Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Roundabout signaling pathway involved in the pathogenesis of COPD by integrative bioinformatics analysis
Authors Lin YZ ![]() , Zhong XN, Chen X, Liang Y, Zhang H, Zhu DL
, Zhong XN, Chen X, Liang Y, Zhang H, Zhu DL
Received 17 May 2019
Accepted for publication 22 August 2019
Published 18 September 2019 Volume 2019:14 Pages 2145—2162
DOI https://doi.org/10.2147/COPD.S216050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Yuan-Zhen Lin, Xiao-Ning Zhong, Xin Chen, Yi Liang, Hui Zhang, Dong-Lan Zhu
Department of Respiratory Medicine, The First Affiliated Hospital, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China
Correspondence: Xiao-Ning Zhong
Department of Respiratory Medicine, The First Affiliated Hospital, Guangxi Medical University, No. 6, Shuangyong Road, Nanning, Guangxi 530021 People’s Republic of China
Tel +86 771 535 1176
Fax +86 771 535 1176
Email [email protected]
Purpose: To explore the potential mechanism underpinning the development of chronic obstructive pulmonary disease (COPD) and to investigate the role of the Roundabout signaling pathway in COPD.
Methods: Three microarray datasets (GSE1650, GSE38974 and GSE76925) including 139 cases of severe COPD and 52 cases of normal smokers without carcinoma, were integrated to screen differentially expressed genes (DEGs) using bioinformatics methods. Gene ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of the DEGs were performed by a DAVID online tool. Finally, a cigarette smoke (CS)- induced emphysema mice model was established, the lung mRNA expression levels of genes associated with Slit guidance ligand 2 (SLIT2) -Roundabout (ROBO) signaling pathway were detected by reverse transcription quantitative polymerase chain reaction (RT-qPCR), and the protein level of SLIT2 was examined by immunohistochemistry staining.
Results: A total of 315 DEGs were identified in three databases. GO and KEGG pathway analyses suggested that the inflammatory response, extracellular matrix disassembly, immune response, the apoptotic signaling pathway, ubiquitination and the Roundabout signaling pathway all together were involved in the development of COPD. The genes SLIT2 and ROBO2 were decreased in patients with COPD and these decreases were significantly negatively correlated with the disease stages of COPD. Consistently, the mRNA expression levels of SLIT2, ROBO1 and ROBO2, and the protein level of SLIT2 were revealed to be lower in the lungs of CS-induced emphysema mice compared with the air-exposed control mice. In addition, the SLIT2 protein level was negatively associated with alveolar mean linear intercept.
Conclusion: Integrated bioinformatics analysis may provide novel insights into the complicated pathogenesis of COPD, and to the best of our knowledge, this study is the first to provide evidence to suggest that the Roundabout signaling pathway may be involved in the pathogenesis of COPD.
Keywords: chronic obstructive pulmonary disease, GEO database, roundabout signaling pathway, SLIT2, ROBO
Introduction
As a typical chronic respiratory disease, chronic obstructive pulmonary disease (COPD), with a mean prevalence rate of 13.1% worldwide,1 is characterized by progressive and irreversible airflow restriction and recurrent respiratory symptoms. As many as 3.2 million patients with COPD succumbed to mortality in 2015-, and COPD was the eighth leading cause of global disability.2 It is widely acknowledged that cigarette smoke (CS) is the predominant risk factor of COPD;3 however, only a few smokers finally develop emphysema, the exact molecular and cellular pathogenesis of this complicated process has yet to be fully elucidated. Additionally, effective prevention and treatment methods available for the disease are still insufficient. Therefore, there is an urgent need to further investigate the molecular mechanisms of COPD to identify novel targets for the treatment of the disease.
In recent decades, mRNA microarrays have been widely applied in cases of multiple tumors, as well as COPD.4–6 Furthermore, an ever-increasing number of chip data recordings are available on public databases, and integrating these microarray datasets should enable us to further explore genes associated with diseases, and thereby to predict the molecular targets for precision therapy.7 Recently, a few investigators have conducted mRNA gene expression profiles on COPD,4,5 and identified hundreds of differentially expressed genes (DEGs) that are likely to be involved in the pathogenesis of COPD. However, the DEGs were found to be inconsistent or even contradictory based on the comparison of the different studies, and the reasons underlying this phenomenon are complex. The lung tissues included in the dataset were mostly from COPD patients that accompanied with lung cancer, which may have introduced bias into the analysis. Besides, different individual samples, different methods of analysis, different disease stages, and different chip platforms also contributed to the difficulties in interpreting the data. However, integrating microarray data from several datasets may circumvent the limitations associated with single datasets, and enable the identification of reliable and efficacious molecular markers.
The Roundabout signaling pathway, consisting predominantly of slit guidance ligand (SLIT) and roundabout (ROBO), was initially shown to be involved in neural development by inhibiting the migration of axons.8 Subsequently, another study demonstrated that SLIT was also able to inhibit the migration of immune cells, including T lymphocytes, neutrophils, macrophages, and dendritic cells,9 and therefore SLIT was regarded as a potential anti-inflammatory molecule.9 T lymphocytes, neutrophils, macrophages and dendritic cells are all involved in the development of COPD,10–12 however, the role of the Roundabout signaling pathway in COPD has yet to be elucidated.
Patients with severe COPD usually suffer from obvious respiratory symptoms that result in high rates of mortality, which places a heavy burden on society. On this basis, the present study focused on the differences in the genes and pathways comparing between cases of severe COPD and smokers to explore the novel mechanism underlying COPD. In the present study, the DEGs from three NCBI Gene Expression Omnibus (GEO) databases were screened for COPD and integrated to explore the potential pathogenesis of COPD according to bioinformatics analysis. By analyzing the biological processes of the differences between COPD and smokers, the present study has attempted to unravel the mechanism to explain how only a few smokers develop COPD, and also to investigate the role of the Roundabout signaling pathway in COPD and cigarette smoke (CS)- induced emphysema mice model.
Materials and methods
mRNA microarray chip datasets
Three original microarray datasets [GSE1650,13 GSE389744 and GSE7692514] were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), which consisted of 139 patients with COPD and 52 healthy smokers.
Searching on the GEO database with the keywords “COPD mRNA”, an emphasis was placed on those datasets which comprised patients with severe COPD and normal smokers without carcinoma. Global initiative for chronic obstructive lung disease (GOLD) stages 3 and 4 were defined as severe COPD. The platform for GSE1650 was GPL96, [HG-U133A] Affymetrix Human Genome U133A array, which contained 18 lung samples from cases with severe COPD, and 3 lung specimens from normal smokers. The platform for GSE38974 was GPL4133, Agilent-014850 Whole Human Genome microarray 4x44K G4112F (feature number version), which comprised 10 lung samples from cases of severe COPD and 9 lung specimens from normal smokers. Finally, the platform for GSE76925 was GPL10558, Illumina HumanHT-12 V4.0 expression bead-chip, which included 111 lung samples from cases of severe COPD and 40 lung specimens from normal smokers. Detailed information of the three datasets is shown in Table 1.
 |
Table 1 Details for GEO COPD data |
Screening for DEGs, and subsequent integration of DEGs in three datasets
The .txt files of the DEGs for the three microarray datasets were obtained using GEO2R online analysis (https://www.ncbi.nlm.nih.gov/geo/geo2r/), and the DEGs in the three datasets were ranked by logFC respectively. The significant DEGs in the three datasets were defined as such when the corrected p-value was <0.05, and the absolute folder change (FC) was >1.5. The raw data were corrected for background noise normalized by the Robust Microarray Analysis (RMA) method. Subsequently, DEGs for the three microarray datasets were integrated using the Robust Rank Aggregation (RRA) method (http://www.icesi.edu.co/CRAN/web/packages/RobustRankAggreg/)15 in R version 3.5.3 (http://cran.rproject.org/). The significant integrated DEGs were defined when the corrected p-value was <0.05.
Protein-protein interaction (PPI) network analysis
PPI networks of integrated DEGs were constructed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) online database (http://stringdb.org/), and the Cytoscape software 3.5.116 and CytoHubba Apps17 were applied to reveal the molecular interactions and to identify the top 20 hub genes associated with COPD.
Gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) pathway analysis for DEGs
GO annotations and KEGG pathway analysis of the integrated DEGs were performed by the database for annotation, visualization, and integrated discovery (DAVID) online tool. The results were downloaded into a .txt file, and subsequently, GOPlot18 and ggplot2 packages in R were used for the visual analysis. The genes associated with the SLIT2- ROBO pathway were further analyzed in COPD patients in the dataset GSE38974.
CS-induced emphysema mice model
Male C57BL/6 mice (6–8 weeks old, 20–25 g body weight) were purchased from the Guangxi Medical University Laboratory Animal Centre (Nanning, China) and were exposed to air (control) or cigarette smoke for 24 weeks (n=6 per group), as previously described.11 Following 24 weeks’ exposure to air or CS, the mice were sacrificed after anesthesia, and the lungs were subsequently removed from the chest cavity, the left lungs were used for RNA analysis, and the right lungs were used for pathological and immunohistochemistry examination. All animal experiments protocols were approved by the Animal Research of Guangxi Medical University Ethics Committee.
Quantifications of mean linear intercept (Lm) and immunohistochemistry staining
The right lungs were fixed in 10% formalin for 24 h before embedding in paraffin; midsagittal sections were stained with hematoxylin and eosin (HE) for histological analysis, we determined enlargement of alveolar spaces by quantifying the Lm as previously described.11 Lung tissue sections were deparaffinized with xylene, hydrated with alcohol, repaired with sodium citrate in high temperature and then incubated with antibody against mouse Slit2 (ab134166; Abcam, Cambridge, UK) overnight at 4 °C. Subsequently, secondary Goat Anti-Rabbit IgG H&L (HRP) antibody (ab205718, Abcam, Cambridge, UK) was stained at room temperature for 20 mins and finally reacted with diaminobenzidine (Solarbio, DA1010, China) for coloration. Fluorescence pathological microscope (Olympus */BX53+DP80, Japan) was used to take photos for all the sections. The mean density of SLIT2 staining was quantified using Image-Pro Plus software 6.0 (Media Cybernetics), and the average score of 2 different viewers was taken for analysis.
RNA extraction and reverse transcription- quantitative polymerase chain reaction (RT-qPCR)
The lung mRNA of air or CS-exposed mice (n=6 per group) were extracted using Eastep® Super Total RNA Extraction kit (Promega Corporation), and then cDNA was prepared using GoScript™ Reverse Transcription mix (Promega Corporation). Quantitative PCR was performed using TB Green® Premix Ex Taq ™ II (Takara Biotechnology Co., Ltd). All experiments were performed by the kit instructions. Data are presented relative to β-actin for target genes based on calculations of 2−ΔΔCt. The mRNA expression results are reported as the quantity of mRNA relative to control. Primers were synthesized by Sangon Biotech Company (Shanghai Co., Ltd.). Primers used are shown in Table 2.
 |
Table 2 Details for primers of mice |
Statistical analysis
The GraphPad Prism 5, and R, version 3.5.3 software were used for statistical analysis. Data are presented as mean ± SD and compared between groups using unpaired Student’s t-tests. Correlation analysis is calculated using Pearson or Spearman rank correlation test. p<0.05 was considered to indicate a statistically significant value.
Results
Identification of DEGs in three datasets
After screening with the threshold of a corrected p-value <0.05 and absolute folder change >1.5, 52 DEGs (37 upregulated and 15 downregulated) were identified in the GSE1650 dataset, 4957 DEGs (1989 upregulated and 2968 downregulated) were identified in GSE38974, and 1649 DEGs (367 upregulated and 1282 downregulated) were identified in GSE76925. Figure 1 shows the heatmaps of the top 200 DEGs in the three datasets.
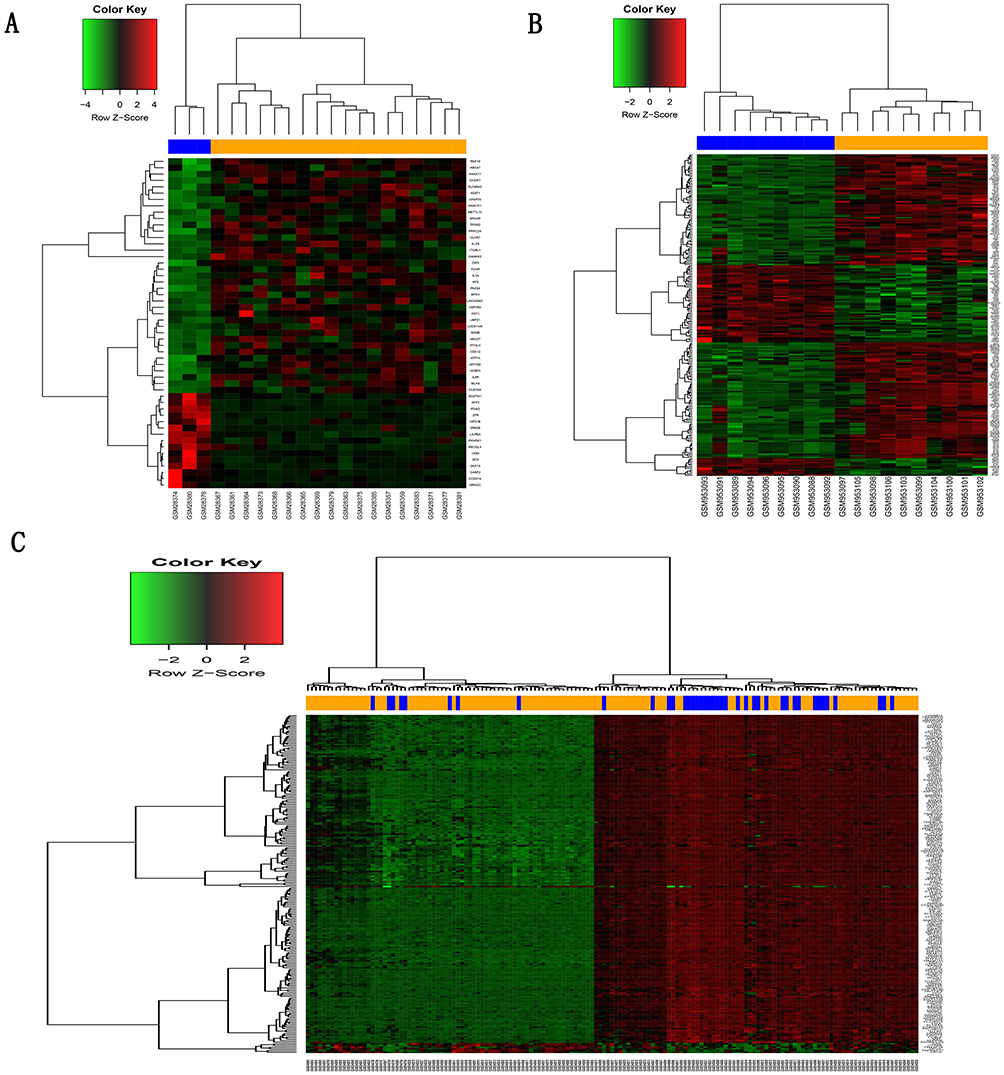 |
Figure 1 Hierarchical clustering heatmap of DEGs screened on the basis of |fold change| >1.5 and a corrected P-value <0.05. Notes: (A) GSE1650 data, (B) GSE38974 data, (C) GSE76925 data. Red indicates that the expression of genes is relatively upregulated, green indicates that the expression of genes is relatively downregulated, and black indicates no significant changes in gene expression. Orange indicates samples from severe COPD patients, and blue indicates samples from normal smokers.Abbreviations: DEGs, differentially expressed genes; COPD, chronic obstructive pulmonary disease. |
Identification of integrated DEGs in severe COPD using the RRA method
The DEGs for the three microarray datasets were sorted according to their logFC values and subsequently were further integrated using the RRA method (adjusted p-value <0.05). At the end of this analysis, 315 DEGs, including 119 upregulated genes and 195 downregulated genes, were identified. Table 3 presents the integrated DEGs in detail. The top 20 up- and down-regulated hub genes were drawn into a heatmap using R–heatmap software, as shown in Figure 2.
 |
Table 3 Integrated DEGs in COPD by RRA |
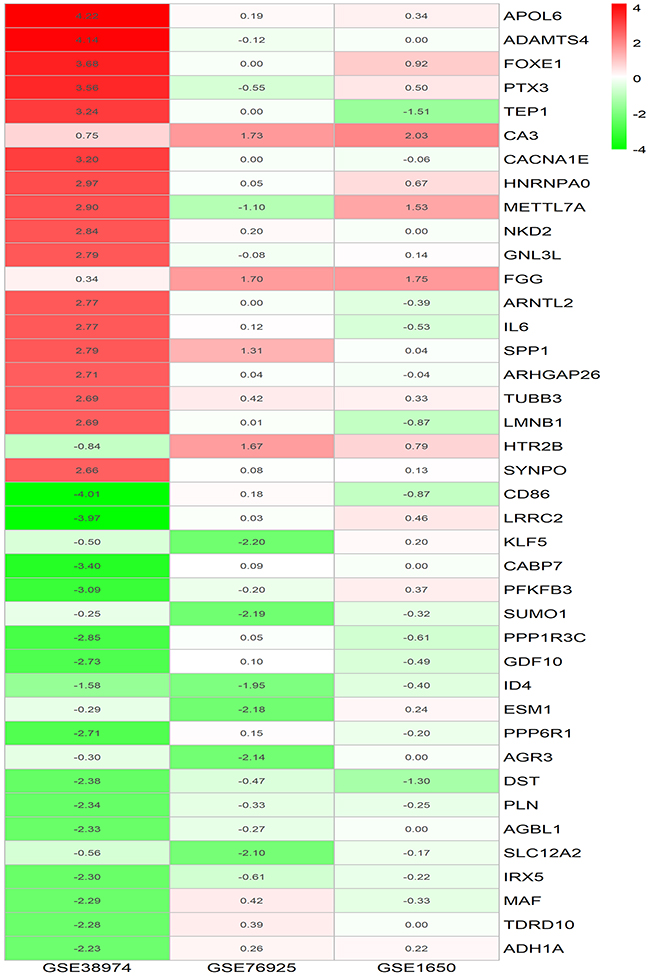 |
Figure 2 LogFC heatmap of top 20 up- and down-regulated DEGs of three microarrays screening by RRA method. Notes: The abscissa is the GEO ID, and the ordinate is the gene name. Red represents logFC >0, green represents logFC <0, and the values in the box represent the logFC values. Abbreviations: FC, fold change; GEO, Gene Expression Omnibus; DEGs, differentially expressed genes. |
PPI network
The PPI network for integrated DEGs is shown in Figure 3. The 20 hub genes that extensively interacted with other genes were IL6, CXCL8, MMP9, SNAI1, CYCS, HIF1A, SPP1, THBS1, MMP1, POSTN, CXCL13, CD86, CD19, SDC1, HMOX1, ATM, CX3CR1, GNG13, ANGPT1 and TNFRSF1A (Figure 4).
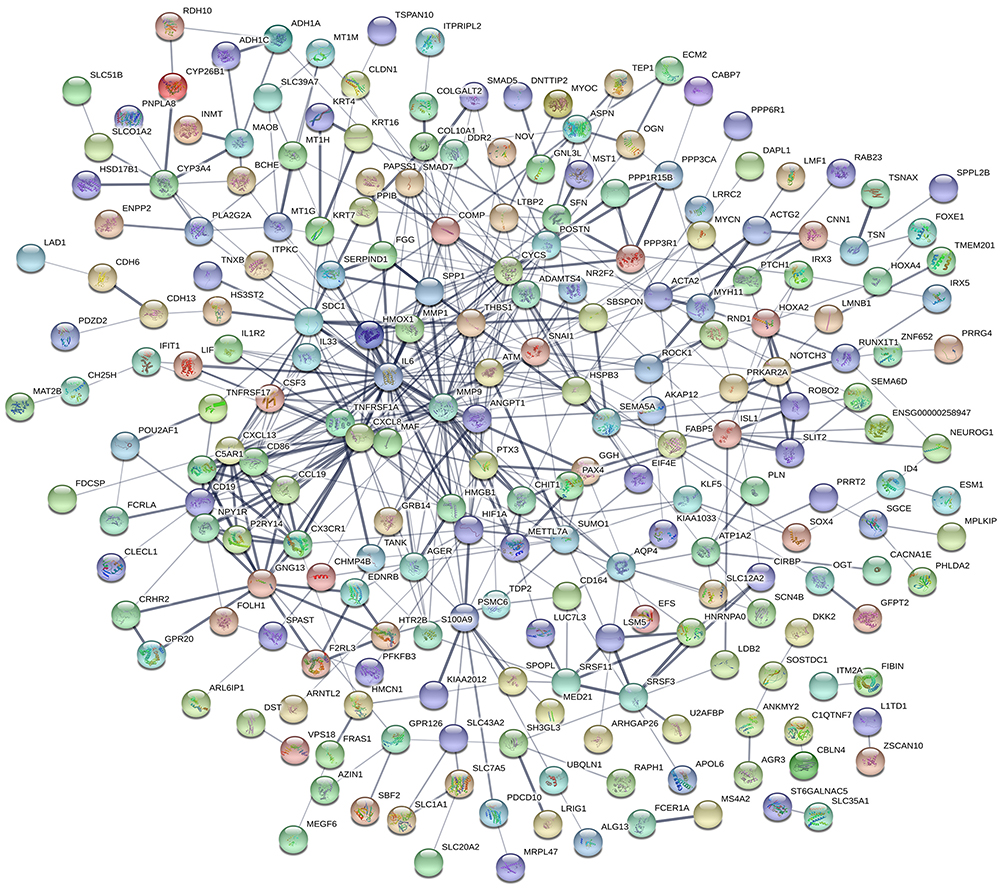 |
Figure 3 PPI network of DEGs. Notes: Circles represent genes, lines represent the interaction of proteins between genes, and the results within the circle represent the structure of proteins.Abbreviations: PPI, protein–protein interaction; DEGs, differentially expressed genes. |
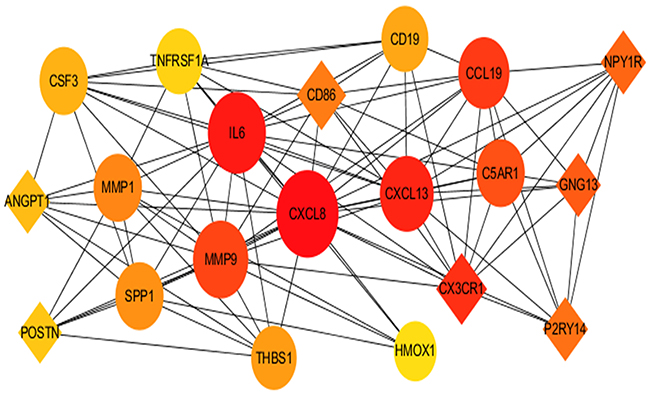 |
Figure 4 The 20 top hub genes in PPI network. Notes: The diamond icon represents the downregulated hub genes, the circle icon represents the upregulated hub genes, the size of the icon represents the strength of nodes that interact with other genes, lines represent the interaction of proteins between genes.Abbreviation: PPI, protein–protein interaction. |
GO analysis of integrated DEGs
Three functional groups in the GO analysis of integrated DEGs were sought after. Namely, cell composition (CC), biological processes (BP), and molecular function (MF). The significant GO enrichment results are shown in Tables 4 and 5, and Figures 5 and 6. With the BP functional group, the up-regulated DEGs were concentrated in the categories of negative regulation of growth, inflammatory response, extracellular matrix (ECM) disassembly, immune response, intrinsic apoptotic signaling pathway in response to DNA damage and positive regulation of the extracellular-signal- regulated (ERK) 1 and ERK2 cascade. By contrast, the downregulated DEGs were concentrated in the categories of negative regulation of protein ubiquitination, Roundabout signaling pathway, cell adhesion, cell fate determination, and negative regulation of neuron differentiation. With the MF functional group, the upregulated DEGs were concentrated in the categories of structural molecule activity, C-C motif chemokine receptor 10 (CCR10) chemokine receptor binding, and chemokine activity. In addition, the downregulated DEGs were mainly associated with the categories of heparin- binding, protein kinase A binding, collagen binding, calcium ion binding, protein serine/threonine phosphatase activity, and integrin binding. Finally, with the CC functional group, the upregulated DEGs were concentrated in the categories of an extracellular region, extracellular exosome, and extracellular space, the downregulated DEGs were mainly associated with the plasma membrane, proteinaceous ECM and ECM. Considering all these results together, the majority of the DEGs were found to be enriched in the categories of the inflammatory response, immune response, apoptotic signaling pathway, ECM disassembly, ubiquitination, and the Roundabout signaling pathway.
 |
Table 4 GO analysis of upregulated genes associated with COPD |
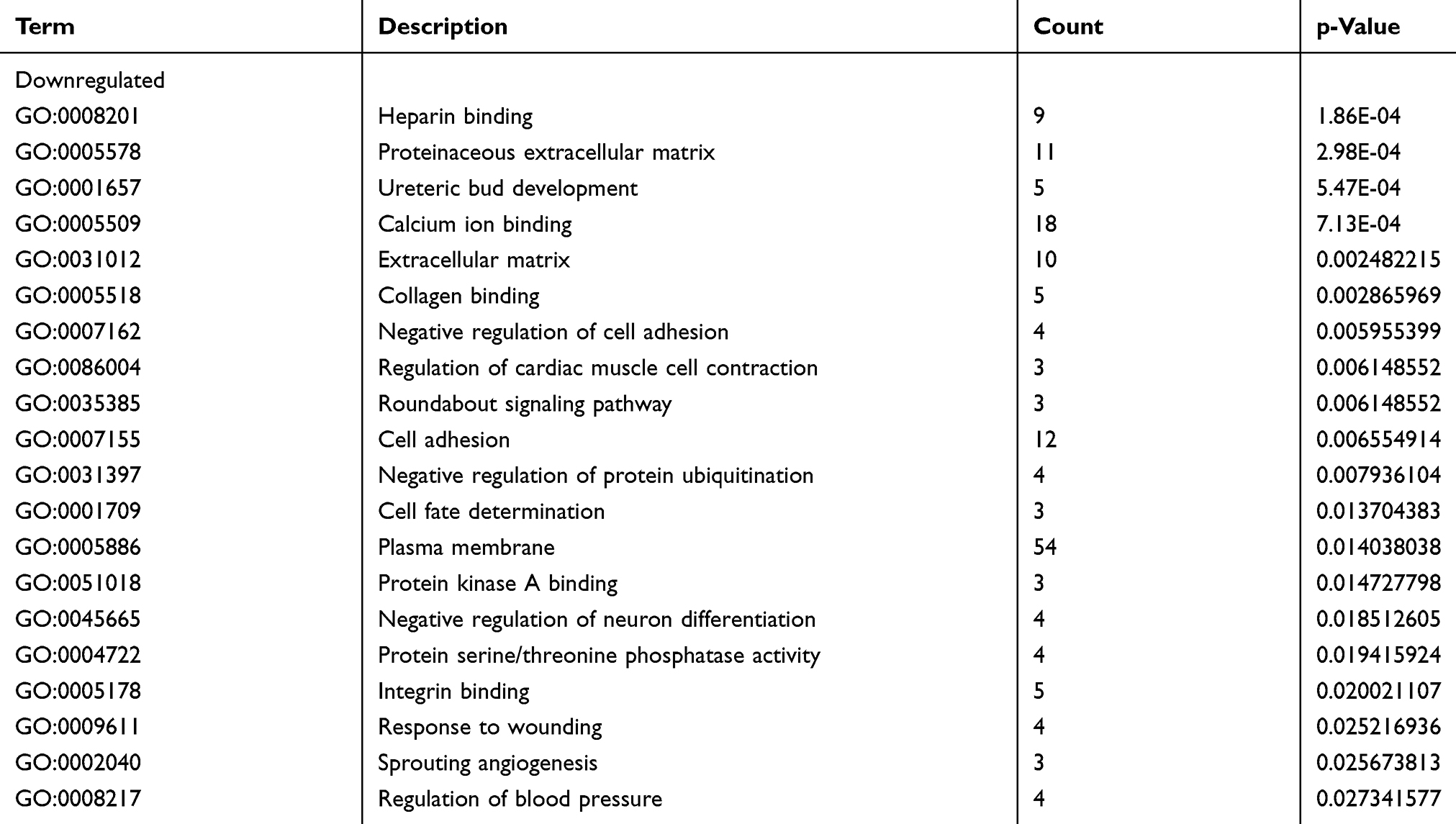 |
Table 5 GO analysis of downregulated genes associated with COPD |
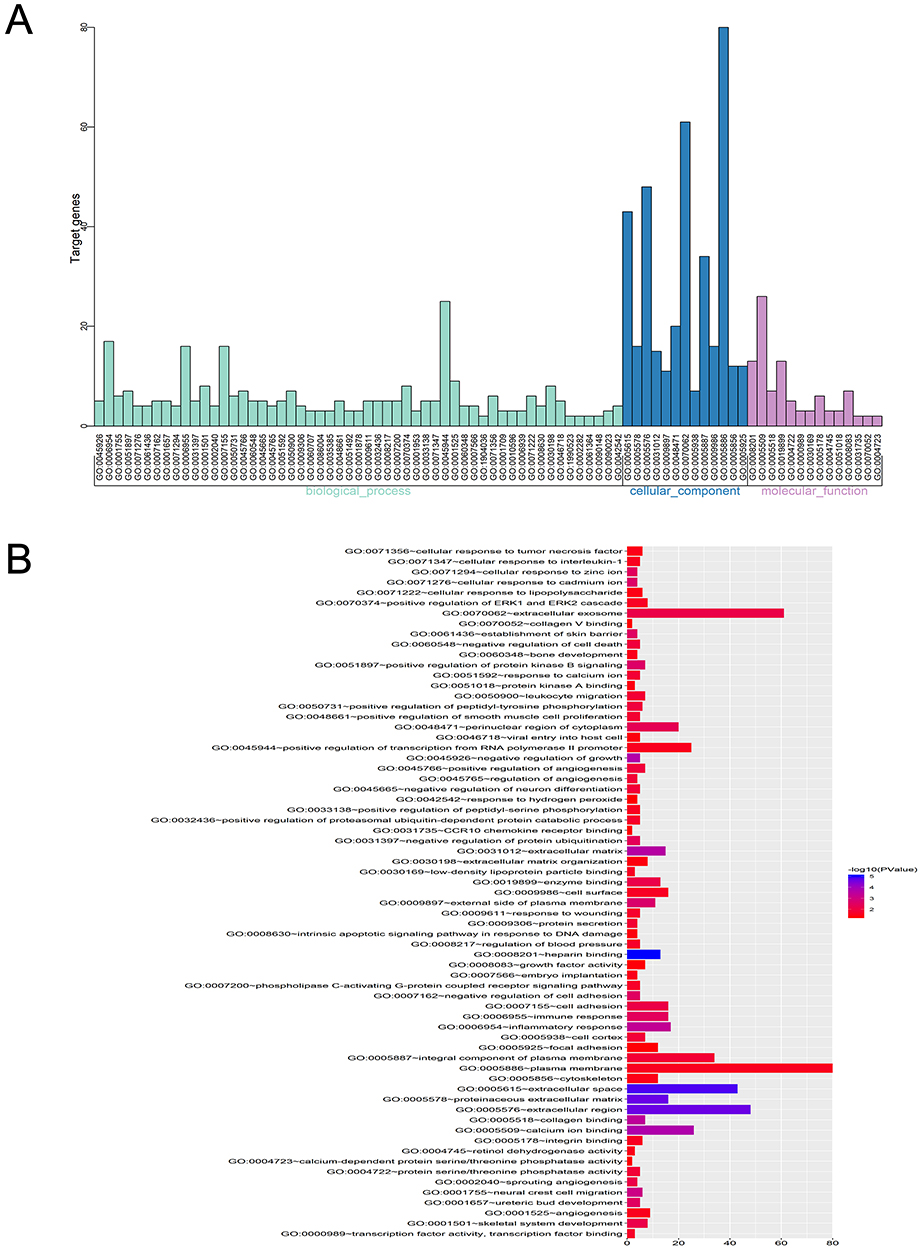 |
Figure 5 GO enrichment analysis of DEGs in severe COPD. Notes: (A) GO analysis divided DEGs into three functional groups: molecular function, biological processes and cell composition. (B) GO enrichment significance items of DEGs in different functional groups.Abbreviations: DEGs, differentially expressed genes; GO, gene ontology; COPD, chronic obstructive pulmonary disease. |
 |
Figure 6 Distribution of DEGs in severe COPD for different GO-enriched functions. Abbreviations: DEGs, differentially expressed genes; GO, gene ontology; COPD, chronic obstructive pulmonary disease. |
KEGG pathway analysis of integrated DEGs
As shown in Figure 7 and Table 6, the KEGG pathway analysis of the DEGs revealed that they were concentrated in the categories of mineral absorption, cytokine-cytokine receptor interaction, axon guidance, transcriptional misregulation in cancer, retinol metabolism and the hypoxia-inducible factor-1 (HIF-1) signaling pathway.
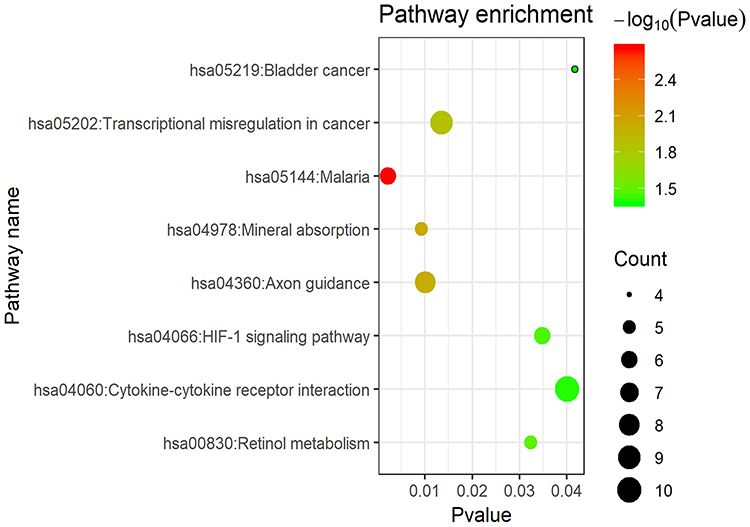 |
Figure 7 KEGG pathway enrichment analysis for DEGs. Notes: The x-axis shows the -log10 (p-value) of each term and y-axis shows the KEGG pathway terms.Abbreviations: DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
The SLIT2-ROBO pathways in COPD
In the GO enrichment analysis, it was revealed that the genes SLIT2 and ROBO2 were enriched in the Roundabout signaling pathway, which has not previously been associated with the pathogenesis of COPD, and furthermore, both genes were downregulated in severe COPD; therefore, the present study sort to investigate the role of the SLIT2-ROBO pathway in all disease stages of the COPD patients. The dataset GSE38974 included not only smokers and patients with severe COPD but also patients with mild and moderate COPD; therefore, the expression matrix of GSE38974, including a total of 31 samples, was downloaded. The mRNA expression levels of SLIT2 and ROBO2 were found to be lower in all stages of COPD compared with smokers (Figure 8A and B), and both were negatively correlated with the GOLD stages in COPD (Figure 8D and E). However, no statistical differences in the expression levels of ROBO1 were identified between COPD and smokers (Figure 8C and F). The expression levels of SLIT2 and ROBO2 were independent of age and smoking status (Figure 8G–J). In addition, the expression of SLIT2 was positively correlated with ROBO2, rather than with ROBO1 (Figure 8K and L). The mRNA expression levels of cell division cycle 42 (Cdc42) and Rac family small GTPase 2 (Rac2) were increased in COPD, and both were significantly positively correlated with the GOLD stages in COPD (Figure 9A–D). The expression of SLIT2 was negatively correlated with Cdc42 and Rac2, whereas the expression of ROBO2 was negatively correlated with Rac2, rather than with Cdc42 (Figure 9E–H).
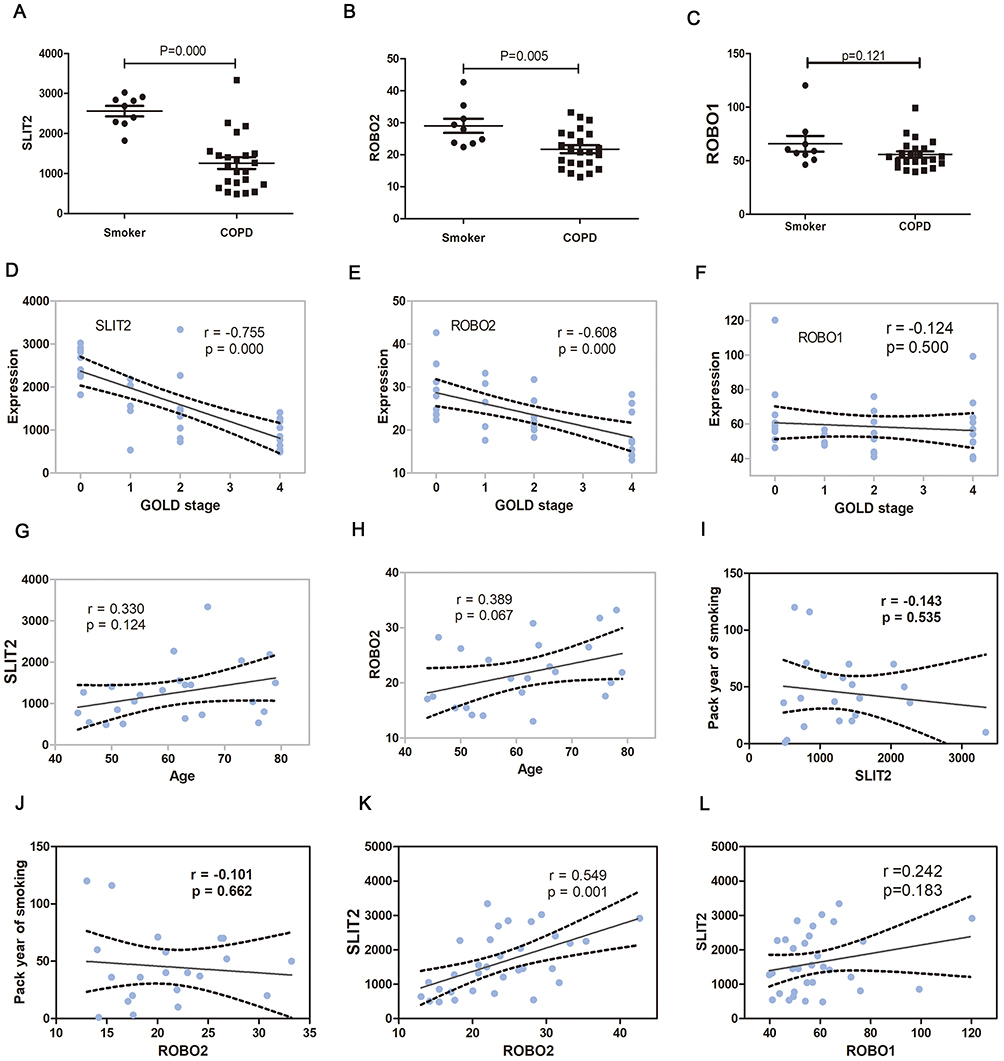 |
Figure 8 The SLIT2 and ROBO expression in GSE38974. Notes: The mRNA expression of SLIT2 (A) ROBO2 (B) and ROBO1 (C) in GSE38974 microarray dataset. (D) Scatter plot of correlation analysis for SLIT2and GOLD stage. (E) Scatter plot of correlation analysis for ROBO2 and GOLD stage. (F) Scatter plot of correlation analysis for ROBO1 and GOLD stage. The GOLD 0 represents smokers without COPD, GOLD 1–4 represents COPD patients with GOLD stages 1–4. (G) Scatter plot of correlation analysis for SLIT2 and age. (H) Scatter plot of correlation analysis for ROBO2 and age. (I) Scatter plot of correlation analysis for SLIT2 and the pack year of smoking. (J) Scatter plot of correlation analysis for ROBO2 and the pack year of smoking. (K) Scatter plot of correlation analysis for SLIT2 and ROBO2. (L) Scatter plot of correlation analysis for SLIT2 and ROBO1.Abbreviations: GOLD, Global initiative for chronic obstructive lung disease; SLIT2, slit guidance ligand 2; ROBO, roundabout guidance receptor; COPD, chronic obstructive pulmonary disease. |
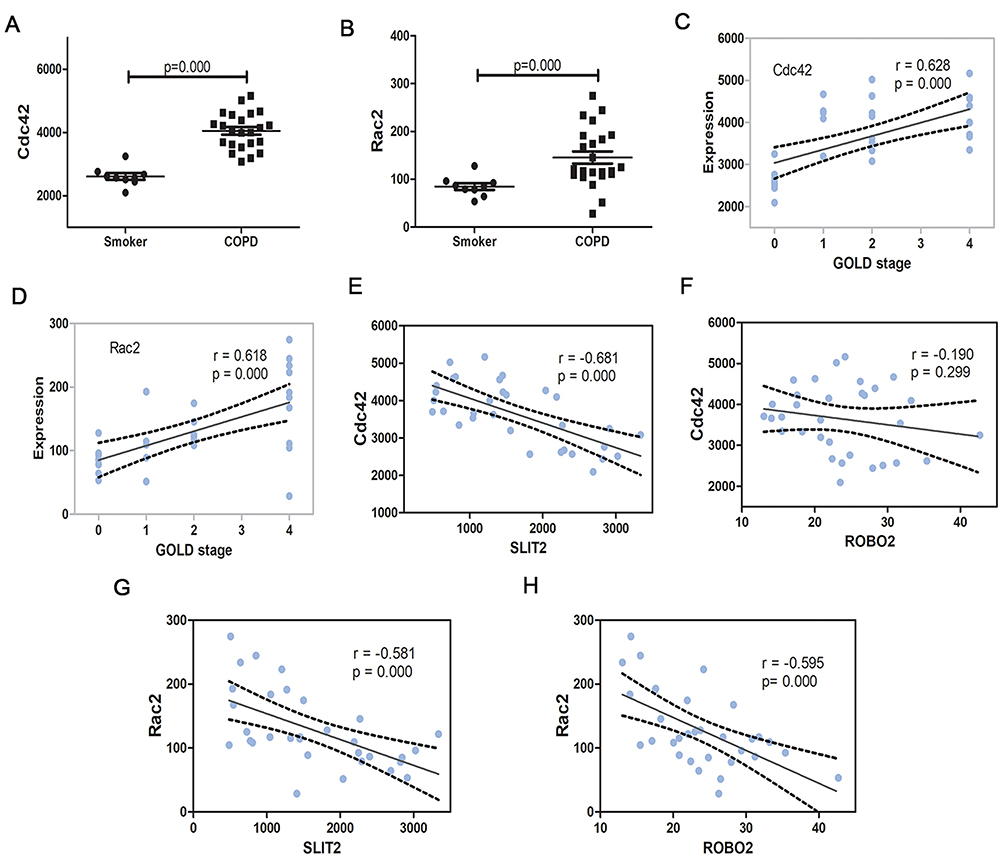 |
Figure 9 The Cdc42 and Rac2 expression in GSE38974. Notes: The mRNA expression of Cdc42 (A) and Rac2 (B) in GSE38974 microarray dataset. (C) Scatter plot of correlation analysis for Cdc42 and GOLD stage. (D) Scatter plot of correlation analysis for Rac2 and GOLD stage. The GOLD 0 represents smokers without COPD, GOLD 1–4 represents COPD patients with GOLD stages 1–4. (E) Scatter plot of correlation analysis for SLIT2 and Cdc42. (F) Scatter plot of correlation analysis for ROBO2 and Cdc42 (G) Scatter plot of correlation analysis for SLIT2 and Rac2. (H) Scatter plot of correlation analysis for ROBO2 and Rac2.Abbreviations: GOLD, Global initiative for chronic obstructive lung disease; SLIT2, slit guidance ligand 2; ROBO, roundabout guidance receptor; COPD, chronic obstructive pulmonary disease; Rac2, Rac family small GTPase 2; Cdc42, cell division cycle 42. |
The SLIT2-ROBO signal pathway is downregulated in CS-induced emphysema mice model
As it proved to be difficult for us to obtain the lungs from COPD patients, CS-induced emphysema mice model was alternatively established to investigate whether the SLIT2–ROBO signal pathway is involved in the pathogenesis of emphysema. In accordance with our previous studies,11 the Lm of CS-exposed mice was significantly higher in than that of control mice (Figure 10A and B), which indicated that the emphysema model had been success established. As shown in Figure11A and B, in accordance with the microarray results, compared with the control mice exposed to only air, the mRNA expression levels of SLIT2 and ROBO2 both were lower in the lungs of CS-exposed mice (0.21 folder change, p=0.000 and 0.03 folder change, p=0.001 respectively). Unexpectedly, the mRNA expression levels of ROBO1 was also lower in CS-induced emphysema mice (0.35 folder change, p=0.004, Figure 11C), however, there were no statistical differences in the mRNA expression levels of Cdc42 and Rac2(Figure 11D and E). Furthermore, immunohistochemistry staining of SLIT2 was recruited to delineate the role of SLIT2 in emphysema; the SLIT2 protein expression was consistently decreased in CS-induced emphysema mice and the SLIT2 was mainly expressed in bronchial epithelial cells (Figure10C and D). In addition, the mean density level of SLIT2 was negatively correlated with Lm (r= −0.645, p=0.024, Figure 10E).
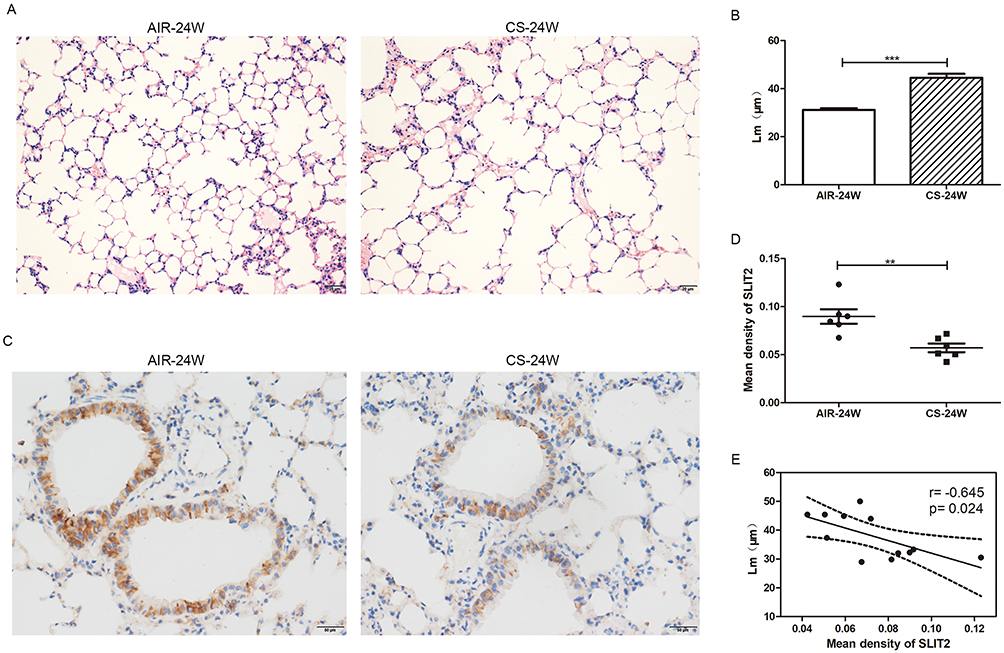 |
Figure 10 The mean linear intercept (Lm) and immunohistochemistry staining of SLIT2 in lung of AIR-exposed and CS-exposed mice. Notes: Representative photomicrographs of hematoxylin and eosin stained lung tissue of 24 weeks AIR-exposed and CS-exposed mice, magnification ×20 (A) The Lm of alveolar in 24 weeks AIR-exposed and CS-exposed mice (B) The photomicrographs for immunohistochemistry staining of SLIT2 in lung of AIR-exposed and CS-exposed mice, magnification ×40 (C) The mean density value of SLIT2 staining in the lung of AIR-exposed and CS-exposed mice (D) Scatter plot of correlation analysis for mean density of SLIT2 staining and Lm in lung of mice (E) AIR-24W represents mice exposed to air for 24 weeks, and CS-24w represents mice exposed to CS for 24 weeks. **p<0.01, ***p<0.001.Abbreviations: Lm, the mean linear intercept; CS, cigarette smoke; SLIT2, slit guidance ligand 2. |
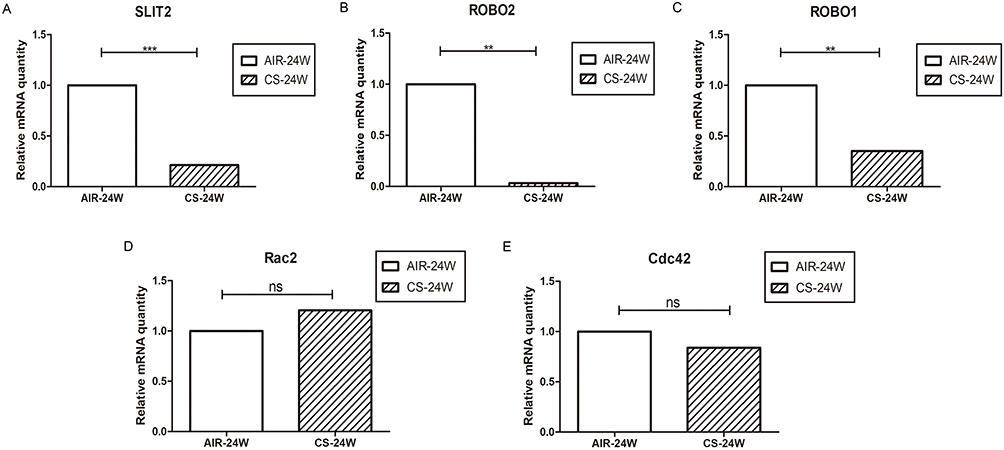 |
Figure 11 The SLIT2-ROBO signal pathway is downregulated in CS-induced emphysema mice model. Notes: The relative mRNA expression of SLIT2 (A) ROBO2 (B) ROBO1 (C) Rac2 (D) and Cdc42 (E) in lungs of mice exposed to air or CS for 24 weeks. AIR-24W represents mice exposed to air for 24 weeks, and CS-24w represents mice exposed to CS for 24 weeks. ** p<0.01, ***p<0.001, ns: not significant.Abbreviations: SLIT2, slit guidance ligand 2; ROBO, roundabout guidance receptor; CS, cigarette smoke; Rac2, Rac family small GTPase 2; Cdc42, cell division cycle 42. |
Discussion
In the present study, 314 DEGs were identified, among which 119 genes were upregulated, and 195 genes were downregulated (Table 2). Subsequently, a PPI network of the DEGs was constructed, and the top 20 hub genes were identified (Figure 4). The top 20 hub genes are mainly associated with the downstream inflammatory response and alveolar destruction that has been widely accepted, however, these hub genes, in themselves, are insufficient to explain the mechanism by which the immune reaction is initiated in patients with COPD. Chemokine (C-X-C motif) ligand 8 (CXCL8) secreted by lung macrophages is able to attract an accumulation of polymorphonuclear neutrophils to the lungs, thereby stimulating the airway epithelium to contract and increase its permeability to inflammatory cells.19 Interleukin-6 (IL-6), CXCL13 and Chemokine (C-C motif) ligand 19 (CCL19), B-cell recruitment and immunoglobulin transcription genes, along with CD19, revealed that the B cell signaling pathway has an important role in the development of COPD. CXCL13 is increased in the pulmonary B-cell regions of lymphoid follicles of patients with COPD and CS-exposed mice, and therapeutic targeting of CXCL13 was shown to absolutely reverse lymphoid follicle formation induced by CS in mice.20 CS is able to induce pulmonary antinuclear antibodies and anti-tissue antibodies, resulting in autoimmune processes and alveolar damage.21 Tumor necrosis factor receptor superfamily member 1A (TNFRSF1A) is associated with symptom burden in COPD,22 which is connected with the apoptotic signals in airway epithelium cells.23 CX3CR1 is essential for the generation of TNF-α and IL-6 by mononuclear phagocytes exposed to CS, and CS-induced emphysema was attenuated in the CX3CR1 knockout mice.24 Increasing expression levels of matrix metalloproteinase (MMP)-1 and MMP-9 were shown to lead to alveolar destruction, and this was demonstrated to be positively correlated with COPD severity.25 HIF1A, C5AR1, heme oxygenase 1 (HMOX1), CXCL8 and thrombospondin 1 (THBS1) are associated with the oxidative stress reaction and are enriched in the positive regulation of angiogenesis (data not shown). In our previous studies, the expression level of CD86 in dendritic cells was elevated both in CS-induced emphysema mice and COPD patients;10 however, in the present study, compared with healthy smokers, the expression of CD86 was reduced in patients with severe COPD, although the reason for this is unclear. Possibly, the patients with severe COPD acquired an impaired immune function following repeated bouts of respiratory infection. As the disease progresses, the ability to dispose of damage signals, including bacteria and viruses, may also be impaired and exhausted. Secreted phosphoprotein 1 (SPP1), which encodes osteopontin, is a pleiotropic molecule that has been previously shown to be linked with Th17 responses in COPD.26 Accumulating evidence has supported the proposal that osteopontin was significantly higher in lung tissue and bronchoalveolar lavage fluid of patients with COPD, and furthermore, this was significantly correlated with the levels of neutrophils in sputum and the extent of emphysema.27 A recently published study identified that osteopontin could competitively bind to several antibacterial proteins in the airways, increasing the susceptibility of COPD patients to infections.28
Subsequently, the GO annotations of 314 DEGs were performed on David database. The enriched functions of these genes were, in part, consistent with the pathogenesis of COPD demonstrated in previous studies.11,29,30 For example, an imbalanced activity of proteases and antiproteases identified in lung parenchyma provide one of the important etiological processes in emphysema,29 and the present study showed that the genes enriched in ECM disassembly were upregulated (Table 4), whereas the genes encoding ECM proteins were downregulated (Table 5). In addition, genes encoding collagen- binding proteins were also downregulated (Table 5). In recent years, an increasing number of studies have suggested that the initiation and imbalance of acquired immunity contribute to the amplification and persistence of chronic inflammation in COPD,11,12 and it has been proposed that COPD may also be an autoimmune disease. Similarly, the results of the present study have also demonstrated that genes associated with the immune response and inflammation were significantly differentially expressed in COPD (Table 4). The apoptosis of both endothelial and epithelial cells has been shown to be involved in the development of emphysema,31 and the apoptosis-associated genes were upregulated in the present study (Table 4). Genes associated with negative regulation of protein ubiquitination were decreased in our analysis. The ubiquitin-proteasome system regulates the degradation and function of proteins. CS can induce cytotoxicity via the ubiquitin-proteasome system,30 in addition, airway epithelial cells and macrophages exposed to CS extract may result in more histone deacetylase 2 (HDAC2) degradation via the ubiquitin-proteasome system,32 which may promote the transcription of inflammatory genes. The ubiquitination of contractile proteins was also shown to be involved in the formation of muscle atrophy in patients with COPD.33 In conjunction with the results reported above, this corroborates the credibility of our method of screening for DEGs.
In the present study, the genes Osteoglycin (OGN), ROBO2 and SLIT2 involved in the Roundabout signaling pathway were downregulated in patients with severe COPD (Table 5), a finding which was unanticipated, and which had not been identified in terms of the etiology of COPD previously. However, the role of ROBO and SLIT2 are poorly understood in terms of COPD. In the present study, it was observed that the expression levels of SLIT2 and ROBO2 were decreased in all stages of COPD, and this was significantly negatively correlated with disease progression (Figure 8A–D). As the SLIT and ROBO genes are associated with tissue morphogenesis,34 the present study investigated whether their expression levels were associated with age, and the findings revealed that the expression changes were independent of age and smoking status (Figure 8G–H). In accordance with the microarray results, the protein level of SLIT2, the mRNA expression levels of SLIT2 and ROBO2, were all decreased in the CS-induced emphysema mice compared to the control mice, and the SLIT2 protein level was negatively associated with the degree of emphysema (Figures 10 and 11). In addition, the mRNA expression level of ROBO1 was also decreased in CS-induced emphysema mice. Therefore, it was possible to confirm that SLIT2-ROBO signaling pathway is involved in the pathogenesis of emphysema induced by CS in the mice model.
The ROBO proteins comprise a group of transmembrane receptors belonging to the immunoglobulin superfamily; SLIT proteins, acting as ligands of ROBO, are secreted glycoproteins.35 Four ROBO proteins (ROBO1, ROBO2, ROBO3, ROBO4) have been identified in humans.36 SLIT proteins are endogenously cleaved into a variety of N- and C-terminal fragments, which can inhibit chemoattraction of many immune cells, including T lymphocytes, dendritic cells, macrophages, and neutrophils.9,37,38 Our previous study identified that CS might promote the formation of neutrophil extracellular traps (NETs), which can activate dendritic cells and initiate acquired immunity.12 Therefore, it was possible to hypothesize that the downregulation of the SLIT2- ROBO signaling pathway may result in the attraction of neutrophils and T lymphocytes, which ultimately destroy lung tissue, and forming emphysema; however, this hypothesizes still requires further investigation to verify.
The SLIT2-ROBO pathway is able to inhibit the migration of neutrophils by suppressing the two small GTPases, Cdc42 and Rac2.9,39 Given this knowledge, the role of Cdc42 and Rac2 in patients with COPD was further explored. Consistently, the expression levels of Cdc42 and Rac2 were significantly increased in COPD, and these increases were positively correlated with disease progression (Figure 9A–D). Furthermore, the expression of SLIT2 was negatively correlated with Cdc42 and Rac2 (Figure 9F and H). However, there were no statistical differences in the mRNA expressions of Cdc42 and Rac2 in the lung of the CS-induced emphysema mice compared to control mice. The reason for this is complicated, as the majority cells in lung tissue are able to express Cdc42 and Rac2, it would be better to isolate the neutrophils and then detect the expressions of Cdc42 and Rac2.
The Roundabout signaling pathway was initially found to be involved in neural development via inhibiting the migration of axons.8 In the KEGG analysis of the DEGs, the downregulated genes were also shown to be enriched in the axon guidance (Table 6 and Figure 7). Branchfield et al40 demonstrated that ROBO inactivation in the lung led to an inability of pulmonary neuroendocrine cells to aggregate into sensory organoids, and induced an increased in neuropeptide production which could attract immune cells, and, as a consequence, the ECM was remodeled, and alveoli numbers were reduced. In the CS-induced emphysema mice, the ROBO1 and ROBO2 mRNA expressions both were decreased; however, whether the pulmonary neuroendocrine cells aggregation is also decreased owing to this phenomenon remains incompletely understood, and further experiments are required to further clarify this matter.
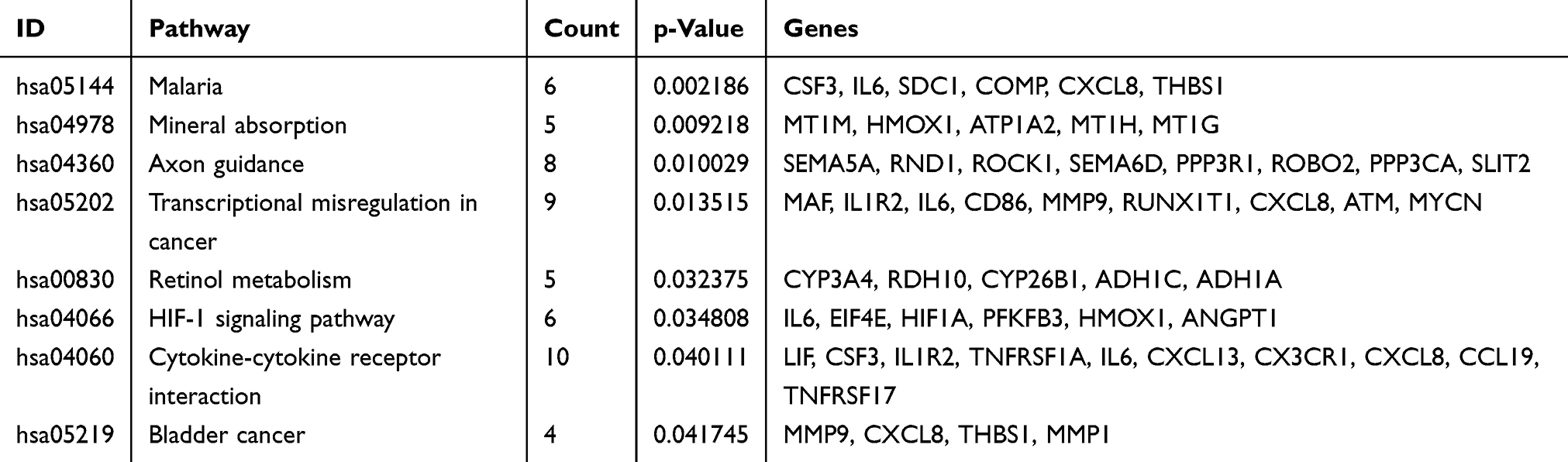 |
Table 6 KEGG pathway analysis of DEGs associated with COPD |
Conclusion
The present study has demonstrated that integrated bioinformatics analysis may provide novel insights into elucidating the complicated pathogenetic processes of COPD. The B cell signaling pathway, osteopontin differentiation, inflammatory response, ECM disassembly, immune response, apoptotic signaling pathway, ubiquitination and the Roundabout signaling pathway all together play an important role in the development of COPD. The SLIT2-ROBO signaling pathway was shown to be downregulated in both patients with COPD and CS-induced emphysema mice, and these changes were significantly negatively correlated with disease progression. To our best known, this study is the first to suggest that the Roundabout signaling pathway may be involved in the pathogenesis of COPD.
Acknowledgment
This study was funded by grants from National Natural Science Foundation of China (grant numbers 81660007 and 81770041).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Blanco I, Diego I, Bueno P, Casas-Maldonado F, Miravitlles M. Geographic distribution of COPD prevalence in the world displayed by geographic information system maps. Eur Respir J. 2019. doi:10.1183/13993003.00610-2019
2. Soriano JB, Abajobir AA, Abate KH, et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet Respir Med. 2017;5(9):691–706. doi:10.1016/S2213-2600(17)30293-X
3. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717. doi:10.1016/S0140-6736(18)30841-9
4. Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67(2):122–131. doi:10.1136/thoraxjnl-2011-200089
5. Tasena H, Faiz A, Timens W, et al. microRNA-mRNA regulatory networks underlying chronic mucus hypersecretion in COPD. Eur Respir J. 2018;52(3). doi:10.1183/13993003.01675-2018.
6. Liu X, Qu J, Xue W, et al. Bioinformatics-based identification of potential microRNA biomarkers in frequent and non-frequent exacerbators of COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:1217–1228. doi:10.2147/COPD.S163459
7. Petryszak R, Burdett T, Fiorelli B, et al. Expression Atlas update–a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42(Database issue):D926–D932. doi:10.1093/nar/gkt1270
8. Li HS, Chen JH, Wu W, et al. Vertebrate slit, a secreted ligand for the transmembrane protein roundabout, is a repellent for olfactory bulb axons. Cell. 1999;96(6):807–818. doi:10.1016/s0092-8674(00)80591-7
9. Tole S, Mukovozov IM, Huang YW, et al. The axonal repellent, Slit2, inhibits directional migration of circulating neutrophils. J Leukoc Biol. 2009;86(6):1403–1415. doi:10.1189/jlb.0609391
10. Qiu SL, Kuang LJ, Tang QY, et al. Enhanced activation of circulating plasmacytoid dendritic cells in patients with Chronic Obstructive Pulmonary Disease and experimental smoking-induced emphysema. Clin Immunol. 2018;195:107–118. doi:10.1016/j.clim.2017.11.003
11. Duan MC, Zhang JQ, Liang Y, et al. Infiltration of IL-17-producing T cells and treg cells in a mouse model of smoke-induced emphysema. Inflammation. 2016;39(4):1334–1344. doi:10.1007/s10753-016-0365-8
12. Qiu SL, Zhang H, Tang QY, et al. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax. 2017;72(12):1084–1093. doi:10.1136/thoraxjnl-2016-209887
13. Spira A, Beane J, Pinto-Plata V, et al. Gene expression profiling of human lung tissue from smokers with severe emphysema. Am J Respir Cell Mol Biol. 2004;31(6):601–610. doi:10.1165/rcmb.2004-0273OC
14. Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep. 2017;7:44232. doi:10.1038/srep44232
15. Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–580. doi:10.1093/bioinformatics/btr709
16. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
17. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi:10.1186/1752-0509-8-49
18. Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–2914. doi:10.1093/bioinformatics/btv300
19. Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics. 2017;7(6):1543–1588. doi:10.7150/thno.15625
20. Bracke KR, Verhamme FM, Seys LJ, et al. Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(3):343–355. doi:10.1164/rccm.201211-2055OC
21. Nunez B, Sauleda J, Anto JM, et al. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(8):1025–1031. doi:10.1164/rccm.201001-0029OC
22. Garudadri S, Woodruff PG, Han MK, et al. Systemic markers of inflammation in smokers with symptoms despite preserved spirometry in SPIROMICS. Chest. 2019. doi:10.1016/j.chest.2018.12.022.
23. Fujita M, Ouchi H, Ikegame S, et al. Critical role of tumor necrosis factor receptor 1 in the pathogenesis of pulmonary emphysema in mice. Int J Chron Obstruct Pulmon Dis. 2016;11:1705–1712.
24. Xiong Z, Leme AS, Ray P, Shapiro SD, Lee JS. CX3CR1+ lung mononuclear phagocytes spatially confined to the interstitium produce TNF-alpha and IL-6 and promote cigarette smoke-induced emphysema. J Immunol. 2011;186(5):3206–3214.
25. Koo HK, Hong Y, Lim MN, Yim JJ, Kim WJ. Relationship between plasma matrix metalloproteinase levels, pulmonary function, bronchodilator response, and emphysema severity. Int J Chron Obstruct Pulmon Dis. 2016;11:1129–1137. doi:10.2147/COPD.S103281
26. Shan M, Yuan X, Song L, et al. Cigarette Smoke Induction of Osteopontin (SPP1) mediates TH17 inflammation in human and experimental emphysema. Sci Transl Med. 2012;4(117):117ra119. doi:10.1126/scitranslmed.3003041
27. Papaporfyriou A, Loukides S, Kostikas K, et al. Increased levels of osteopontin in sputum supernatant in patients with COPD. Chest. 2014;146(4):951–958. doi:10.1378/chest.13-2440
28. Gela A, Bhongir RK, Mori M, et al. Osteopontin that is elevated in the airways during COPD impairs the antibacterial activity of common innate antibiotics. PLoS One. 2016;11(1):e0146192. doi:10.1371/journal.pone.0146192
29. Pandey KC, De S, Mishra PK. Role of proteases in chronic obstructive pulmonary disease. Front Pharmacol. 2017;8:512. doi:10.3389/fphar.2017.00512
30. Kim SY, Lee JH, Huh JW, et al. Cigarette smoke induces Akt protein degradation by the ubiquitin-proteasome system. J Biol Chem. 2011;286(37):31932–31943. doi:10.1074/jbc.M111.267633
31. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol. 2019;81:375–402. doi:10.1146/annurev-physiol-020518-114320
32. Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40(4):464–473. doi:10.1165/rcmb.2008-0255OC
33. Debigare R, Cote CH, Maltais F. Ubiquitination and proteolysis in limb and respiratory muscles of patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2010;7(1):84–90. doi:10.1513/pats.200906-051JS
34. Dickinson RE, Duncan WC. The SLIT-ROBO pathway: a regulator of cell function with implications for the reproductive system. Reproduction. 2010;139(4):697–704. doi:10.1530/REP-10-0017
35. Huang T, Kang W, Cheng AS, Yu J, To KF. The emerging role of Slit-Robo pathway in gastric and other gastro intestinal cancers. BMC Cancer. 2015;15:950. doi:10.1186/s12885-015-1584-3
36. Barak R, Yom-Tov G, Guez-Haddad J, et al. Structural principles in robo activation and auto-inhibition. Cell. 2019;177(2):272–285 e216. doi:10.1016/j.cell.2019.02.004
37. Pilling D, Chinea LE, Consalvo KM, Gomer RH. Different isoforms of the neuronal guidance molecule slit2 directly cause chemoattraction or chemorepulsion of human neutrophils. J Immunol. 2019;202(1):239–248. doi:10.4049/jimmunol.1800681
38. Guan H, Zu G, Xie Y, et al. Neuronal repellent Slit2 inhibits dendritic cell migration and the development of immune responses. J Immunol. 2003;171(12):6519–6526. doi:10.4049/jimmunol.171.12.6519
39. Ye BQ, Geng ZH, Ma L, Geng JG. Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation. J Immunol. 2010;185(10):6294–6305. doi:10.4049/jimmunol.1001648
40. Branchfield K, Nantie L, Verheyden JM, Sui P, Wienhold MD, Sun X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science (New York, N Y). 2016;351(6274):707–710. doi:10.1126/science.aad7969
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.