Back to Journals » Drug Design, Development and Therapy » Volume 20
Rotenone Inhibited Osteosarcoma Cell Growth Through USP47-Induced Decreases in FEN1 Stability and DNA Integrity
Authors Li Z, Ma X, Ma H, Chen B, Jiang K, Zhu Z, Wang J, Wang B, Wang Y ![]() , Dong S
, Dong S ![]()
Received 20 December 2025
Accepted for publication 25 April 2026
Published 4 May 2026 Volume 2026:20 590613
DOI https://doi.org/10.2147/DDDT.S590613
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leonidas D. Panos
Zhen Li,1 Xiang Ma,2 Hengwei Ma,3 Bao Chen,3 Kun Jiang,3 Ziqiang Zhu,3 Jianqiang Wang,3 Baoqing Wang,1 Yunqing Wang,3 Suwei Dong3
1Department of Medical Oncology, The Second Affiliated Hospital of Xuzhou Medical University, Xuzhou, 221000, People’s Republic of China; 2Department of Orthopaedics, The Third Affiliated Hospital of Kunming Medical University, Kunming, 650118, People’s Republic of China; 3Department of Orthopaedics, The Second Affiliated Hospital of Xuzhou Medical University, Xuzhou, 221000, People’s Republic of China
Correspondence: Suwei Dong, Email [email protected]
Background: Osteosarcoma (OS) is the most prevalent primary malignant bone tumor in children. We previously showed that rotenone suppressed OS cell metastasis. However, its effects on OS cell growth and the underlying mechanisms remain unclear. The purpose of this study was to investigate the role of rotenone in OS and identify its direct target.
Methods: Molecular dockingand Biacore assay were used to confirm the interaction between rotenone and USP47. Co-immunoprecipitation, immunofluorescence, and cycloheximide assays were used to verify the relationship betweenUSP47 and FEN1. Cell cycle and apoptosis were examined by flow cytometry. Comet analyses were used to determine DNA damage. Deubiquination and Ub-VME assays were carried out to assess ubiquitination status and properties. Immunohistochemistry and a xenograft mouse model were utilized to validate the effects of various proteins and rotenone in vivo.
Results: Knockdown of FEN1 and USP47 in OS cell lines induced cell cycle arrest and apoptosis through the induction of DNA damage. FEN1 exhibited a direct interaction with USP47. Mechanistically, wild-type USP47 regulated FEN1 protein stability through deubiquitination modification, whereas mutated Cys109Ser USP47 did not. Furthermore, rotenone modulated USP47 protein expression and the combined quantity of Ub-USP47 conjugation through physical interaction. Xenograft studies further confirmed the anti-OS activity of rotenone in vivo.
Conclusion: Rotenone is a potential therapeutic agent for OS due to its direct targeting of USP47 and resultant decrease in FEN1 stability and DNA integrity.
Keywords: osteosarcoma, rotenone, USP47, FEN1, DNA damage
Introduction
Osteosarcoma (OS) is the most frequently occurring malignant tumor in children and adolescents, especially in teenagers during their pubertal growth spurts.1 In the last two decades, the combination of surgery and neoadjuvant chemotherapy has improved the 5-year survival rate to 60–70% in patients with localized tumors.2 However, as OS is a highly heterogeneous tumor, standard chemotherapy does not exert robust antineoplastic effects in some patients.3 OS generally exhibits aberrant gene expression and molecular regulation mechanisms.4 Therefore, the identification of novel targets and effective therapeutic agents is important to improving the management of patients with OS.
Flap structure-specific endonuclease 1 (FEN1) is a structure-specific and multifunctional endonuclease5 that plays a crucial role in double-strand break (DSB) repair,6 long-patch base excision repair (BER),7 and non-homologous end joining (NHEJ).8 As radiotherapy and chemotherapy are based on the induction of DNA damage, enhanced expression of FEN1 in tumors may lead to chemoresistance.9,10 In concordance with this, our previous studies have confirmed that FEN1 participates in chemosensitivity modulation in OS cells.11,12 Thus, there is an urgent need to elucidate the modulating mechanisms of FEN1.
Ubiquitin-specific peptidase 47 (USP47) is a member of the USP family of deubiquitinating enzymes (DUBs).13 It is a post-translational modification enzyme that exerts regulatory effects on target protein activity and many biological processes such as cell growth,14 differentiation,15 and apoptosis.16 Previous reports have demonstrated that silencing of USP47 inhibited the BER pathway and increased DNA double-strand break (DSB) agent sensitivity.17,18 However, identification of USP47 deubiquitylation target proteins during the DNA-damage process has not been achieved. In addition, whether USP47 is required for FEN1 protein stability has also not been determined. Previous studies have indicated the significance of USP47 as a therapeutic target for treating cancer.
Rotenone is a strong inhibitor of the mitochondrial electron transport chain complex I. It is derived from the roots and stems of the Lonchocarpus and Derris species. Mechanistically, rotenone causes a blockade of oxidative phosphorylation, limiting the synthesis of ATP2 and resulting in the generation of reactive oxygen species (ROS). Recent studies have also found that rotenone can induce apoptosis in various cancer cells.19,20 Consistent with these findings, our recent study confirmed that rotenone inhibited OS cell metastasis in vivo and in vitro.21 However, the direct targets interacting with rotenone are not well known.
In the present study, USP47 expression was associated with FEN1 in OS tissues. Moreover, USP47 modulation of OS cell proliferation, apoptosis, and DNA damage was dependent on the ubiquitylation of FEN1. We further confirmed a direct interaction between rotenone and USP47 and that USP47 is required for rotenone-induced apoptosis and DNA damage. Finally, rotenone inhibited tumor growth and USP47/FEN1 expression in mouse model. Together, our data highlight a mechanism by which USP47 deubiquitylated FEN1 to modulate OS cell proliferation, apoptosis, and the DNA damage response. Additionally, rotenone directly inhibited USP47 deubiquitinating enzyme activity. Therefore, rotenone may be a potential anticancer treatment strategy for use in OS patients.
Materials and Methods
Human Tissues
Forty-six OS and age- and sex-matched 38 benign bone tumor (26 osteochondroma, 8 enchondroma, and 4 osteoid osteoma) samples were obtained at random from the Third Affiliated Hospital of Kunming Medical University, Yunnan, China. The Medical Research Ethics Committee of the Third Affiliated Hospital of Kunming Medical University approved this study. The study was conducted following the ethical guidelines of the Declaration of Helsinki, and informed consent was obtained from each participant or their legally authorized representatives.
Bioinformatic Analysis
The gene expression profiling interactive analysis (GEPIA) database (http://gepia.cancer-pku.cn/) was used to analyze the correlation between USP47 and FEN1.
Molecular Docking Model
Binding between rotenone and USP47 was assessed via molecular docking analysis in which the rotenone structure file was downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and the crystal structure of USP47 was obtained from the Protein Database (PDB, https://www.rcsb.org/). AutoDock 4.0 software was used to conduct docking analyses, the docking score was calculated by ITScorePro, and the docking results were visualized with PyMOL 2.6.
Cell Culture, Plasmid Transfection, and Protein Purification
The MG63, U2OS, and 143B human osteosarcoma cell lines and the HEK-293 T human embryonic kidney cell line were obtained from the National Certified Cell Culture Collection Center (Shanghai, China). Cell lines were cultured in MEM (Gibco, USA) containing 10% FBS (Hyclone, USA) and 1% antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin, Solarbio, China). All cells were incubated at 37°C with 5% CO2. The cells were seeded in 6-well plates and cultured overnight to form a 60% monolayer and then transfected using lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s instructions. The cells were harvested after 24 h for subsequent assays. The synthesized plasmids were obtained from RiboBio (China) and verified by direct DNA sequencing. Recombinant protein purification was carried according to the methods described in a previous study.22 Briefly, the cDNAs encoding wild-type USP47 were subcloned into pGEX6P1-N-His vector and expressed in Escherichia coli BL21(DE3). Total protein was obtained following lysis of E. coli, and the proteins were then incubated with Anti-His-tag beads (Beyotime, China). The beads were washed with RIPA buffer and then eluted with His Peptide (Beyotime, China). The purified proteins were collected by centrifugation.
Western Blotting
Cells were subjected to the indicated treatments and lysed with RIPA lysis buffer (Pierce, USA) containing protease inhibitors to extract total protein. A total of 30 μg of protein was separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked before being incubated with the following primary antibodies: USP47 (1:1000, ab72143, Abcam, USA), FEN1 (1:800, ab314001, Abcam, USA), γH2AX (1:1000, 9718, CST, USA), Caspase-3 (1:1000, ab32042, Abcam, USA), Cleaved Caspase-3 (1:1000, ab13847, Abcam, USA), GAPDH (1:1000, ab9485, Abcam, USA), and Lamin B1(1:1000, ab16048, Abcam, USA) overnight at 4°C. Subsequently, the membranes were washed three times with TBST and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10000, 7074, CST, USA) for 1 h. Finally, the membranes were subjected to chemiluminescence using ECL reagent (Millipore, USA). Experimental condition was independently repeated quintuplicate.
Flow Cytometric Analysis
The cell cycle and apoptosis were analyzed using a BD LSRFortessa flow cytometer (BD Biosciences, USA) according to the manufacturer’s protocols. Apoptosis was determined with an annexin V-FITC/PI apoptosis detection kit (Beyotime, China). The cells were washed with cold PBS three times and resuspended in binding buffer, followed by the addition of Annexin V-FITC and PI. The cells were then gently stirred and incubated in the dark at room temperature for 10 min. Cell suspensions were immediately analyzed by flow cytometry. For cell cycle detection, the cells were harvested, cell debris was removed by centrifugation (500×g, 5 min), resuspended in 70% ethanol, washed with PBS, stained with PI/RNase staining buffer for 30 min at room temperature in the dark, and then subjected to flow cytometry. Experimental condition was independently repeated at least 5 times.
CCK‑8 Assay
CCK-8 cell proliferation kit (Beyotime, China) was used for cell viability assessment. Briefly, the cells were seeded in a 96-well plate and incubated under the indicated conditions, followed by the addition of 10 μL of CCK-8 reagent to each well at 24 h, 48 h, 72 h, and 96 h. The optical density (OD) values were detected using a microplate reader (Thermo, USA) at 450 nm. Experimental condition was independently repeated at least 5 times.
Comet Assay
A comet assay kit (Beyotime, China) was used to measure DNA damage following different cell treatments. Single-cell suspensions were prepared in low melting point agarose (Invitrogen, USA) and pipetted onto normal melting point agarose pre-treated slides. The slides were placed in an electrophoresis tank containing chilled electrophoresis buffer and subjected to electrophoresis. Then, the slides were rinsed with Tris HCl and stained with PI, and the comets were photographed under a fluorescence microscope (Olympus, Japan). Tail-moment was used to evaluate the degree of DNA damage. Experimental condition was independently repeated at least 5 times.
Immunofluorescence Analysis
Cells were fixed with 4% formaldehyde, permeabilized with Triton X-100, and blocked with BSA. The slides were incubated with the primary antibody overnight and incubated with secondary antibodies (Alexa Fluor 488and 594, Thermo Fisher, USA), and nuclei were stained with DAPI (Beyotime, China). Images were obtained with a confocal microscope (Zeiss Axiovert 200 M, Germany) with magnification at 400. Experimental condition was independently repeated quintuplicate.
Co-IP Assay
Co-IP assay was conducted to explore the interaction between USP47 and FEN1. The U2OS or 293T cell lysates were collected, and IP lysate (50 mM Tris HCl, 1 mM EDTA, 150 mM NaCl) was added. Protein A/G beads (Santa Cruz, USA) were then added, followed by the antibodies (IgG, ab133470, Abcam, UK; His, 2365, CST, USA; Flag, 14793, CST, USA), and incubated overnight. The beads were subjected to boiling in a loading buffer. Finally, protein complexes and input proteins were ultimately assessed via Western blot analysis. Experimental condition was independently repeated quintuplicate.
De-Ubiquitination Assay
To perform the ubiquitination assays, indicated plasmids were transfected into 293T cells. After transfection for 48 h, 293T cells were treated with MG132 (BML-PI102, Enzo Life Science, USA) at a concentration of 20 μM for 12 h, and collected for Co-IP experiments. The levels of FEN1 ubiquitination were determined by immunoprecipitation (IP) using an anti-Myc antibody (1:1000, ab9106, Abcam, UK), followed by immunoblotting (IB) utilizing an anti-Ub antibody (1:1000, 58395, CST, USA). Experimental condition was independently repeated quintuplicate.
Cycloheximide (CHX) Chase Assay
To measure the half-life of the FEN1 protein, 293T cells were treated with or without rotenone (0.1 μM). After 48 h, the cells were treated with 50 μg/mL of the protein synthesis inhibitor cycloheximide (CHX, 239764, Sigma-Aldrich, USA) for the indicated times. The expression level of FEN1 in the cells was evaluated by Western blotting. Experimental condition was independently repeated quintuplicate.
Ubiquitin Vinylmethyl Ester (Ub-VME) Assay
Ub-VME assay was employed to evaluate drug occupancy on USP47 in U2OS cell lysates, based on a previous study.22 Purified USP47 was incubated with rotenone at the indicated concentration (0.05, 0.1 and 0.2μM) in labeling buffer at room temperature for 20 min. Ub-VME (005, UbiQ, Netherlands) was then added and incubated at 37 °C for 20 min. Protein complexes were ultimately assessed via Western blot analysis using an anti-USP47 antibody. Experimental condition was independently repeated quintuplicate.
Biacore Assay for Surface Plasmon Resonance (SPR) Analysis
To validate the direct interaction between rotenone and USP47, the Biacore S200 biosensor detector (GE Healthcare, USA) and CM5 sensor chip (Xantec; Duesseldorf, Germany) were employed. For SPR analysis, purified USP47 was bound to the parallel-flow channels of the CM5 sensor chip. Rotenone was injected at various concentrations into the flow system at a rate of 20 μL/min. The protein binding period was set to 3 min, and the decomposition period was adjusted to 250 s. Experimental condition was independently repeated quintuplicate.
Immunohistochemistry
All dissected tissues were fixed in 4% PFA and embedded in paraffin. Tissue slices were deparaffinized with xylene, rehydrated with ethanol, and blocked with goat serum. The slices were incubated with primary antibodies against USP47 (1:200, ab84531, Abcam, USA) and FEN1 (1:150, ab314001, Abcam, USA) and then with the appropriate secondary antibodies (ab205718, Abcam, USA). A diaminobenzidine (DAB) tetrahydrochloride kit (Beyotime, China) was used for chromogenic detection, and the sections were subjected to hematoxylin staining and sealed.
Tumor Xenograft Model
BALB/c nude female mice (4–5 weeks old) were obtained from Charles River Laboratories (China). The 143B cells (5 × 106) were suspended in 100 μL saline and injected into the mice via the right flank. When the tumor volume reached approximately 200 mm3, the mice were randomly divided into two groups and treated with DMSO (100 μL) or rotenone (1 mg/kg) via intraperitoneal injection every 3 days. Mouse weight and tumor size were recorded every 3 days. Tumor volumes were calculated using the formula: volume (mm3) = 0.5 × longest tumor diameter × (shortest tumor diameter)2. The mice were euthanized when the tumor diameter exceeded 2.0 cm in any direction, body weight loss exceeded 20%, or tumor burst occurred according to the NIH Guidelines for Endpoints in Animal Study Proposals. IHC staining and Western blotting of tumor tissues were performed according to standard procedures. All procedures followed ethical guidelines for animal experiments of the Experimental Animal Ethics Committee of the Kunming Medical University.
Statistical Analysis
Statistical analyses and mapping were conducted using SPSS Statistics (version 20.0), and GraphPad Prism (version 8.0.2). Two-tailed Student’s t test was used to compare individual data between two groups, and Pearson’s correlation coefficient was used for the linear correlation between two different parameters. The Kaplan-Meier method and Log rank test were used to analyze the survival curves. Data are presented as mean ± SD. A P-value < 0.05 was considered statistically significant for all analyses.
Results
FEN1 Modulated OS Cell Cycle, Apoptosis, and DNA Damage
To understand the potential role of FEN1 in OS, we first revealed that knockdown of FEN1 in the MG-63 and U2OS OS cell lines induced cell cycle G1 phase arrest (siFEN1 vs. Scramble, 67.42 ± 1.69% to 57.08 ± 0.34% for MG-63 and 68.01 ± 0.76% to 61.22 ± 1.03% for U2OS. Figure 1A and B). Flow cytometric analysis also showed that FEN1 knockdown induced apoptosis (siFEN1 vs. Scramble, 16.57 ± 0.16% to 4.65 ± 0.48 for MG-63 and 29.12 ± 0.28% to 4.79 ± 0.24% for U2OS. Figure 1C). It has been confirmed that FEN1 plays an important role in DNA repair and replication through the removal of 5′-flap structures formation in Okazaki fragment maturation and long-patch base excision repair by cleaving within the apurinic/apyrimidinic site-terminated flap.23,24 Therefore, we investigated whether FEN1 modulated OS cell proliferation and apoptosis via its DNA damage repair function. Comet assay showed significant DNA damage after FEN1 knockdown (Figure 1D). In concordance with this, protein expression of the DNA damage marker γH2AX was markedly elevated (Figure 1E). These observations suggest that down regulation of FEN1 induced OS cell cycle G1 phase arrest, apoptosis, and DNA damage.
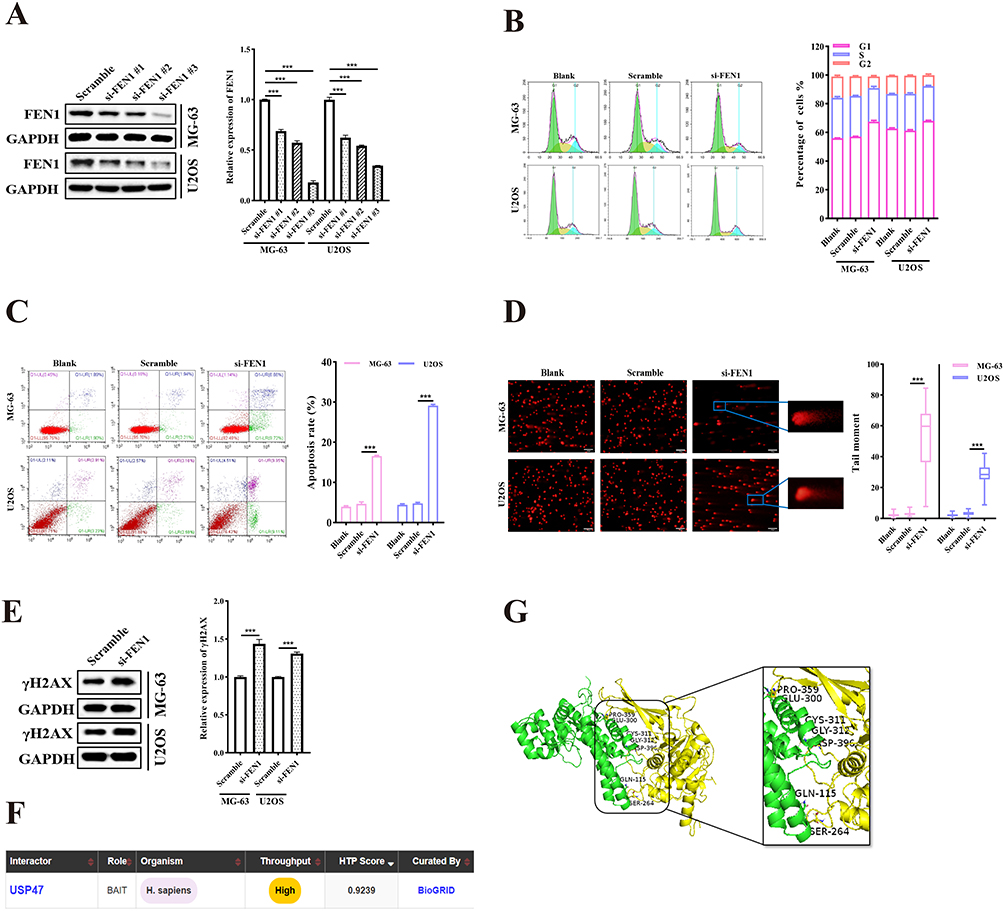 |
Figure 1 FEN1 modulated OS cell viability. (A and B) Cell cycle distribution of MG-63 and U2OS cells with or without FEN1 knockdown using specific siRNAs (n = 5). (C) Apoptosis rates of MG-63 and U2OS cells transfected with FEN1-siRNAs (n = 5). (D and E) Comet assay and γH2AX protein levels demonstrating DNA damage after FEN1 knockdown in MG-63 and U2OS cells. (Scale bar: 100 μm, n = 5). (F) USP47 was predicted to bind to FEN1 using the BioGrid database. (G) The combination of USP47 and FEN1 was verified by HDock software. Results are the mean ± SD. ***P < 0.001. |
To illustrate FEN1 protein expression modulatory mechanisms, the BioGRID database (https://thebiogrid.org/) was utilized to screen for potential FEN1 interacting proteins. This analysis predicted that USP47 may bind with FEN1 (Figure 1F). Moreover, HDock (http://hdock.phys.hust.edu.cn/) was utilized to verify the binding of FEN1 to USP47 (Figure 1G). Thus, we speculated that USP47 may participate in FEN-1-mediated OS cell cycle arrest, apoptosis induction, and DNA damage.
USP47 Modulated OS Cell Cycle Arrest, Apoptosis, and DSB
USP47 expression was knocked down in the OS cell lines via USP47 siRNA to assess the role of USP47 in OS cells. The results showed that USP47 knockdown inhibited OS cell cycle G1 phase arrest (siUSP47 vs. Scramble, 73.09 ± 0.21% to 56.75 ± 0.22% for MG-63 and 71.15 ± 0.73% to 62.56 ± 0.56% for U2OS. Figure 2A and B). In addition, flow cytometric analysis demonstrated a notable increase in apoptosis following transfection with siRNA-USP47 compared with the negative control group (siUSP47 vs. Scramble, 26.56 ± 1.36% to 12.21 ± 0.47 for MG-63 and 18.11 ± 0.64% to 8.2 ± 0.22% for U2OS. Figure 2C). This result was consistent with the Western blotting results showing that cleaved-caspase 3 was elevated in OS cells after USP47 and FEN1 siRNA transfection (Figure 2D). Furthermore, comet assay confirmed that silencing of USP47 resulted in DNA damage (Figure 2E), and γH2AX expression was subsequently upregulated (Figure 2F). These findings confirmed that USP47 participates in OS cell cycle arrest, apoptosis, and DNA damage.
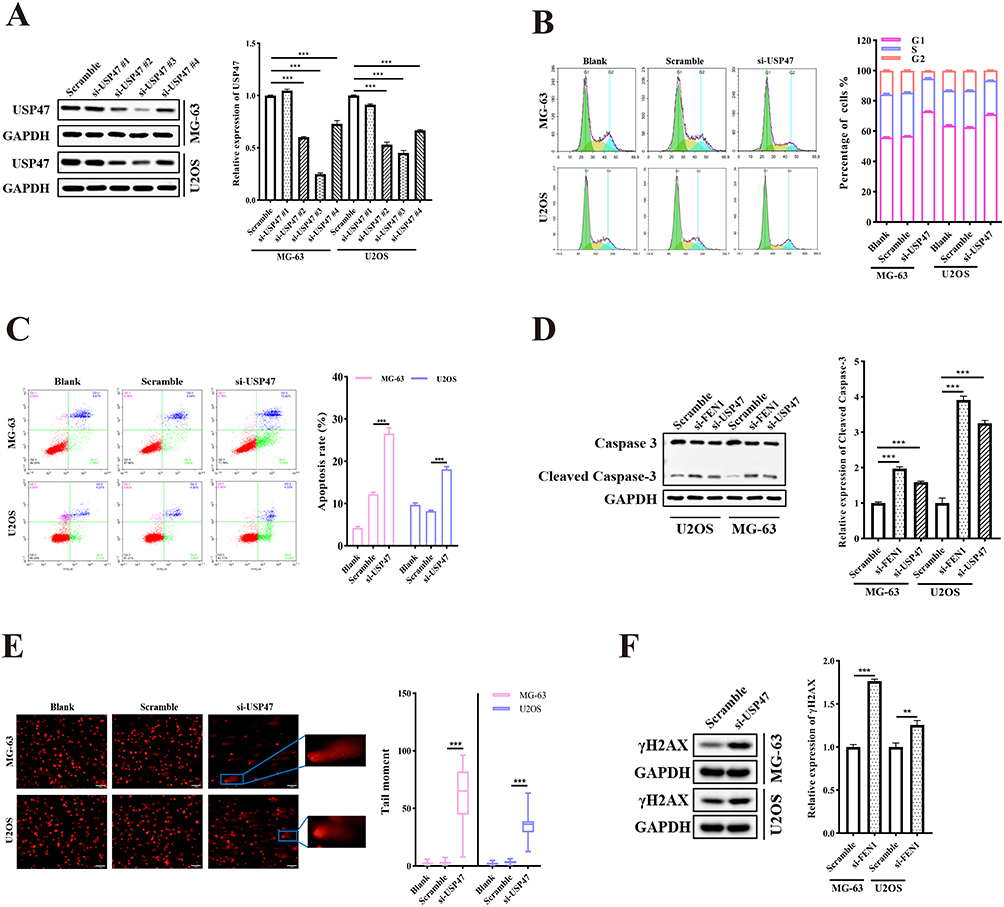 |
Figure 2 USP47 modulated OC cell viability. (A and B) Cell cycle distribution of MG-63 and U2OS cells with or without USP47 knockdown via specific USP47-siRNAs (n = 5). (C) Apoptosis rates in MG-63 and U2OS cells transfected with siRNAs (n = 5). (D) Cleaved-caspase3 protein levels demonstrating apoptosis induced by FEN1 or USP47 knockdown. (E and F) Comet assay and γH2AX protein levels showing DNA damage after USP47 knockdown in MG-63 and U2OS cells. (Scale bar: 100 μm, n = 5). Results are the mean ± SD. **P < 0.01, and ***P < 0.001. |
USP47 Interacted with and Deubiquitinated FEN1
To assess the potential role of USP47 in regulating FEN1 protein expression, we transfected USP47 siRNA or expression plasmids into OS cells. Western blotting results showed that USP47 knockdown decreased the total and nuclear FEN1 protein levels, and USP47 expression resulted in a contrary result (Figure 3A). To validate the interaction between USP47 and FEN1, plasmids were co-transfected into 293T cells. Confocal microscopy data exhibited co-localization of the USP47 and FEN1 proteins (Figure 3B). Moreover, Co-IP assay verified their direct interaction with each other (Figure 3C). To explore the consequence of the interaction between USP47 and FEN1, we analyzed the deubiquitinating function of USP47 and its effect on FEN1 protein stability. The results showed that the transfection of USP47 dramatically reduced the polyubiquitination levels of FEN1 (Figure 3D). To verify if the deubiquitinase activity of USP47 is required for FEN1 protein stability, we synthesized a mutated Cys109Ser USP47 protein lacking deubiquitinase activity, as previously described.13 The 293T cells were co-transfected with a Myc-tagged FEN1-expressing vector containing a Flag-tagged wild-type or mutant USP47 expressing vector. As expected, the wild-type but not the mutant USP47 decreased the polyubiquitination levels of FEN1 (Figure 3E). This indicated that USP47 regulated FEN1 protein stability through deubiquitination modification.
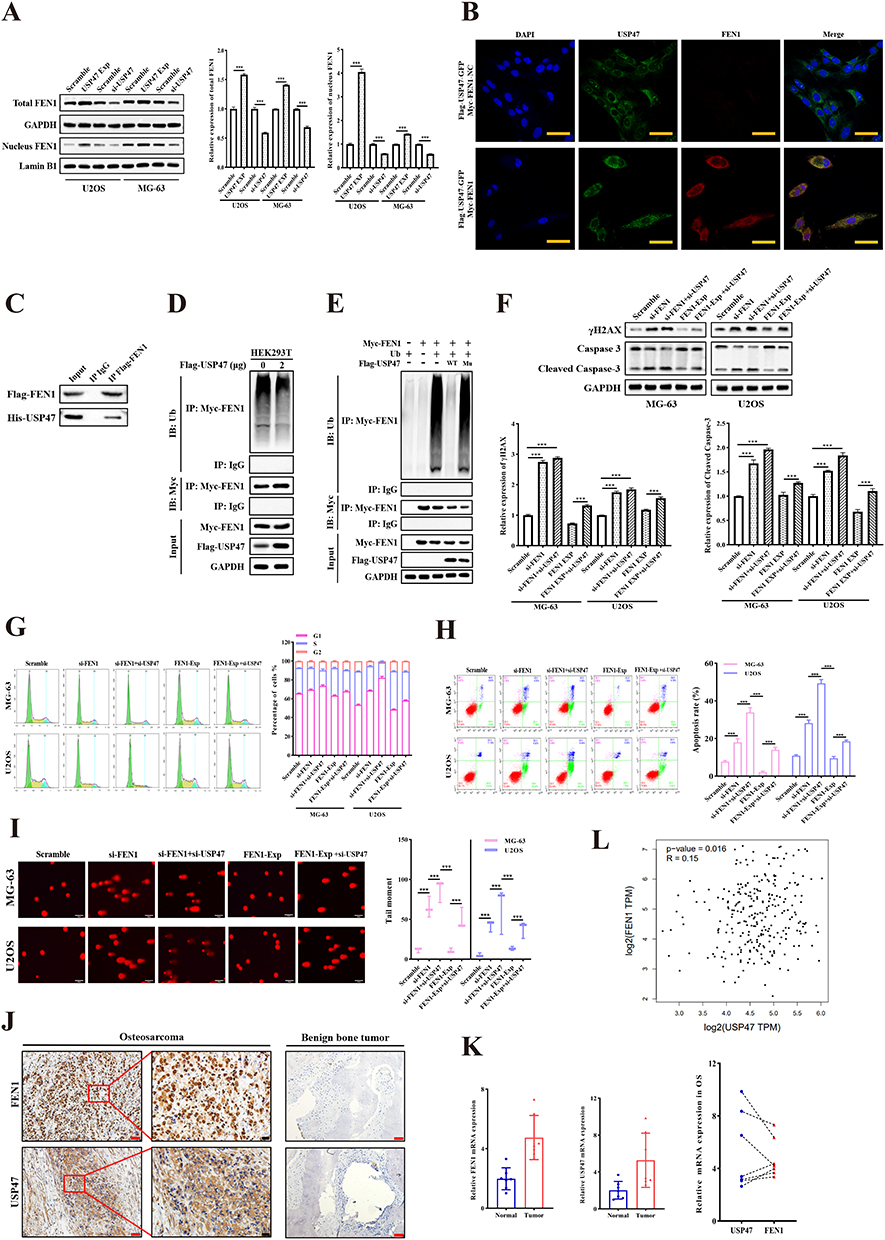 |
Figure 3 USP47 interacted with and deubiquitinated FEN1. (A) FEN1 protein levels with USP47 siRNA or expression plasmid transfection. (B) Confocal microscopy was used to clarify the co-location of FEN1 and USP47. (Scale bar: 50 μm, n = 5). (C) Co-IP analysis was performed to test the interaction between FEN1 and USP47. (D and E) 293T cells were co-transfected with Myc-FEN1, Flag-USP47, and Flag-USP47 mutant plasmids, and polyubiquitination analysis of FEN1 was carried out. (F) γH2AX and cleaved-caspase 3 protein levels after USP47 knockdown in FEN1 low/high-expression OS cells. (G) Cell cycle distribution of MG-63 and U2OS cells treated with indicated conditions (n = 5). (H) Apoptosis rates in MG-63 and U2OS cells transfected with indicated plasmids (n = 5). (I) Comet assay showing DNA damage after treatment. (Scale bar: 200 μm, n = 5). (J and K) USP47 and FEN1 protein (n = 46 for OS, n = 38 for control) and mRNA (n = 7) expression in human OS and control group tissues. Scale bar: 50 μm (Red) or 20 μm (Black). (L) Correlation between USP47 and FEN1 expression based on the GEPIA database. Results are the mean ± SD. ***P < 0.001. |
More importantly, FEN1 knockdown minimized the increased γH2AX and cleaved-caspase 3 protein levels induced by USP47 down-regulation. In contrast, FEN1 overexpression neutralized the effect of USP47-siRNA on OS cells (Figure 3F). This result further confirmed by flow cytometry-based cell cycle and apoptosis detection and comet assay (Figure 3G–I). These indicate that USP47-modulated OS cell DNA damage and apoptosis may be dependent on FEN1 to some extent. Furthermore, we detected USP47 and FEN1 expression in OS and benign bone tumor tissues in the xenograft mouse model. IHC and QPCR results showed that USP47 and FEN1 expression was upregulated in and associated with tumor size in human OS tissues compared to controls (Figure 3J, K and Tables 1, 2). Moreover, statistical analysis revealed a positive correlation between the expression of FEN1 and USP47 (Table 3). Consistent with this, GEPIA database (http://gepia.cancer-pku.cn/) results showed a positive correlation between these two proteins in sarcoma (Figure 3L). Taken together, these results demonstrate that FEN1 is a substrate of the USP47 deubiquitinase.
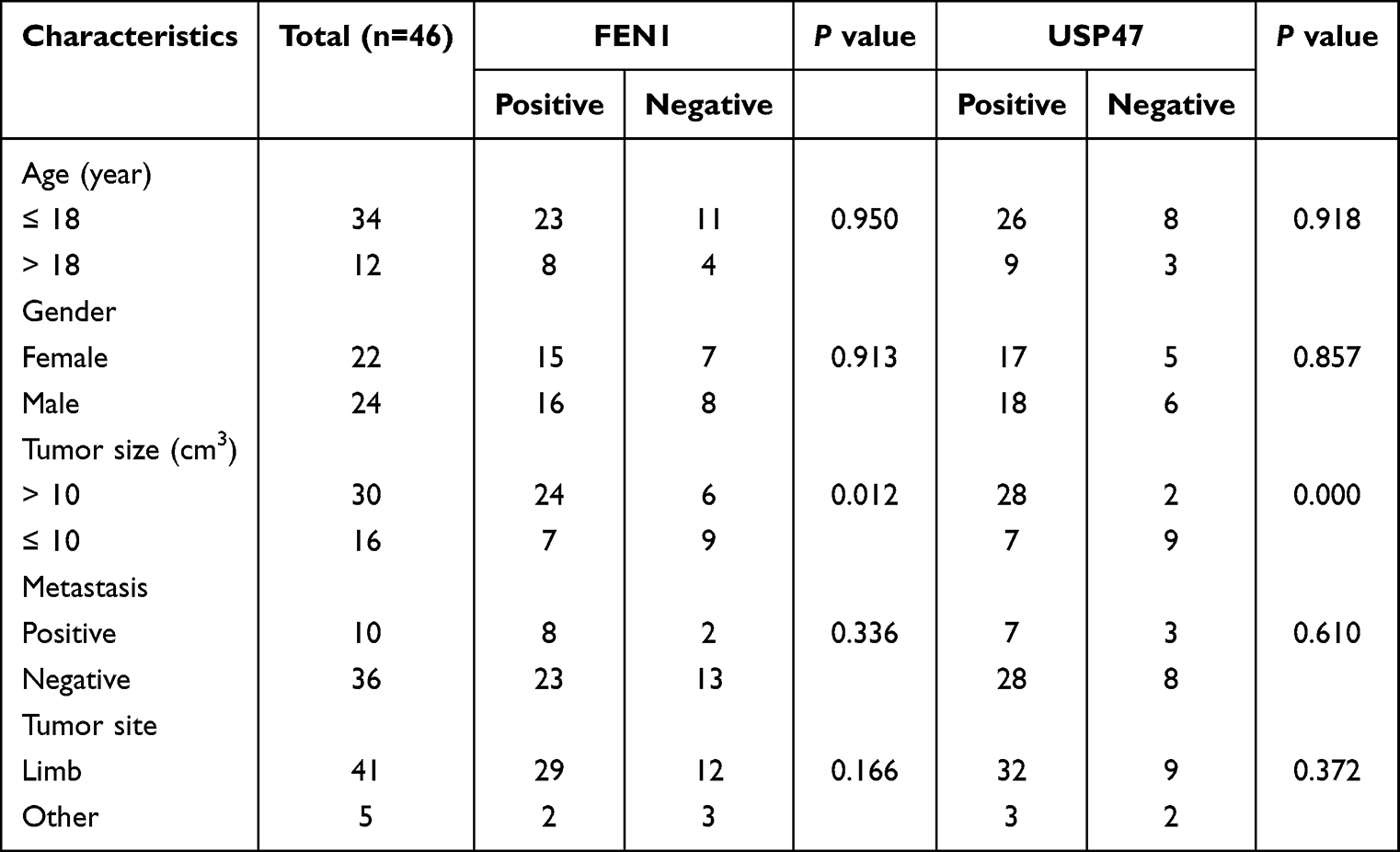 |
Table 1 Correlations Between the FEN1 and USP47 Expression Levels and Clinical Characteristics |
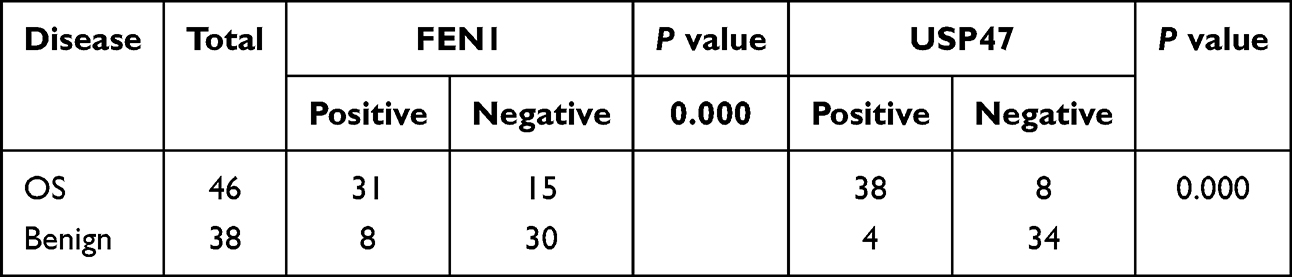 |
Table 2 Expression of FEN1 and USP47 in OS and Benign Bone Tumor |
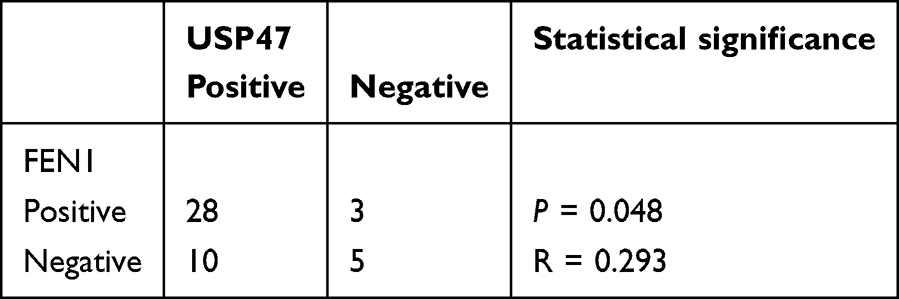 |
Table 3 Correlation Between FEN1 and USP47 Expression in OS |
Rotenone Modulated OS Cell Cycle Arrest, Apoptosis, and DSB
Our recent study suggested rotenone (Figure 4A) as a potential treatment for OS due to its cytotoxic and anti-metastatic effects.21 To assess the ability of rotenone to induce cell cycle arrest, apoptosis, and DNA damage, OS cells were exposed to rotenone for 48 h at its IC50 concentration.21 Then, cell cycle distribution was analyzed via flow cytometry. Western blotting detected upregulated levels of cleaved-caspase 3 protein. The results also demonstrated that rotenone induced OS G1 phase arrest (Rotenone vs. DMSO, 71.28 ± 1.34% to 57.24 ± 0.39% for MG-63, 68.87 ± 0.92% to 61.88 ± 0.81% for U2OS and 82.91 ± 1.36 to 70.12 ± 2.81% for 143B) and apoptosis (Rotenone vs. DMSO, 50.54 ± 1.34% to 8.76 ± 0.39% for MG-63, 50.71 ± 7.62% to 10.87 ± 0.14% for U2OS and 40.56 ± 4.66 to 8 ± 3.17% for 143B). CCK-8 results also showed that rotenone or FEN1/USP47 knockdown significantly inhibited OS cell proliferation. Further, rotenone also induced significant DNA damage (Figure 4B–G. Subsequent Western blotting analysis showed that rotenone significantly decreased USP47 and FEN1 protein levels (Figure 4H). Importantly, USP47 knockdown reinforced rotenone-induced γH2AX and cleaved-caspase 3 expression. Alternatively, USP47 overexpression compromised the effects of rotenone treatment (Figure 4I). These results suggest that rotenone-induced DNA damage and apoptosis in OS cells may be dependent on USP47 expression.
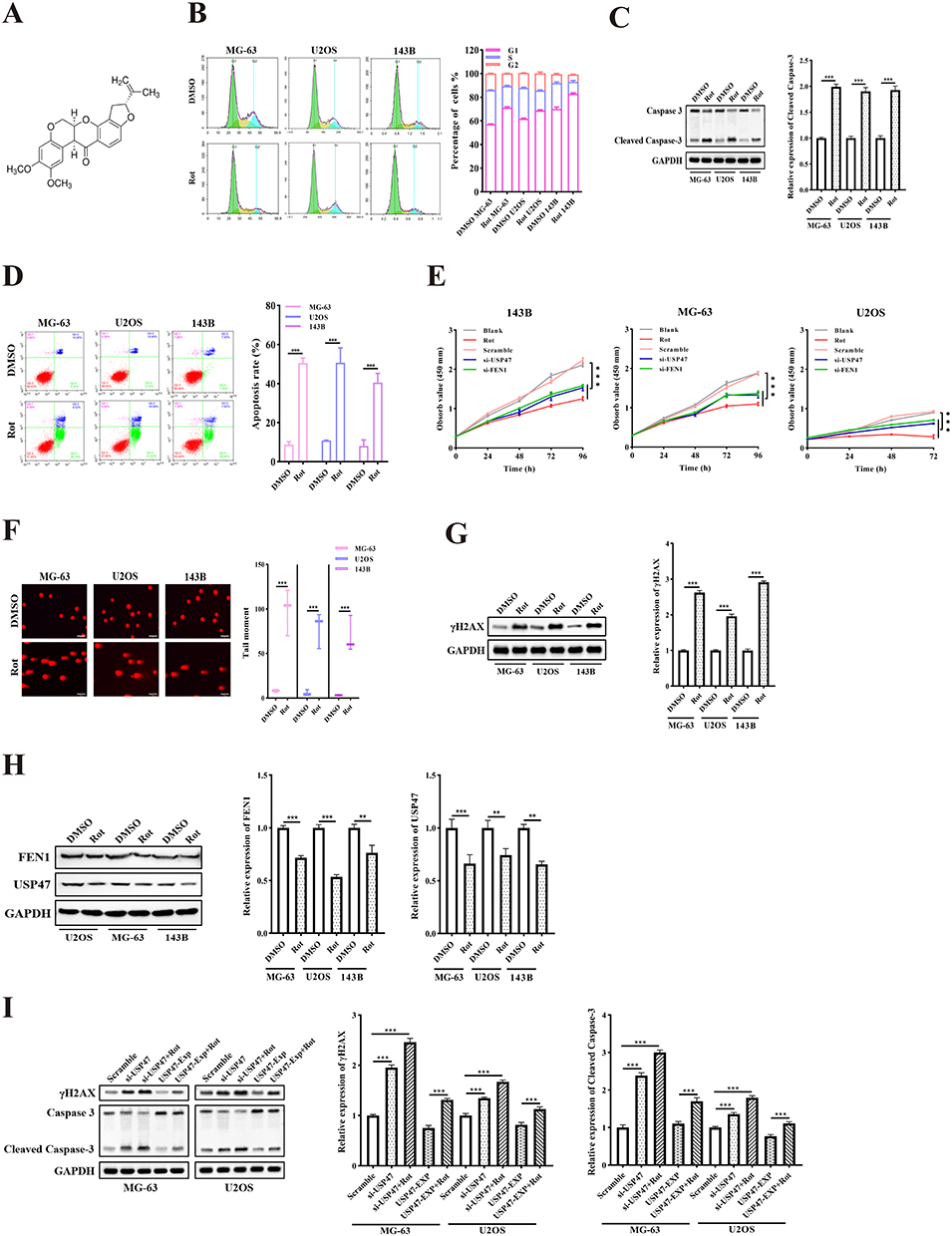 |
Figure 4 Rotenone modulated OS cell viability. (A) Chemical structure of rotenone. (B) Cell cycle distribution of OS cells treated with rotenone (n = 5). (C) Cleaved-caspase 3 protein expression of OS cells treated with rotenone. (D) Apoptosis rates of different groups were measured by flow cytometric analysis (n = 5). (E) OS cell proliferation measured by MTT after treatment with rotenone, si-FEN1, and si-USP47 (n = 5). (F) Comet assay showing DNA damage after rotenone treatment. (Scale bar: 200 μm, n = 5). (G) γH2AX protein levels showing DNA damage in cells treated with rotenone. (H) USP47 and FEN1 protein expression in OS cells treated with rotenone. (I) γH2AX and cleaved-caspase 3 protein levels after rotenone treatment in USP47 low/high-expression OS cells. Rotenone treatment at 0.02, 1.09, and 1.35 μM in MG-63, U2OS, and 143B cells, respectively. Results are the mean ± SD. **P < 0.01, and ***P < 0.001. |
Rotenone Induced USP47 Protein Inhibition and Degradation
The underlying mechanisms through which USP47 modulates rotenone-induced deregulation of OS cells remain largely unknown. First, we determined the half-life of the FEN1 protein in 293T cells. Analysis showed that rotenone treatment markedly accelerated FEN1 protein degradation in the presence of cycloheximide, which is used to inhibit protein biosynthesis (Figure 5A). However, rotenone did not block the interaction between USP47 and FEN1 (Figure 5B). This indicated that rotenone can efficiently degrade USP47.
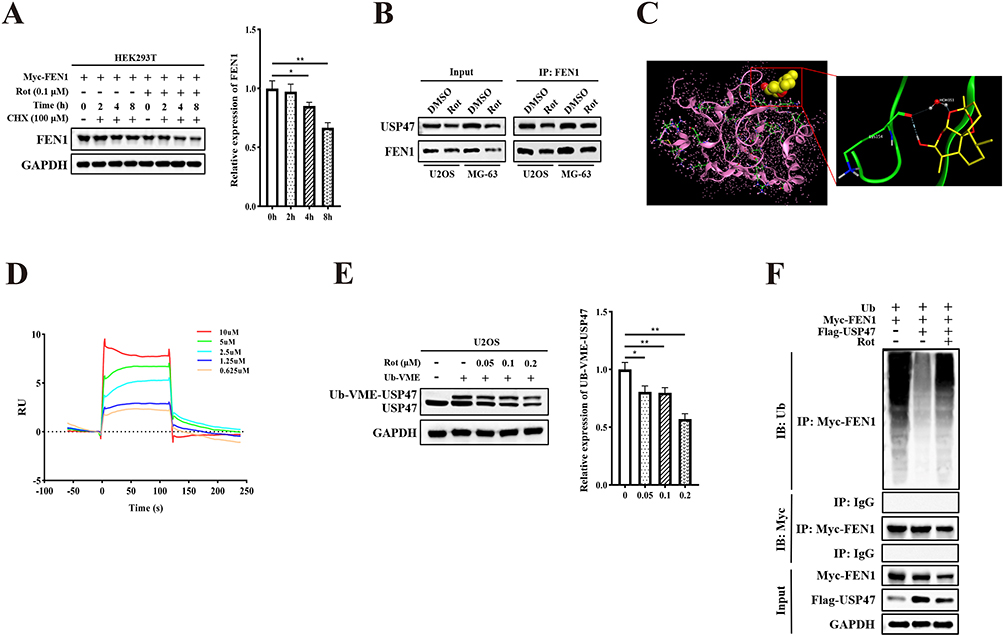 |
Figure 5 Effects of rotenone on USP47 and molecular docking analysis. (A) 293T cells treated with rotenone for the indicated times; FEN1 protein was detected as shown. (B) Co-IP analysis showed the interaction between FEN1 and USP47 when treated with rotenone. (C) Rotenone was predicted to bind to USP47 using molecular docking. (D) The combination of rotenone and USP47 was verified by Biacore analysis. (E) U2OS cells were treated with rotenone and the Ub-VME probe was added. Samples were subsequently analyzed by Western blotting using anti-USP47 antibody to detect the Ub-USP47 conjugate formation. (F) 293T cells were co-transfected with Myc-FEN1, Flag-USP47, and treated with or without rotenone; polyubiquitination analysis of FEN1 was carried out. Results are the mean ± SD. *P < 0.05, and **P < 0.01. |
Next, we tested whether USP47 is a direct target of rotenone. To validate this hypothesis, we analyzed the potential interaction between USP47 and rotenone through molecular docking (Figure 5C). Biacore analysis further demonstrated that rotenone interacts with the USP47 protein directly (KD value 2.252E-6, Figure 5D). Then, we performed competition assays between rotenone and the ubiquitin-vinyl methyl ester Ub active site probe (Ub-VME). The results showed that recombinant USP47 incubated with Ub-VME exhibited a Ub-USP47 conjugate formation. In contrast, the Ub-USP47 conjugate formation was dose-dependently inhibited in rotenone-treated samples (Figure 5E). Because rotenone degrades USP47, this could impact the combination quantity of Ub-USP47 conjugation. We then co-transfected Flag-USP47 and Myc-FEN1 into 293T cells with or without rotenone treatment. The results showed that rotenone dramatically increased the polyubiquitination levels of FEN1 (Figure 5F). These results illustrated that rotenone degraded and inhibited the USP47 protein to modulate its expression and deubiquitinase activity.
Rotenone Exerted Anti-OS Activity in vivo
To further confirm the inhibitory effect of rotenone on OS in vivo, a BALB/c xenograft model was employed; DMSO was used as a control group. The mice were intraperitoneally injected with DMSO (100 μL) or rotenone (1 mg/kg) every 3 days. The results indicated that rotenone significantly suppressed tumor volume (Rotenone vs. DMSO, 1053.59 ± 253.52 mm3 to 1389.87 ± 175.47 mm3) and weight (Rotenone vs. DMSO, 0.76 ± 0.26 g to 1.17 ± 0.16 g) compared to the control group (Figure 6A and 6B). Notably, we found that the mice receiving rotenone treatment survived significantly longer than those in the control group (Figure 6C). In addition, IHC staining and Western blotting showed that USP47 and FEN1 expression was decreased following rotenone treatment (Figure 6D and E). These results validated the anti-OS effects and mechanisms of rotenone in vivo.
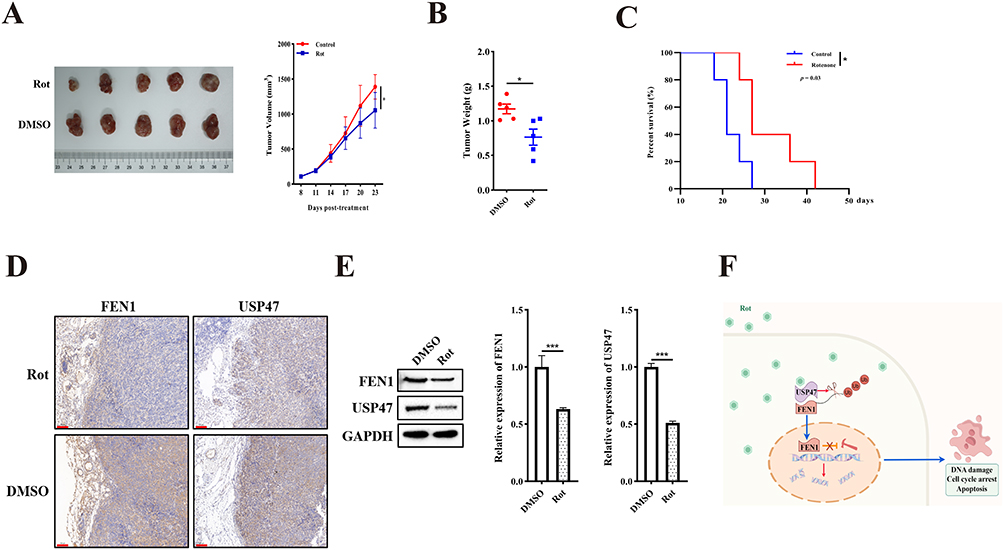 |
Figure 6 Rotenone suppressed the USP47/FEN1 pathway in vivo. (A) Tumor growth of subcutaneously implanted mice was measured every 3 days (n = 5). (B) Tumor weight was measured. (C) The survival rate of tumor-bearing mice (time to 20 mm in maximum diameter) was determined by Kaplan-Meier analysis; statistical differences were analyzed with Log rank test and survival. Scale bar: 100 μm. (D) Representative IHC images of USP47 and FEN1 in xenograft tissues. (E) Protein expression of USP47 and FEN1 in xenograft tissues. (F) Working model illustrating Rotenone inhibited OS cell growth through USP47-induced decreases in FEN1 stability and DNA integrity. Results are the mean ± SD.*P < 0.05, and ***P < 0.001. |
Taken together, our findings prove that Rotenone inhibited OS cell growth through USP47-induced decreases in FEN1 stability and DNA integrity (Figure 6F).
Discussion
Our previous study showed that FEN1 may serve as a novel therapeutic target in OS, mainly at the induction of autophagy and apoptosis.12 FEN1 is involved in eukaryotic DNA replication and repair.25 Which is considered as an anti-tumor factor, as it is involved in DSB repair and thus supports genetic stability.26,27 Dysregulated FEN1 expression has been shown to exhibit pro-tumorigenesis functions in various cancer types.28–31 Previously, we confirmed that FEN1 is associated with OS cell apoptosis, autophagy, and chemosensitivity.12 The present study further evidenced the functions of FEN1 in DNA damage rectification, cell cycle modulation, and apoptosis. These results confirmed that FEN1 is needed for OS malignant phenotype by sustain the DNA stability. It is known that tumors acquire increased levels of FEN1 protein to maintain genetic stability and escape from DNA damage-induced apoptosis.10,32 However, the underlying mechanism that modulates the abnormal expression of FEN1 remains largely unknown.
Protein quality control depends on the balance between synthesis and degradation. Ubiquitination is an important post-translational modification. Deubiquitinating enzymes (DUBs) are proteases in the ubiquitin-proteasome system (UPS) that can protect target proteins from degradation by the 26S proteasome. Approximately 80% of cellular proteins are degraded via UPS.33 Thus, DUBs have recently emerged as a drug target in cancer management.34 USP47 acts as a deubiquitylation enzyme, and its silencing has been shown to inhibit cell survival and sensitize cells to DNA-damaging agents.18 However, the underlying mechanism remains unclear. In the present study, we showed that FEN1 is a substrate of USP47 and that USP47-induced apoptosis and DNA damage are dependent on FEN1. Furthermore, we demonstrated a positive correlation between the expression of USP47 and FEN1 in OS tissues. In present study, we collected 46 OS and 38 control samples, an expanded sample size and multicenter cooperation will provide powerful additional subgroup analysis. In addition to this, the normal tissues adjacent OS are also needed. Of note, as a DUB, USP47 could modulates stability of various proteins. Thus, FEN1 might not be the only or most important substrate. Then, the function of USP47 is dependent on the protein expression pattern of different cancers. Present data suggest that USP47 could be a potential target in OS treatment.
Rotenone is a natural product derived from the Lonchocarpus and Derris species. Rotenone is a mitochondrial electron transport chain complex (ETC) I inhibitor, and it exhibits multiple anti-cancer pharmacological activities.19,35 Our previous study showed that rotenone-induced ROS triggered Ca2+/AMPK/ZO-2 signaling, contributing to inhibition of OS cell metastasis.21 However, the direct targets interacting with rotenone remained unclear. In this study, we confirmed that rotenone directly interacted with USP47 and inhibited its deubiquitinating enzyme activity. It is regrettable that present data did not prove the precise binding sites between rotenone and USP47. Present study indicates that rotenone holds potential as a therapeutic drug against USP47 over expression OS.
However, chronic rotenone administration has been shown to result in Parkinson’s disease (PD)-like biochemical and behavioral changes in rats, which is why it is widely used to induce a rat model of neurodegeneration mimicking PD in humans. This is achieved with various doses as well as frequencies and administration routes (orally, subcutaneous injection, and intraperitoneal injection).36–39 Neurotoxicity analysis showed that rotenone did not induce toxicity at a concentration of 1 μM, where a dose of 25 μM was cytotoxic.40 Of note, the management dosage in the current study was lower and dosing was less frequent than that used to induce the PD model (1.5–2.5 mg/kg/day for intraperitoneal injection).41,42 Although we did not measure the in vivo rotenone concentration, no significant PD-like symptoms were observed in the present study.
Undeniably, rotenone poisoning is fatal and with no antidote.43 For humans, ingested 1.14–2.28 mg/kg rotenone would be safe.43 As USP47 might not be the unique target of rotenone, off-target effects would cause serious side effects, and limited the toxic dose. To minimize the adverse reactions, intratumoral injection could be a considerable treatment for accessible tumors. Moreover, synthetize a novel small molecule compound based on rotenone crystal structure that contain USP47 binding sites could target USP47 specificity.
It’s interesting that liver, bone marrow and bone are the most important target organs for rotenone. Moreover, administration of rotenone through the diet did not lead to obvious neurotoxicity in rats model.44 Thus, we speculate that rotenone could be a high affinity drug for bone tumors treatment. However, for further application, systematic safety and toxicology investigation of rotenone are needed, especially oral administration.
Rotenone can penetrate into the central nervous system (CNS), and recent studies have shown an anti-glioma effect of rotenone in vitro.45,46 Thus, rotenone may be a candidate for use in patients with glioma or brain metastasis. However, it is difficult to measure the drug concentration in the CNS. Thus, further investigation is needed on rotenone pharmacokinetics and toxicology before it can be used clinically. Additionally, as radiotherapy and chemotherapy are based on the induction of DNA damage,47 we speculate that a combination of low-dose rotenone with chemotherapy may promote chemosensitivity, and this may be a more feasible strategy for cancer treatment.Collectively, our study demonstrated that USP47 inhibitory activity was a novel bioactivity of rotenone and elucidated a potential molecular mechanism of rotenone in FEN1 stability and DNA integrity. Therefore, rotenone holds promise as a candidate therapeutic agent for USP47-aberrant OS.
Data Sharing Statement
The data used to support the conclusions of this study are included in the article.
Ethics Approval and Consent to Participate
The study protocol was approved by the Medical Ethics Committee of the Third Affiliated Hospital of Kunming Medical University. The procedures for the collection and use of tissues were performed in accordance with the guidelines of the Helsinki Declaration of 2013 (No. KYCS-011, 26 Feb 2024). Animal-welfare was following the NIH Guidelines for Endpoints in Animal Study Proposals (2019).
Acknowledgments
We thank LetPub (www.letpub.com.cn) for its linguistic assistance during the preparation of this manuscript. Figure 6F was created using Figdraw (www.figdraw.com, ID: WWWYYb3033).
Funding
This study was supported in part by grants from the National Natural Science Foundation of China (82203396), the Natural Science Foundation of Jiangsu Province (BK20220235, BK20231164), the Provincial Clinical Medicine Center Construction Project of Yunnan Province (2024YNLCYXZX0397), and the Pengcheng Yingcai Medical Youth Reserve Talent Training Project (XWRCHT20220028).
Disclosure
The authors declare no conflicts of interest.
References
1. Bielack SS, Kempf-Bielack B, Delling G, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002;20:776–16. doi:10.1200/JCO.2002.20.3.776
2. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer. 2009;115:1531–1543. doi:10.1002/cncr.24121
3. Bernthal NM, Federman N, Eilber FR, et al. Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer. 2012;118:5888–5893. doi:10.1002/cncr.27651
4. Zhou C, Balmer L, Song M, et al. Identification of circRNA biomarkers in osteosarcoma: an updated systematic review and meta-analysis. Noncoding RNA Res. 2024;9:341–349. doi:10.1016/j.ncrna.2024.01.007
5. Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–138. doi:10.1146/annurev-biochem-072511-122603
6. Wu X, Wilson TE, Lieber MR. A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. Proc Natl Acad Sci U S A. 1999;96:1303–1308. doi:10.1073/pnas.96.4.1303
7. Ranalli TA, Tom S, Bambara RA. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem. 2002;277:41715–41724. doi:10.1074/jbc.M207207200
8. Mengwasser KE, Adeyemi RO, Leng Y, et al. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol Cell. 2019;73:885–899.e6. doi:10.1016/j.molcel.2018.12.008
9. He L, Yang H, Zhou S, et al. Synergistic antitumor effect of combined paclitaxel with FEN1 inhibitor in cervical cancer cells. DNA Repair. 2018;63:1–9. doi:10.1016/j.dnarep.2018.01.003
10. Lu X, Liu R, Wang M, et al. MicroRNA-140 impedes DNA repair by targeting FEN1 and enhances chemotherapeutic response in breast cancer. Oncogene. 2020;39:234–247. doi:10.1038/s41388-019-0986-0
11. Dong S, Xiao Y, Zhu Z, et al. Metformin sensitises osteosarcoma to chemotherapy via the IGF-1R/miR-610/FEN1 pathway. Eur J Histochem. 2023;67:3612.
12. Dong S, Xiao Y, Ma X, et al. miR-193b increases the chemosensitivity of osteosarcoma cells by promoting FEN1-mediated autophagy. Onco Targets Ther. 2019;12:10089–10098. doi:10.2147/OTT.S219977
13. Piao J, Tashiro A, Nishikawa M, et al. Expression, purification and enzymatic characterization of a recombinant human ubiquitin-specific protease 47. J Biochem. 2015;158:477–484. doi:10.1093/jb/mvv063
14. Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi:10.1146/annurev.biochem.78.082307.091526
15. Bernassola F, Karin M, Ciechanover A, et al. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21. doi:10.1016/j.ccr.2008.06.001
16. Melino G. Discovery of the ubiquitin proteasome system and its involvement in apoptosis. Cell Death Differ. 2005;12:1155–1157. doi:10.1038/sj.cdd.4401740
17. Parsons JL, Dianova II, Khoronenkova SV, et al. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase β. Mol Cell. 2011;41:609–615. doi:10.1016/j.molcel.2011.02.016
18. Peschiaroli A, Skaar JR, Pagano M, et al. The ubiquitin-specific protease USP47 is a novel beta-TRCP interactor regulating cell survival. Oncogene. 2010;29:1384–1393. doi:10.1038/onc.2009.430
19. Hu W, Tian H, Yue W, et al. Rotenone induces apoptosis in human lung cancer cells by regulating autophagic flux. IUBMB Life. 2016;68:388–393. doi:10.1002/iub.1493
20. Deng YT, Huang HC, Lin JK. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol, Carcinog. 2010;49:141–151. doi:10.1002/mc.20583
21. Ma X, Li Z, Ma H, et al. Rotenone inhibited osteosarcoma metastasis by modulating ZO-2 expression and location via the ROS/Ca(2+)/AMPK pathway. Redox Rep. 2025;30(2493556). doi:10.1080/13510002.2025.2493556
22. Li X, Kong L, Yang Q, et al. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth. J Biol Chem. 2020;295:3576–3589. doi:10.1074/jbc.RA119.011396
23. Andronikou C, Burdova K, Dibitetto D, et al. PARG-deficient tumor cells have an increased dependence on EXO1/FEN1-mediated DNA repair. EMBO j. 2024;43:1015–1042. doi:10.1038/s44318-024-00043-2
24. Varshney GK, Burgess SM. DNA-guided genome editing using structure-guided endonucleases. Genome Biol. 2016;17(187). doi:10.1186/s13059-016-1055-4
25. Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO j. 1997;16:3341–3348. doi:10.1093/emboj/16.11.3341
26. Qiu J, Li X, Frank G, et al. Cell cycle-dependent and DNA damage-inducible nuclear localization of FEN-1 nuclease is consistent with its dual functions in DNA replication and repair. J Biol Chem. 2001;276:4901–4908. doi:10.1074/jbc.M007825200
27. Zheng L, Jia J, Finger LD, et al. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011;39:781–794. doi:10.1093/nar/gkq884
28. Song DY, Park YJ, Kim DM. A one-pot transcriptional assay method that detects the tumor biomarker FEN1 based on its flap cleavage activity. Anal Chim Acta. 2023;1282:341928.
29. Berfelde J, Hildebrand LS, Kuhlmann L, et al. FEN1 inhibition as a potential novel targeted therapy against breast cancer and the prognostic relevance of FEN1. Int J Mol Sci. 2024;25:2024. doi:10.3390/ijms25042024
30. Peng Z, Wang S, Wen D, et al. FEN1 upregulation mediated by SUMO2 via antagonizing proteasomal degradation promotes hepatocellular carcinoma stemness. Transl Oncol. 2024;44(101916):101916. doi:10.1016/j.tranon.2024.101916
31. Zhang X, Dan S, Pan X, et al. Identification of VPS34-PI(3)P-FEN1-mediated DNA repair pathway as a potential drug target to overcome chemoresistance. Biochem Biophys Res Commun. 2023;674:27–35. doi:10.1016/j.bbrc.2023.06.079
32. Zhang K, Keymeulen S, Nelson R, et al. Overexpression of flap endonuclease 1 correlates with enhanced proliferation and poor prognosis of non-small-cell lung cancer. Am J Pathol. 2018;188:242–251. doi:10.1016/j.ajpath.2017.09.011
33. Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843:182–196. doi:10.1016/j.bbamcr.2013.06.031
34. Poondla N, Chandrasekaran AP, Kim KS, et al. Deubiquitinating enzymes as cancer biomarkers: new therapeutic opportunities? BMB Rep. 2019;52:181–189. doi:10.5483/BMBRep.2019.52.3.048
35. Xue W, Men S, Liu R. Rotenone restrains the proliferation, motility and epithelial-mesenchymal transition of colon cancer cells and the tumourigenesis in nude mice via PI3K/AKT pathway. Clin Exp Pharmacol Physiol. 2020;47:1484–1494. doi:10.1111/1440-1681.13320
36. Bakhsh HT, Abu-Baih DH, Abu-Baih RH, et al. Unveiling Lobophytum sp. the neuroprotective potential of Parkinson’s disease through multifaceted mechanisms, supported by metabolomic analysis and network pharmacology. Sci Rep. 2024;14(21810). doi:10.1038/s41598-024-66781-9
37. Radad K, Al-Shraim M, Al-Emam A, et al. Rotenone: from modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019;57:317–326. doi:10.5114/fn.2019.89857
38. Khodir SA, Sweed EM, Faried MA, et al. Neuroprotective effect of maresin-1 in rotenone-induced parkinson’s disease in rats: the putative role of the JAK/STAT pathway. Neurochem Res. 2024;50(30). doi:10.1007/s11064-024-04282-x
39. Kulikova O, Troshev D, Berezhnoy D, et al. Neuroprotective efficacy of a nanomicellar complex of carnosine and lipoic acid in a rat model of rotenone-induced parkinson’s disease. Antioxidants. 2023;12:2023. doi:10.3390/antiox12122023
40. Pamies D, Block K, Lau P, et al. Rotenone exerts developmental neurotoxicity in a human brain spheroid model. Toxicol Appl Pharmacol. 2018;354:101–114. doi:10.1016/j.taap.2018.02.003
41. Wang C, Su T, Xiao L, et al. Right vagus nerve stimulation improves motor behavior by exerting neuroprotective effects in Parkinson’s disease rats. Ann Transl Med. 2022;10(1314):1314. doi:10.21037/atm-22-5366
42. Latham AS, Rocha SM, McDermott CP, et al. Neuroprotective efficacy of the glucocorticoid receptor modulator PT150 in the rotenone mouse model of parkinson’s disease. bioRxiv. 2024. doi:10.1101/2024.04.12.589261
43. Tat J, Heskett K, Boss GR. Acute rotenone poisoning: a scoping review. Heliyon. 2024;10:e28334. doi:10.1016/j.heliyon.2024.e28334
44. Heinz S, Freyberger A, Lawrenz B, et al. Mechanistic investigations of the mitochondrial complex i inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep. 2017;7(45465). doi:10.1038/srep45465
45. Lee M, Yoo JH, Kim I, et al. The compartment-specific manipulation of the NAD(+)/NADH ratio affects the metabolome and the function of glioblastoma. Sci Rep. 2024;14:20575.
46. Immanuel SRC, Ghanate AD, Parmar DS, et al. Integrated genetic and metabolic landscapes predict vulnerabilities of temozolomide resistant glioblastoma cells. NPJ Syst Biol Appl. 2021;7(2). doi:10.1038/s41540-020-00161-7
47. Alhmoud JF, Woolley JF, Al Moustafa AE, et al. DNA damage/repair management in cancers. Cancers. 2020;12:2020. doi:10.3390/cancers12082020
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.