Back to Journals » Nursing: Research and Reviews » Volume 6
Rufinamide in patients with Lennox-Gastaut syndrome
Received 30 June 2016
Accepted for publication 1 September 2016
Published 2 November 2016 Volume 2016:6 Pages 51—59
DOI https://doi.org/10.2147/NRR.S116114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Cindy Hudson
Peggy O Clark,1 Patricia A Gibson2
1Cincinnati Children’s Hospital Medical Center, The University of Cincinnati College of Medicine, Cincinnati, OH, 2Epilepsy Information Service, Wake Forest University School of Medicine, Winston-Salem, NC, USA
Objectives: Lennox–Gastaut syndrome (LGS) is an epileptic encephalopathy with an onset at the age of ~4 years. LGS is notoriously difficult to manage, as most patients experience multiple seizures per day, despite their concomitant use of several antiepileptic drugs (AEDs). Rufinamide (BANZEL®) is an AED approved by the US Food and Drug Administration for the adjunctive treatment of seizures associated with LGS in pediatric patients aged ≥1 year and in adults. The expert care of nurses knowledgeable in the treatment options for LGS is valuable to patients and caregivers. This review summarizes the existing knowledge on LGS and data from clinical and real-world studies on the use of rufinamide in patients with LGS.
Methods: Recent review articles and information from the Epilepsy Foundation Website were reviewed for data on LGS treatment. Primary articles on rufinamide were also selected for review.
Results and conclusion: The efficacy and safety of rufinamide have been evaluated in children and adults by using double-blind, open-label, and observational studies. In general, these studies indicate that rufinamide effectively reduces the frequency and severity of multiple seizure types associated with LGS and has tolerable side effects, the most common being vomiting and somnolence. Dosing modifications based on age, weight, and concomitant AED usage are recommended for patients using rufinamide.
Keywords: Lennox–Gastaut syndrome, anticonvulsants, rufinamide
Introduction: Lennox–Gastaut syndrome
Lennox–Gastaut syndrome (LGS) is an epileptic encephalopathy with onset during childhood.1 LGS is characterized by a triad of symptoms: multiple seizure types, slow spike-and-wave patterns on an interictal electroencephalogram (EEG), and cognitive impairment.2 The majority of patients with symptomatic LGS experience more than 3 seizures per day.3 The seizure types that are most frequently observed in LGS are tonic, atypical absence, atonic, myoclonic, generalized tonic–clonic, and focal onset (previously referred to as partial onset) seizures.3–5 Patients with LGS experience frequent seizures, usually during sleep, with 10–20 Hz fast rhythms.6,7
LGS is thought to comprise as much as 10% of all childhood epilepsies.4,8,9 Onset of LGS usually occurs before 8 years of age, and peaks between ages 3 and 5 years.2 The median age of LGS onset is ~4 years.3 Many patients (46.9%, by one estimate) who develop LGS during childhood continue to present the complete disease profile into adulthood.10
For the majority of patients with LGS, seizures remain difficult to treat,11–13 and most of the patients cannot live independently as adults.12 Support from a multidisciplinary team, including epilepsy specialists, physicians, nurses, psychiatrists, psychologists, physiotherapists, education specialists, social workers, family members, and friends, is important in the treatment and management of LGS.2,13,14 Frequent “drop attacks” caused by multiple seizure types lead to injuries,2 as well as burdensome medical costs.15 Although patients can use headgear to prevent some injuries, this can interfere with the quality of life (QoL), as it is highly stigmatizing and may deter participation in activities.15,16 Furthermore, with epileptic encephalopathies, such as LGS, seizure activity exacerbates patients’ cognitive and behavioral impairments.17 The behavioral and cognitive symptoms of LGS are notoriously difficult to manage and have not been well studied.4
The relative risk of mortality in patients with LGS is quite high. In the Atlanta metropolitan area, the relative risk of mortality in children with LGS compared to children in the general population was found to be 13.92 (95% confidence interval: 7.19–24.31).18 In many cases, death results from accidents related to seizures.19 In addition, the risk of sudden unexpected death in epilepsy is higher in patients with complicated forms of epilepsy (those associated with the presence of known structural brain lesion, abnormal neurologic examination, or significant intellectual disability), such as LGS, relative to patients with uncomplicated epilepsy (ie, in the absence of these factors).20,21 Overall, LGS has a significant impact on the health-related QoL (HRQoL) of patients and their caregivers.22
LGS is difficult to diagnose, in part due to its varied etiologies. Indeed, many other epileptic syndromes share features with LGS.16 Due to the difficulty in distinguishing between tonic and atonic seizures, these 2 seizure types are sometimes grouped as “drop attacks.”23,24 Aside from the challenge of discriminating among seizure types, tonic and atypical absence seizures are often subtle, and clinically, they are not easily identified. In addition, the frequency of each type of seizure tends to fluctuate, sometimes being brought on by the treatments themselves.6,16 A correct diagnosis of LGS must include a detailed medical history, along with waking and sleeping EEGs and may require years of follow-up to confirm.16
Misdiagnosis of LGS could lead to futile attempts to treat patients with drugs that are not effective for their actual diagnosis. At the same time, because LGS is resistant to most antiepileptic drugs (AEDs), even patients who are accurately diagnosed with LGS often use multiple AEDs concurrently.4,23,24 Unfortunately, such “polytherapy” can increase the incidence of adverse events (AEs).2 In some cases, the pursuit of seizure reduction should be reconsidered, for example, when the patient’s QoL is reduced more by treatment-related AEs than by the seizures.2 Often, epilepsy specialist nurses are the main providers of skilled and individualized care to meet the intricate needs of their patients25 and can help with these decisions.
This review summarizes the existing knowledge on LGS and data from clinical and real-world studies on the use of rufinamide (BANZEL®; Eisai Inc., Woodcliff Lake, NJ, USA) in patients with LGS. Recent review articles and information from the Epilepsy Foundation Website were reviewed for data on LGS treatment.26–28 Primary articles focused on the use of rufinamide in patients with LGS were also selected to gather information on pharmacokinetics and data on clinical trial and real-world efficacy and safety.
Treatment for LGS
Rufinamide, an orphan drug, is indicated for the adjunctive treatment of seizures associated with LGS, both in pediatric patients aged ≥1 year and in adults.29 Other AEDs approved for LGS include clonazepam, felbamate, lamotrigine, topiramate, and clobazam (Table 1).26,27 Several other AEDs, while not specifically indicated for LGS, are often used to treat the seizures associated with the syndrome (Table 1). Patients with LGS should avoid carbamazepine, gabapentin, oxcarbazepine, pregabalin, and tiagabine, due to the risk of seizure aggravation.28 Data on the use of vigabatrin to treat seizures associated with LGS are inconsistent.6,30
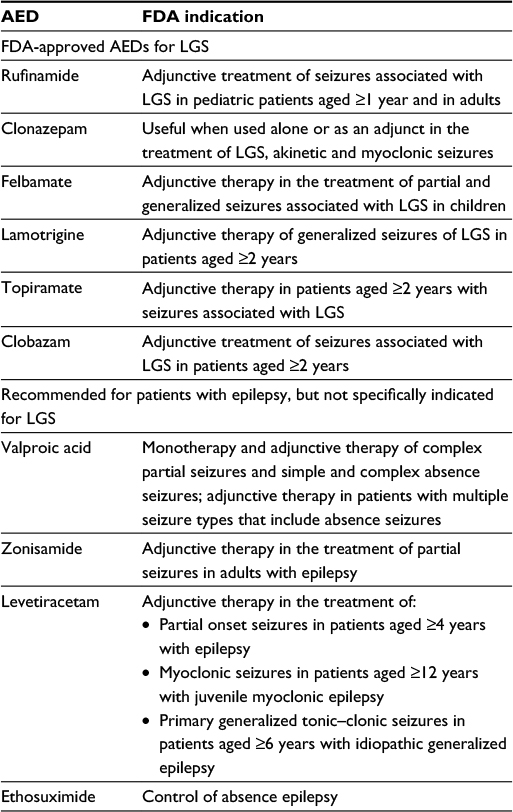 | Table 1 Antiepileptic drug treatments for LGS Note: Data from Benbadis and Sheth,26 Epilepsy Foundation Website,27 and Carmant and Whiting.28 Abbreviations: AED, antiepileptic drug; FDA, the US Food and Drug Administration; LGS, Lennox–Gastaut syndrome. |
Besides these pharmacologic treatments, several nonpharmacologic treatments may be used in the treatment of LGS (Table 2). Based on the clinical experience of the authors, the ketogenic diet should be implemented as early as possible, as this diet is easier to control in younger children.
 | Table 2 Nonpharmacologic treatments for Lennox–Gastaut syndrome Note: Data from Arzimanoglou et al,2 Carmant and Whiting,28 and Schachter and Sirven.54 |
Rufinamide
The European Medicines Agency and the US Food and Drug Administration granted orphan status to rufinamide in 2004.31 Although rufinamide is approved for the adjunctive treatment of seizures associated with LGS,29,32 its use in the treatment of drug-resistant partial seizure conditions has also been investigated.33,34
Rufinamide is a triazole derivative that differs structurally from other AEDs,31 and its mechanism of action is not fully understood.29 Rufinamide prevents the hyperexcitation of neurons by extending the inactivation phase of voltage-gated sodium channels,31 which may stabilize the cell membrane.35,36 Rufinamide may also prevent seizures by having an inhibitory effect at the human recombinant metabotropic glutamate receptor subtype 5.31
Rufinamide is formulated as a film-coated tablet (200 or 400 mg) or a 40 mg/mL oral suspension and should be taken with food.29 The tablets can be administered as whole, half, or crushed. The oral suspension, which has the same bioavailability as the tablet formulation, is also available, which is suitable for those who have trouble swallowing.37 The oral suspension has also been used with endoscopic gastrostomy or nasogastric tube.38 The dosing recommendations for rufinamide in Europe depend on the patient’s body weight and concomitant use of valproic acid.31,39 In the USA, the recommended starting daily dose of rufinamide in pediatric patients (age ranging from 1 year to <17 years) with LGS is ~10 mg/kg, administered in 2 equally divided doses. The dose should be increased by ~10 mg/kg every other day until a maximum daily dose of 45 mg/kg (not to exceed 3,200 mg), administered in 2 equally divided doses, is reached. The recommended starting daily dose of rufinamide in adults aged ≥17 years with LGS is 400–800 mg per day, administered in 2 equally divided doses. The dose should be increased by 400–800 mg every other day until a maximum daily dose of 3,200 mg, administered in 2 equally divided doses, is reached. Patients taking valproic acid should begin rufinamide at a dose <10 mg/kg per day (pediatric patients) or 400 mg per day (adults).29
Pharmacokinetics
Rufinamide is largely metabolized to an inactive product.31 The pharmacokinetic properties of rufinamide were investigated in a 5-week multiple-dose study. Among people with epilepsy, the half-life (t1/2) of rufinamide was ~7 hours and did not change over time.31
Studies evaluating the use of rufinamide in pediatric, adult, and elderly populations revealed no significant age-related differences in the pharmacokinetics of the drug.29 Similarly, in a double-blind Japanese study of children and adults with LGS, body weight and age were not found to affect the circulating rufinamide concentration.40 Rufinamide clearance appears to be 6%–14% lower in females than in males.29 Race and renal function also do not seem to affect the pharmacokinetics of rufinamide;31 however, the drug is not recommended for patients with severe hepatic impairment.29
Concomitant use of certain AEDs was found to be associated with reduced plasma rufinamide concentrations; these AEDs included carbamazepine (rufinamide concentration decreased by 19%–26%), vigabatrin (by 14%–30%), phenytoin (by 25%–46%), phenobarbital (by 25%–46%), and primidone (by 25%–46%).31 On the other hand, valproic acid has been shown to reduce rufinamide clearance, which may cause plasma rufinamide concentrations to increase by ~55%–70% in children, 24%–26% in adolescents, and 15% in adults.31 Concomitant use of lamotrigine, topiramate, or benzodiazepines did not significantly affect plasma concentrations of rufinamide.31
Regarding its metabolic effects, no inhibition of the 8 major cytochrome P (CYP) isozymes has been associated with rufinamide, and therefore, it is not expected to affect the metabolism of drugs that are CYP substrates.31 However, clearance of the AEDs carbamazepine and lamotrigine increased by 8%–16% during concomitant rufinamide use, whereas clearance of phenobarbital and phenytoin decreased by 7%–18%.31 In another study, rufinamide did not affect the serum levels of valproic acid, lamotrigine, or clobazam.40 A similar lack of effect was also reported regarding valproic acid, lamotrigine, topiramate, clonazepam, and carbamazepine23 and for carbamazepine, phenytoin, and valproic acid.41 However, rufinamide was found to increase the metabolism of triazolam and an oral contraceptive containing norethindrone and ethinyl estradiol.31
Rufinamide use in the treatment of LGS
The development of tolerance to AEDs in patients with epilepsy42 and the finding that AEDs may aggravate rather than alleviate seizures43 are important considerations for drug resistance. LGS remains resistant to most AEDs, and many patients take several drugs concomitantly in an attempt to control seizures.4,23,24 One study reported that the median number of AEDs that patients had tried before rufinamide was 7.5.44 Rufinamide is an appealing treatment option for LGS because it has been shown to significantly reduce seizure frequency. Rufinamide is rapidly absorbed, and thus, it can be efficiently titrated to the effective dose. Moreover, rufinamide is tolerable and effective in both children and adults and typically leads to only mild or moderate AEs.23,29,45 Specific studies on the efficacy and safety of rufinamide are described in the following sections.
Efficacy
The safety and efficacy of rufinamide adjunctive therapy in adult and pediatric patients with LGS (aged 4–30 years, n=139) were evaluated in a double-blind, randomized, placebo-controlled study.23 This short-term study included 14 days of drug titration, followed by 70 days of maintenance, amounting to a total of 12 weeks. The target dose of rufinamide was ~45 mg/kg per day, with the dose ranging from 1,000 to 3,200 mg per day, depending on the patient’s weight. Specifically, the actual doses by weight were as follows: 1,000 mg per day for patients who weighed 18.0–29.0 kg, 1,800 mg per day for those who weighed 29.1–50.0 kg, 2,400 mg per day for those who weighed 50.1–70.0 kg, and 3,200 mg per day for those who weighed more than 70.0 kg. All patients were taking 1–3 other AEDs during the study, primarily valproic acid, lamotrigine, and topiramate.23
The median number of total seizures per 28 days was found to be reduced in the rufinamide group (290 at baseline and 204 during the double-blind phase), but not in the placebo group (205 both at baseline and in the double-blind phase).23 The median percentage reduction in total seizure frequency was greater in the rufinamide group (32.7%) than in the placebo group (11.7%). Median tonic–atonic (drop attack) seizure frequency was reduced by 42.5% in the rufinamide group, which represents a significantly greater improvement when compared with the placebo group, which experienced a 1.4% increase in seizure frequency (P<0.0001). More patients in the rufinamide group than in the placebo group experienced a 50% reduction in seizure frequency (both total and tonic–atonic). For total seizures, a 50% reduction occurred in 31.1% of rufinamide-treated patients and 10.9% of placebo-treated patients, whereas for tonic–atonic seizures, a 50% reduction occurred in 42.5% of rufinamide-treated patients and 16.7% of placebo-treated patients. The median percent reduction in absence/atypical absence and atonic seizure frequencies also was greater in the rufinamide-treated group. Seizure severity was reduced in 53.4% of rufinamide-treated patients and 30.6% of placebo-treated patients, consistent with the improved parental evaluation scores for this parameter (0.88 for rufinamide and 0.27 for placebo). Other parameters in the parental evaluation did not differ between the groups. The plasma concentrations and dosages of other AEDs that the patients used were unaffected by the rufinamide treatment.23
Patients who completed this study were eligible to be included in a long-term, open-label extension study, with cohorts being followed for up to 3 years.24 This study included both children and adults (age range, 4–37 years; n=124) and was preceded by a double-blind conversion phase, such that all patients in the open-label extension were treated with rufinamide. The dose of the drug was 10–60 mg/kg per day, at the investigator’s discretion (median, 52.9 mg/kg per day). Of the 124 patients participating, 42 (33.9%) patients were still receiving rufinamide at study termination and 82 (66.1%) had withdrawn: 51 patients had withdrawn due to dissatisfaction with treatment efficacy, 12 due to AEs, and 19 for other reasons.24 Despite this patient withdrawal, the seizure frequency decreased at every time point for each cohort of patients treated with rufinamide for at least 6, 12, 18, 24, 30, and 36 months.
In both the original rufinamide treatment arm and the placebo conversion group, the median frequencies of total and tonic–atonic seizures continued to decline throughout the study.24 Seizure frequency reductions of at least 50% were observed for total seizures in 45.1% of patients and for tonic–atonic seizures in 47.9% of patients, during the last 6 months of treatment. Two patients (1.6%) became free of all seizures, whereas 11 patients (9.4%) became free of tonic–atonic seizures within the last 6 months. During the first 6 months of the open-label extension, nearly half of the patients (47.7%) were able to reduce their daily dosages of concomitant AEDs. This trend continued for >50% of patients during months 6–36 of the open-label extension. Overall, the long-term efficacy of rufinamide in the open-label extension was comparable to its short-term efficacy and tolerability in the double-blind study.24
Another double-blind, placebo-controlled study assessed the use of rufinamide in children and adults with LGS in Japan (age range, 4–30 years; n=59).40 A similar weight-dependent dosing scheme was employed in this study (1,000 mg per day for those weighing 15–30 kg, 1,800 mg per day for those weighing 30.1–50 kg, 2,400 mg per day for those weighing 50.1–70 kg, and 3,200 mg per day for those weighing ≥70.1 kg), and the same periods of drug titration and maintenance were used. In this study, rufinamide was associated with a median total seizure frequency reduction of 32.9% (vs 3.1% for placebo) and a median tonic–atonic seizure frequency reduction of 24.4% (vs 3.3% for placebo). Tonic, myoclonic, and partial seizure frequencies all were reduced in patients treated with rufinamide versus placebo. In the rufinamide group, 25.0% of patients responded with ≥50% fewer tonic–atonic seizures versus 6.7% of the placebo group. Clinical global impressions of the patients’ conditions were significantly better in the rufinamide group than in the placebo group (P=0.007).40
Efficacy: real-world experience
The efficacy of rufinamide has also been explored in observational studies. Coppola et al conducted an Italian multicenter, prospective, add-on, open-label study in children and adults with LGS (age range, 4–34 years; median =15 years; n=43).44 The mean dose of rufinamide was 33.5 mg/kg per day for those taking valproic acid and 54.5 mg/kg per day for those not taking valproic acid. A reduction in total seizures of ≥50% was observed in 60.5% of patients (responders) after 12 months, and 9.3% of patients became seizure-free, whereas 30.2% experienced no change. Among the 26 responders, a response ≥50% was observed specifically for drop attacks in 78.9% of patients and for tonic seizures in 57.7% of patients; 10 patients became free of either drop attacks or tonic seizures; tonic–clonic seizures were reduced by ≥50% in 11.6% of responders; 2 patients (4.7%) had an increased seizure frequency in response to rufinamide.44
A European (German and Austrian) 12-week observational study was conducted by using retrospective data from 60 patients with epilepsy taking rufinamide, 31 of whom had LGS.46 Among the patients with LGS, 54.8% responded to the treatment with 50% fewer seizures than at baseline. In an 18-month extension of this study using retrospective data from 52 patients with epilepsy taking rufinamide (age range, 1–50 years; mean =14.9 years; median maintenance dose =35.5 mg/kg per day), 31 of whom had LGS, 35.5% of patients with LGS experienced at least 50% fewer seizures in the last 4 weeks of the study than at baseline.45
Vendrame et al retrospectively analyzed the use of rufinamide in children at a single US epilepsy center.47 Of the 77 patients receiving rufinamide in the study population (age range, 1–27 years; median =12 years), 26 patients had LGS, and all were receiving concomitant AEDs. In the LGS group, 10 of the 26 patients (38.4%) were responders (>50% reduction in seizure frequency), and the median seizure frequency reduction was 50%.47
A single-center retrospective cohort study by Kessler et al demonstrated the effectiveness of rufinamide in patients with LGS compared to those with other epilepsy types by using retention rate as the outcome measure.48 For patients not having LGS in this analysis (n=94), the probability of remaining on rufinamide treatment with or without additional therapy was 49% and 40%, respectively, at 12 months compared to 78% and 64% in patients with LGS (n=39). The median time to discontinuation of rufinamide or additional therapy was 18 months for patients with LGS versus 6 months for those with other epilepsy types.48
Overall, in double-blind, open-label, and observational studies, rufinamide has proven to be efficacious in reducing seizure frequency and/or severity in children and young adults, allowing a large proportion of patients to live with 50% fewer seizures, including the drop attacks that so frequently reduce QoL. Although rufinamide overall dosing tends to be lower with slower titration schedules in real-world settings compared with those used in clinical studies, real-world clinical experience still reinforces the efficacy of rufinamide seen in the double-blind clinical studies.49
Safety
In all of the aforementioned studies of the efficacy of rufinamide, the safety of the treatment was also assessed. In the short-term (12-week), double-blind study of children and adults with LGS, drug-related AEs were observed in 55.4% of rufinamide-treated patients and 43.8% of placebo-treated patients.23 Somnolence and vomiting were more frequent in the rufinamide group (24.3% and 21.6%, respectively) than in the placebo group (12.5% and 6.3%, respectively). Six patients withdrew from the study because of AEs, and all of these patients were in the rufinamide-treated group. Status epilepticus occurred in 3 patients, all from the rufinamide-treated group. Serious AEs occurred in an equal number of patients from the rufinamide- and placebo-treated groups (n=2, each). Cognitive/psychiatric AEs were less prevalent among rufinamide-treated patients (17.6%) than among placebo-treated patients (23.4%).23
In the open-label extension study on rufinamide in children and adults with LGS, 70.2% of patients experienced AEs that were attributed to the treatment.24 The most common AEs were vomiting, pyrexia, upper respiratory tract infection, somnolence, diarrhea, nasopharyngitis, reduced appetite, fatigue, anorexia, constipation, aggression, and headache. Serious AEs occurred more frequently during the open-label extension study (13.7%) than in the double-blind study (2.7%), 9 of which were attributed to the treatment: rash, constipation, esophagitis, weight loss, gastritis, anorexia, vomiting, lethargy, and status epilepticus. Overall, the tolerability of rufinamide in the open-label extension study was thought to be similar to that in the double-blind study.24
In the aforementioned Japanese study, 62.1% of patients on rufinamide experienced AEs related to treatment versus 16.7% of placebo-treated patients.40 The most common AEs were reduced appetite, somnolence, and vomiting. Seizures worsened transiently in 27.6% of rufinamide-treated patients and 16.7% of placebo-treated patients, but the majority of these were not attributed to the study treatment. Status epilepticus occurred in 17.2% of patients in the rufinamide-treated group and in 6.7% of patients in the placebo-treated group.40 Of the 59 patients randomized, 54 (91.5%) patients completed the trial, and 5 (8.5%) discontinued early due to AEs; 4 of these 5 patients were in the rufinamide-treated group. The AEs responsible for withdrawal in the rufinamide group were rash, purpura, decreased appetite, vomiting, dizziness, and headache.
The safety of rufinamide is also being evaluated in an ongoing Phase III open-label study, specifically in children (aged 1–4 years), in which rufinamide is administered at a target dose of 45 mg/kg per day.50 The interim data show that treatment-emergent AEs were similar between the rufinamide and any other AED groups, with most events considered mild or moderate.
In general, the AE profile for rufinamide in children aged between 1 and 4 years has been found to be similar to that for older children and adults.29 AEs observed in at least 8% of patients treated with rufinamide and at a higher frequency than in the control group include vomiting (24%), somnolence (16%), bronchitis (12%), constipation (12%), cough (12%), reduced appetite (12%), rash (12%), otitis media (8%), pneumonia (8%), reduced weight (8%), gastroenteritis (8%), nasal congestion (8%), and pneumonia aspiration (8%).29
Safety: real-world experience
In the Italian study by Coppola et al, 23.2% of patients reported AEs, including vomiting, irritability/aggressiveness, drowsiness, skin rash, and reduced appetite.44 In the 18-month European study of patients with various epilepsy syndromes (not only LGS), 37 of 60 participants (61.7%) experienced AEs when taking rufinamide, including fatigue, vomiting, and loss of appetite.45 In both of these studies, most of the AEs occurred during drug titration.44,45 In the retrospective study analyzing the use of rufinamide in children at a US epilepsy center, AEs were reported by 23 (29.9%) of the 77 patients in the total population. AEs included drowsiness in 10 patients (13.0%), rash in 5 (6.5%), dizziness in 4 (5.2%), nausea and vomiting in 3 (3.9%), anorexia in 2 (2.6%), headache in 1 (1.3%), and visual disturbance in 1 (1.3%) patient.47
The safety of rufinamide in certain patients deserves special attention. For instance, rufinamide is contraindicated in patients with familial short QT syndrome, which is associated with an increased risk of sudden death and ventricular arrhythmias, especially ventricular fibrillation.29 In a placebo-controlled study of the QT interval, QT shortening of >20 ms at the time of peak plasma concentration (Tmax) was observed in a higher percentage of rufinamide-treated patients (46% at 2,400 mg, 46% at 3,200 mg, and 65% at 4,800 mg) than placebo-treated patients (5%–10%).29 There is no known clinical risk associated with this degree of QT shortening; however, caution is advised when rufinamide is administered with other drugs that shorten the QT interval, such as digitalis.29,51 A small, open-label study (n=19) evaluating the effect of adjunctive rufinamide on QT interval found that concomitant treatment with lamotrigine or valproic acid resulted in greater QT shortening than that observed with rufinamide alone, although no symptomatic cardiac arrhythmias were reported by any patients.51
Overall, the AEs associated with rufinamide are mild and transient, and in many cases, they do not outweigh the benefits of the drug or lead to discontinuation. Many patients with LGS experience AEs during drug titration and improve after continued use of rufinamide. As shown in clinical practice, the tolerability of rufinamide improves when lower starting doses and slower titration schedules are followed.49
Conclusion: management of LGS and resources for support
Rufinamide is efficacious as an adjunctive treatment for children and adults with LGS. Rufinamide treatment has been investigated in children and adults (aged 1–50 years) in randomized, double-blind, and real-world studies. The frequency of seizures is reduced by ≥50% in many cases, and in a few rare cases, patients become seizure-free.
The AEs associated with rufinamide are tolerated by many patients but may be serious enough to recommend discontinuation in some patients. Common AEs include vomiting and somnolence, which can occur during the titration of rufinamide in the early stages of treatment. Steady-state plasma rufinamide concentrations appear to correlate with a reduction in seizure severity, but also with the incidence of AEs.31 The dose of rufinamide should be based on the age, body weight, and medication profile of an individual patient and may need to be adjusted to reduce the likelihood of AEs or to reflect the progression of the disease. Whenever possible, the authors recommend considering a reduction in the number of medications to minimize drug interactions and side effects. Long-term follow-up of patients using rufinamide is needed to determine whether their prognoses continue to improve over time.
Support from a multidisciplinary team, including epilepsy specialists, physicians, nurses, psychiatrists, psychologists, physiotherapists, education specialists, social workers, family members, and friends, is important in the treatment and management of LGS.2,13,14 By knowing how to classify seizures, as well as the efficacy and side effects of various AEDs, including rufinamide, nurses can assist patients and their caregivers with the treatments prescribed to patients. Table 3 lists the recommended resources for families affected by LGS.52
 | Table 3 Resources and support for families affected by LGS Note: Data from Gibson.52 Abbreviation: LGS, Lennox–Gastaut syndrome. |
Given the high burden of care associated with LGS, social support and resources available to help patients, families, and caregivers to manage this condition are very important. Families affected by LGS may benefit from appropriate resources and support services, improving the HRQoL of the caregivers and the patient.53 Ultimately, the management of LGS should be multifaceted and seek to reduce seizures while also considering intellectual and behavioral comorbidities of the patient, potential AEs associated with AED therapy, and the need for social and developmental support.20
Acknowledgment
Editorial support was funded by Eisai Inc. and provided by Imprint Science, New York, NY, USA.
Disclosure
Peggy O Clark serves on an Advisory Board and Speakers Program of both Eisai and Supernus. Patricia A Gibson serves on the UCB Scholarship committee and has been a consultant to Sunovion Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
Gastaut H, Roger J, Soulayrol R, et al. Childhood epileptic encephalopathy with diffuse slow spike-waves (otherwise known as “petit mal variant”) or Lennox syndrome. Epilepsia. 1966;7(2):139–179. | ||
Arzimanoglou A, French J, Blume WT, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 2009;8(1):82–93. | ||
Goldsmith IL, Zupanc ML, Buchhalter JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia. 2000;41(4):395–399. | ||
Hancock EC, Cross JH. Treatment of Lennox-Gastaut syndrome. Cochrane Database Syst Rev. 2013;2:CD003277. | ||
Berg AT, Millichap JJ. The 2010 revised classification of seizures and epilepsy. Continuum (Minneap Minn). 2013;19(3 Epilepsy):571–597. | ||
van Rijckevorsel K. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat. 2008;4(6):1001–1019. | ||
Genton P, Guerrini R, Dravet C. The Lennox-Gastaut syndrome. In: Meinardi H, editor. Handbook of Clinical Neurology. 29th ed. Amsterdam: Elsevier Science B.V.; 2000:211–222. | ||
Kramer U, Nevo Y, Neufeld MY, Fatal A, Leitner Y, Harel S. Epidemiology of epilepsy in childhood: a cohort of 440 consecutive patients. Pediatr Neurol. 1998;18(1):46–50. | ||
Trevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997;38(12):1283–1288. | ||
Roger J, Remy C, Bureau M, Oller-Daurella L, Beaumanoir A, Favel P, Dravet C. Le syndrome de Lennox-Gastaut de l’adulte [Lennox-Gastaut syndrome in the adult]. Rev Neurol (Paris). 1987;143(5):401–405. French. | ||
Camfield P, Camfield C. Long-term prognosis for symptomatic (secondarily) generalized epilepsies: a population-based study. Epilepsia. 2007;48(6):1128–1132. | ||
Camfield C, Camfield P. Twenty years after childhood-onset symptomatic generalized epilepsy the social outcome is usually dependency or death: a population-based study. Dev Med Child Neurol. 2008;50(11):859–863. | ||
Crumrine PK. Lennox-Gastaut syndrome. J Child Neurol. 2002;17(Suppl 1):S70–S75. | ||
Gallop K, Wild D, Verdian L, et al. Lennox-Gastaut syndrome (LGS): development of conceptual models of health-related quality of life (HRQL) for caregivers and children. Seizure. 2010;19(1):23–30. | ||
Skornicki M, Clements KM, O’Sullivan AK. Budget impact analysis of antiepileptic drugs for Lennox-Gastaut syndrome. J Manag Care Spec Pharm. 2014;20(4):400–406. | ||
Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011;52(Suppl 5):3–9. | ||
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676–685. | ||
Autry AR, Trevathan E, Van Naarden Braun K, Yeargin-Allsopp M. Increased risk of death among children with Lennox-Gastaut syndrome and infantile spasms. J Child Neurol. 2010;25(4):441–447. | ||
Beaumanoir A, Blume W. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th ed. Paris, France: John Libbey Eurotext Ltd; 2005:125–148. | ||
Kerr M, Kluger G, Philip S. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord. 2011;13(Suppl 1):S15–S26. | ||
Berg AT, Nickels K, Wirrell EC, et al. Mortality risks in new-onset childhood epilepsy. Pediatrics. 2013;132(1):124–131. | ||
Gallop K, Wild D, Nixon A, Verdian L, Cramer JA. Impact of Lennox-Gastaut syndrome (LGS) on health-related quality of life (HRQL) of patients and caregivers: literature review. Seizure. 2009;18(8):554–558. | ||
Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology 2008;70(21):1950–1958. | ||
Kluger G, Glauser T, Krauss G, Seeruthun R, Perdomo C, Arroyo S. Adjunctive rufinamide in Lennox-Gastaut syndrome: a long-term, open-label extension study. Acta Neurol Scand. 2010;122(3):202–208. | ||
Hopkins J, Irvine F. Qualitative insights into the role and practice of epilepsy specialist nurses in England: a focus group study. J Adv Nurs. 2012;68(11):2443–2453. | ||
Benbadis SR, Sheth RD. Advances in identifying and treating Lennox-Gastaut syndrome: do you know LGS when you see it? Neurol Rev. 2012;(Suppl):S1–S8. | ||
Epilepsy Foundation Website. Lennox-Gastaut Syndrome (LGS). 2014. Available from: http://www.epilepsy.com/learn/types-epilepsy-syndromes/lennox-gastaut-syndrome-lgs. Accessed February 1, 2016. | ||
Carmant L, Whiting S. Lennox-Gastaut syndrome: an update on treatment. Can J Neurol Sci. 2012;39(6):702–711. | ||
BANZEL (rufinamide) Prescribing Information. Woodcliff Lake (NJ): Eisai Inc.; 2015. | ||
Appleton RE. Vigabatrin in the management of generalized seizures in children. Seizure. 1995;4(1):45–48. | ||
Perucca E, Cloyd J, Critchley D, Fuseau E. Rufinamide: clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia. 2008;49(7):1123–1141. | ||
Epilepsy Foundation Website 2015. Rufinamide. Available from: http://www.epilepsy.com/medications/rufinamide/advanced. Accessed February 1, 2016. | ||
Elger CE, Stefan H, Mann A, Narurkar M, Sun Y, Perdomo C. A 24-week multicenter, randomized, double-blind, parallel-group, dose-ranging study of rufinamide in adults and adolescents with inadequately controlled partial seizures. Epilepsy Res. 2010;88(2–3):255–263. | ||
Biton V, Krauss G, Vasquez-Santana B, Bibbiani F, Mann A, Perdomo C, Narurkar M. A randomized, double-blind, placebo-controlled, parallel-group study of rufinamide as adjunctive therapy for refractory partial-onset seizures. Epilepsia. 2011;52(2):234–242. | ||
Wisniewski CS. Rufinamide: a new antiepileptic medication for the treatment of seizures associated with Lennox-Gastaut syndrome. Ann Pharmacother. 2010;44(4):658–667. | ||
McClean M, Schmutz M, Pozza M, Wamil M. The influence of rufinamide on sodium currents and action potential firing in rodent neurons. In: Abstracts from the joint annual meeting of the American Epilepsy Society and the American Clinical Neurophysiology Society. Washington, DC, USA, December 2–6, 2005. Abstract 3.062. Epilepsia. 2005;46(Suppl 8):1–373. | ||
Critchley DJ, Aluri J, Boyd P, Whayman M, Narurkar M, Delargy H, Bibbiani F. Bioavailability of three rufinamide oral suspensions compared with the marketed 400-mg tablet formulation: results from a randomized-sequence, open-label, four-period, four-sequence crossover study in healthy subjects. Clin Ther. 2011;33(1):146–157. | ||
la Marca G, Rosati A, Falchi M, Malvagia S, Della Bona ML, Pellacani S, Guerrini R. A pharmacokinetic study and correlation with clinical response of rufinamide in infants with epileptic encephalopathies. Pharmacology. 2013;91(5–6):275–280. | ||
Inovelon Tablets and Oral Suspension Summary of Product Characteristics. Hatfield, Hertfordshire, UK: Eisai Ltd.; 2013. | ||
Ohtsuka Y, Yoshinaga H, Shirasaka Y, Takayama R, Takano H, Iyoda K. Rufinamide as an adjunctive therapy for Lennox-Gastaut syndrome: a randomized double-blind placebo-controlled trial in Japan. Epilepsy Res. 2014;108(9):1627–1636. | ||
Palhagen S, Canger R, Henriksen O, van Parys JA, Riviere ME, Karolchyk MA. Rufinamide: a double-blind, placebo-controlled proof of principle trial in patients with epilepsy. Epilepsy Res. 2001;43(2):115–124. | ||
Loscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia. 2006;47(8):1253–1284. | ||
Gayatri NA, Livingston JH. Aggravation of epilepsy by anti-epileptic drugs. Dev Med Child Neurol. 2006;48(5):394–398. | ||
Coppola G, Grosso S, Franzoni E, et al. Rufinamide in children and adults with Lennox-Gastaut syndrome: first Italian multicenter experience. Seizure. 2010;19(9):587–591. | ||
Kluger G, Haberlandt E, Kurlemann G, et al. First European long-term experience with the orphan drug rufinamide in childhood-onset refractory epilepsy. Epilepsy Behav. 2010;17(4):546–548. | ||
Kluger G, Kurlemann G, Haberlandt E, et al. Effectiveness and tolerability of rufinamide in children and adults with refractory epilepsy: first European experience. Epilepsy Behav. 2009;14(3):491–495. | ||
Vendrame M, Loddenkemper T, Gooty VD, et al. Experience with rufinamide in a pediatric population: a single center’s experience. Pediatr Neurol. 2010;43(3):155–158. | ||
Kessler SK, McCarthy A, Cnaan A, Dlugos DJ. Retention rates of rufinamide in pediatric epilepsy patients with and without Lennox-Gastaut Syndrome. Epilepsy Res. 2015;112:18–26. | ||
Coppola G, Besag F, Cusmai R, et al. Current role of rufinamide in the treatment of childhood epilepsy: literature review and treatment guidelines. Eur J Paediatr Neurol. 2014;18(6):685–690. | ||
Arzimanoglou A, Ferreira JA, Satlin A, et al. Safety and pharmacokinetic profile of rufinamide in pediatric patients aged less than 4 years with Lennox-Gastaut syndrome: an interim analysis from a multicenter, randomized, active-controlled, open-label study. Eur J Paediatr Neurol. 2016;20(3):393–402. | ||
Schimpf R, Veltmann C, Papavassiliu T, et al. Drug-induced QT-interval shortening following antiepileptic treatment with oral rufinamide. Heart Rhythm. 2012;9(5):776–781. | ||
Gibson PA. Resources for caregivers and families of patients with Lennox-Gastaut syndrome. Epilepsia. 2014;55(Suppl 4):34–36. | ||
Gibson PA. Lennox-Gastaut syndrome: impact on the caregivers and families of patients. J Multidiscip Healthc. 2014;7:441–448. | ||
Schachter SC, Sirven JI. Vagus Nerve Stimulation (VNS). Epilepsy Foundation Website; 2013. Available from: www.epilepsy.com/learn/treating-seizures-and-epilepsy/devices/vagus-nerve-stimulation-vns. Accessed February 1, 2016. | ||
LGS Foundation Website. Available from: http://www.lgsfoundation.org/. Accessed February 1, 2016. | ||
Epilepsy Foundation Website. Available from: http://www.epilepsy.com. Accessed February 1, 2016. | ||
Citizens United for Research in Epilepsy (CURE) Website. Available from: http://www.cureepilepsy.org/aboutCURE/. Accessed February 1, 2016. | ||
Wake Forest Baptist Health Website. Available from: http://www.wakehealth.edu/Neurosciences/Comprehensive-Epilepsy-Center/Epilepsy-Resources.htm. Accessed February 1, 2016. | ||
Child Neurology Foundation Website. Available from: http://www.childneurologyfoundation.org/. Accessed February 1, 2016. | ||
Child Neurology Society Website. Available from: http://www.childneurologysociety.org/. Accessed February 1, 2016. | ||
The Charlie Foundation for Ketogenic Therapies Website. Available from: http://www.charliefoundation.org/. Accessed February 1, 2016. | ||
Family Caregiver Alliance Website. Available from: https://www.caregiver.org/. Accessed February 1, 2016. | ||
National Alliance for Caregiving Website. Available from: http://www.caregiving.org/. Accessed February 1, 2016. | ||
BANZEL (rufinamide) Website. Available from: https://www.banzel.com/. Accessed February 1, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.