetic regulation, the JAK-STAT pathway and splicing but marginally carried RAS pathway-associated mutations.6 In addition, RAS pathway mutations often coexist with RUNX1, GATA2, and STAG2 mutations.7 The patient harboured KRAS, EZH2, RUNX1, STAG2, PLCG1 and CREBBP mutations that were combined epigenetic regulator, transcription factor, and growth factor signalling pathway-associated mutations.
MDS/MPN-U is a disease that exhibits both dysplastic and proliferative features, and accurate risk stratification is undetermined.1 To date, the prognostic model of MDS/MPN-U refers to MDS or MPN, such as the International Prognostic Scoring System (IPSS) or Revised IPSS (R-IPSS). Previous studies suggested four established MDS prognostic models, which included IPSS,8 R-IPSS,9 Global MD Anderson (MDA)10 model and low-risk MD Anderson Risk Model (LR-MDAS).11 Those models successfully stratified MDS/MPN-U patients for OS and leukaemia-free survival (LFS), and R-IPSS was more effective than others.12 In this case, the patient was stratified as intermediate by R-IPSS.
MDS/MPN-U is an incurable disease with poor outcome, and treatment continues to be challenging. Ruxolitinib and azacytidine were used in a phase II trial in 35 MDS/MPN patients, demonstrating a response rate of 57%.13 After four cycles of ruxolitinib plus decitabine, the symptom response, spleen response, peripheral blood improvement, and decreased variant allele fraction of KRAS implied that this regimen was effective for MDS/MPN-U with KRAS mutation. RAS genes encode a family of 21-kDa proteins that belong to small guanosine triphosphate hydrolase enzymes and regulate cell proliferation and differentiation by activating the Raf/Mek/Erk and PI3K/Akt pathways, which are strongly associated with myeloid malignancies.14,15 In MDS/MPN, the RAS pathway promoted cell proliferation by causing granulocyte-macrophage colony stimulating factor hypersensitivity.16 In this case, the incremental VAF of the KRAS mutation might play an important role in disease progression. However, drugs targeting KRAS have not been applied in the clinic at present. As a Janus kinase (JAK)1/2 inhibitor, ruxolitinib was also useful in haematological malignancies with RAS pathway hyperactivation because ruxolitinib could reduce STAT5 activation.17,18 STAT5 is important to maintain the overactivated RAS pathway, and ruxolitinib could reverse the expansion of immature myeloid cells and decrease autonomous colony-forming unit-granulocyte-macrophage formation to alleviate symptoms and reduce the spleen size by reducing STAT5 activation.17,18 Furthermore, previous research demonstrated that ruxolitinib markedly reduced the tumour cell proliferation of KRAS-mutated mice and decreased the KRAS activation gene signature.19 In addition, as a DNA hypomethylating drug, decitabine could upregulate microRNA-181c, which is downregulated by DNA methylation, to suppress K-RAS protein expression.20 Reduced KRAS mutation expression induced by ruxolitinib plus decitabine likely contributed to the alleviation of the patient’s symptoms, splenomegaly and peripheral blood cell counts.
Other drugs might be effective in the disease. Patients presenting with proliferative features, including leukocytosis and splenomegaly, can be treated with hydroxyurea to manage symptoms and control leukocytosis.21 However, the coexistence of proliferative features and cytopenia made HMAs more appropriate.21 Trametinib, an oral selective MEK1 and MEK2 inhibitor,22 acts downstream of KRAS to suppress signalling through the Raf/Mek/Erk pathway.23 This treatment potentially suppresses the Raf/Mek/Erk pathway to restrain cell proliferation.
Conclusion
In conclusion, our research indicated that the ruxolitinib plus decitabine regimen was effective in JAK2-negative but KRAS-positive patients by reducing STAT5 activation and upregulating microRNA-181c to decrease the VAF of KRAS. However, additional investigations of this regimen are warranted.
Funding
The research was supported by the Key Project of Jinhua Science and Technology Plan, China (Nos. 2020XG-29 and 2020-3-011); the 2018-2020 Key Medical Discipline (Haematology) Fund of Yiwu, China; the 2019-2021 Key Medical Discipline (Haematology) Fund of Jinhua, China; and the 2019-2024 Academician Workstation Fund of the Forth Affiliated Hospital of Zhejiang University School of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Smith BN, Savona M, Komrokji RS. Challenges in myelodysplastic/myeloproliferative neoplasms (MDS/MPN). Clin Lymphoma Myeloma Leuk. 2019;19(1):1–8. doi:10.1016/j.clml.2018.11.019
2. Myelodysplastic/myeloproliferative neoplasms unclassified (MDS/MPN-U) overlap can we alter the natural history? Blood. 2016;128(22).
3. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
4. Savona MR, Malcovati L, Komrokji R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125(12):1857–1865. doi:10.1182/blood-2014-10-607341
5. Tanaka TN, Bejar R. MDS overlap disorders and diagnostic boundaries. Blood. 2019;133(10):1086–1095. doi:10.1182/blood-2018-10-844670
6. Meggendorfer M, Jeromin S, Haferlach C, Kern W, Haferlach T. The mutational landscape of 18 investigated genes clearly separates four subtypes of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2018;103(5):e192–e195. doi:10.3324/haematol.2017.183160
7. Zhang H, Wilmot B, Bottomly D, et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood. 2019;134(11):867–879. doi:10.1182/blood.2019000611
8. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. doi:10.1182/blood.V89.6.2079
9. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. doi:10.1182/blood-2012-03-420489
10. Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original international prognostic scoring system. Cancer. 2008;113(6):1351–1361. doi:10.1002/cncr.23697
11. Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22(3):538–543. doi:10.1038/sj.leu.2405070
12. Mangaonkar AA, Swoboda DM, Coltro G, et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center study of 135 consecutive patients. Leukemia. 2020;34(2):656–661. doi:10.1038/s41375-019-0574-x
13. Assi R, Kantarjian HM, Garcia-Manero G, et al. A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am J Hematol. 2018;93(2):277–285.
14. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120(17):3397–3406.
15. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17–33. doi:10.1016/j.cell.2017.06.009
16. Deininger MWN, Tyner JW, Solary E. Turning the tide in myelodysplastic/myeloproliferative neoplasms. Nat Rev Cancer. 2017;17(7):425–440. doi:10.1038/nrc.2017.40
17. Sachs Z, Been RA, DeCoursin KJ, et al. Stat5 is critical for the development and maintenance of myeloproliferative neoplasm initiated by Nf1 deficiency. Haematologica. 2016;101(10):1190–1199. doi:10.3324/haematol.2015.136002
18. Geissler K, Jager E, Barna A, et al. Is ruxolitinib a potentially useful drug in hematological malignancies with RAS pathway hyperactivation? Haematologica. 2016;101(12):E492. doi:10.3324/haematol.2016.156448
19. Mohrherr J, Haber M, Breitenecker K, et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int J Cancer. 2019;145(12):3376–3388.
20. Go H, Jang J-Y, Kim C-W, Huh J, Kim P-J, Jeon YK. Identification of microRNAs modulated by DNA hypomethylating drugs in extranodal NK/T-cell lymphoma. Leuk Lymphoma. 2020;61(1):66–74. doi:10.1080/10428194.2019.1654096
21. Hunter AM, Zhang L, Padron E. Current management and recent advances in the treatment of chronic myelomonocytic leukemia. Curr Treat Options Oncol. 2018;19(12):67. doi:10.1007/s11864-018-0581-6
22. Zeiser R, Andrlová H, MeissF. Trametinib (GSK1120212).Recent Results Cancer Res. 2018;211:91–100. doi:10.1007/978-3-319-91442-8_7.
23. Manchado E, Weissmueller S, Morris JP, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534(7609):647–651. doi:10.1038/nature18600
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.