")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Role of pirfenidone in the management of pulmonary fibrosis
Received 25 October 2016
Accepted for publication 15 December 2016
Published 3 April 2017 Volume 2017:13 Pages 427—437
DOI https://doi.org/10.2147/TCRM.S81141
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Keith C Meyer,1,2 Catherine A Decker3,4
1University of Wisconsin Lung Transplant and Advanced Lung Disease Program, 2Department of Medicine, Section of Allergy, Pulmonary and Critical Care Medicine, Clinical Sciences Center, University of Wisconsin School of Medicine and Public Health, 3Department of Pharmacy, 4Department of Pulmonary Medicine, University of Wisconsin Hospital and Clinics, Madison, WI, USA
Abstract: Pulmonary fibrosis is associated with a number of specific forms of interstitial lung disease (ILD) and can lead to progressive decline in lung function, poor quality of life, and, ultimately, early death. Idiopathic pulmonary fibrosis (IPF), the most common fibrotic ILD, affects up to 1 in 200 elderly individuals and has a median survival that ranges from 3 to 5 years following initial diagnosis. IPF has not been shown to respond to immunomodulatory therapies, but recent trials with novel antifibrotic agents have demonstrated lessening of lung function decline over time. Pirfenidone has been shown to significantly slow decline in forced vital capacity (FVC) over time and prolong progression-free survival, which led to its licensing by the United States Food and Drug Administration (FDA) in 2014 for the treatment of patients with IPF. However, pirfenidone has been associated with significant side effects, and patients treated with pirfenidone must be carefully monitored. We review recent and ongoing clinical research and experience with pirfenidone as a pharmacologic therapy for patients with IPF, provide a suggested approach to incorporate pirfenidone into a treatment algorithm for patients with IPF, and examine the potential of pirfenidone as a treatment for non-IPF forms of ILD accompanied by progressive pulmonary fibrosis.
Keywords: idiopathic pulmonary fibrosis, treatments, pirfenidone, interstitial lung disease
Introduction
Many forms of interstitial lung disease (ILD) lead to pulmonary fibrosis that can distort and obliterate terminal airways, alveoli, and the vascular structures that comprise gas exchange units as abnormal injury-response lung tissue remodeling occurs.1–3 Idiopathic pulmonary fibrosis (IPF) is the most frequently diagnosed type of idiopathic interstitial pneumonia (IIP) and the most commonly encountered form of progressive pulmonary fibrosis (Figure 1). Although IPF is characterized by a usual interstitial pneumonia (UIP) pattern on lung histopathologic specimens,2,4 a UIP or UIP-like pattern can be seen with other forms of ILD such as lung disease associated with connective tissue disease (CTD-ILD) or chronic fibrosing hypersensitivity pneumonitis (HP).5–7 Therefore, alternative causes of UIP must be ruled out before a confident diagnosis of IPF is established.4 Non-IPF forms of fibrotic ILD (Table 1) can also progress to end-stage disease,5,7 and some of these entities may be difficult to discern from IPF, despite a comprehensive clinical evaluation, high-resolution computed tomographic (HRCT) imaging of the thorax, invasive surgical procedures to obtain diagnostic lung tissue, and a multidisciplinary team evaluation.8
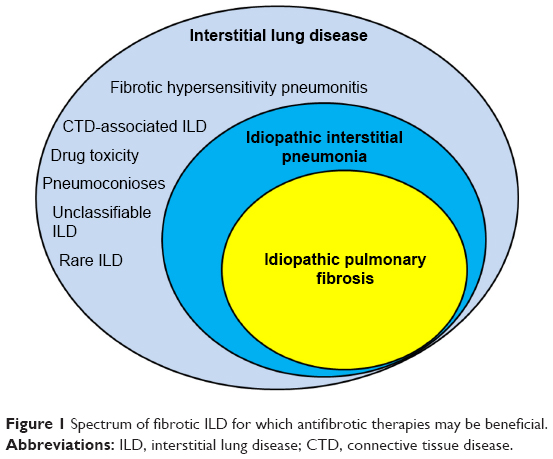 | Figure 1 Spectrum of fibrotic ILD for which antifibrotic therapies may be beneficial. |
 | Table 1 Types of interstitial lung disease (ILD) usually or potentially associated with progressive fibrosis |
IPF accounts for approximately one-fifth of all cases of ILD and has a median survival that is estimated to range from 3 to 5 years with an annual death rate of >30,000 per year in the United States.9 Historically, IPF was considered a disease of uncontrolled inflammation. As such, systemic glucocorticoids and immunosuppressive therapies were thought to be the mainstay of treatment.10 More current research, however, has identified alveolar epithelial cell (AEC) injury with damage to alveolar basement membranes as the insult that leads to deposition of abnormal matrix tissue by myofibroblasts as aberrant wound healing responses remodel and distort lung architecture.11,12 Although immune responses are also involved, currently available anti-inflammatory, immunomodulatory, or cytotoxic pharmacologic therapies are generally ineffective and do not have a significant impact on disease progression. Furthermore, treatment with azathioprine has recently been associated with significantly increased risk of mortality and other complications.13 IPF is highly associated with advanced age, and numerous genetic variants, such as age-associated changes in telomere function,14 MUC5B gene polymorphisms,15 and epigenetic factors that affect gene function,16 have now been linked to IPF risk and pathogenesis. Advancing age is also associated with some forms of CTD-ILD, such as rheumatoid arthritis, and confounding environmental factors such as tobacco use, exposures to metal or dust particulates, silica, and farm environments have been linked to an increased risk of developing IPF and some other forms of pulmonary fibrosis.
Prior to 2014, there were no US Food and Drug Administration (FDA)-approved medications available for clinicians to use to manage IPF. However, two antifibrotic agents, pirfenidone and nintedanib, were approved for the treatment of patients with IPF in October 2014 on the basis of large clinical trials that supported efficacy.17,18 These newly approved drugs, which significantly lessened the decline in forced vital capacity (FVC) in Phase III clinical trials that led to FDA approval, have changed the clinical management of patients with IPF and are now perceived as standard-of-care medications. This manuscript will focus on the current use of pirfenidone to treat IPF and will also discuss the potential for using pirfenidone to treat other progressive forms of fibrotic ILD that do not adequately respond to anti-inflammatory, immunomodulatory pharmacologic therapies.
Natural history and diagnosis of IPF
A considerable variability in survival rates has been found by various investigators.19,20 Earlier studies have estimated survival at 2–3 years from the time of diagnosis, but more recent investigations and examination of data from large clinical trials suggest that median survival is in the range of 3–5 years following diagnosis. Some patients can progress rapidly from the time of diagnosis, some can have relative stability over a prolonged period of time, some can have a substantial period of disease stability followed by an accelerated disease course, and some patients experience an unpredictable acute exacerbation of IPF (AEIPF) that usually leads rapidly to death.19,21 Interestingly, a substantial number of patients can experience long-term survival without undergoing lung transplantation,22 and epidemiologic studies performed with similar methodologies have found considerable geographic variability, suggesting that ethnic background or environmental exposures may affect disease progression.20 Although respiratory failure is the usual cause of death, a substantial number of patients succumb to other causes that include complications of coronary artery disease, pulmonary embolism, or lung cancer. Additionally, certain clinical phenotypes, such as those patients with significant pulmonary hypertension (PH) or those with combined pulmonary fibrosis and emphysema (CPFE), may have worse survival than patients who lack significant emphysema or PH.23 Finally, numerous comorbidities that frequently affect patients with IPF, such as gastroesophageal reflux and sleep-disordered breathing, may affect the clinical course of the disease.24,25
A number of clinical, radiographic, physiologic, histopathologic, and biomarker variables have been devised and shown to correlate with survival,21 but no single clinical variable accurately predicts prognosis or disease behavior by itself. A recently devised mortality risk prediction model (gender–age–physiology [GAP]) that is based upon simple baseline variables (age, gender, FVC, and diffusion capacity for carbon monoxide [DLCO]) has been validated for patients with IPF26 and could be used in the clinical setting to inform prognosis, facilitate life planning, and help guide management decisions such as timing of lung transplantation. Additionally, when applied to chronic ILD other than IPF, the GAP model (ILD-GAP) also performed well,27 and the addition of a 24-week change in FVC and history of a respiratory hospitalization (longitudinal GAP model) appears to improve the estimation of risk for patients with IPF.28
Making a correct diagnosis of IPF by carefully excluding non-IPF forms of ILD29,30 is a critical step to ensure that appropriate principles of disease management will be followed, and the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association consensus statement provides an algorithm for evaluating patients with a potential diagnosis of IPF.4 Most patients present with new onset of dyspnea on exertion, cough, and/or fatigue, but asymptomatic or relatively asymptomatic patients with earlier stages of IPF may be identified when interstitial abnormalities are an incidental finding on thoracic imaging. A careful and comprehensive interview helps to exclude potential alternative causes of pulmonary fibrosis that can have a UIP pattern, such as the presence of a CTD, chronic fibrotic HP, occupational/environmental exposures, or drug-induced fibrosis.30 Chest auscultation usually reveals basilar “Velcro-like” crackles on lung auscultation, and digital clubbing may be present. Pulmonary function testing usually shows a restrictive pattern of lung dysfunction with reduced FVC, total lung capacity (TLC), and DLCO, although patients with early disease may have values for FVC and TLC that are still in the range of normal predicted values, and patients with CPFE may also lack a restrictive pattern due to the opposing forces of emphysematous hyperinflation and restrictive fibrosis.
Arriving at a confident diagnosis of IPF includes the exclusion of non-IPF forms of ILD (eg, chronic HP, non-IPF forms of IIP, CTD-ILD, or drug-induced pulmonary fibrosis) plus having either a definite UIP pattern on thoracic HRCT scanning or a combination of specific HRCT patterns (possible/probable UIP) combined with a lung biopsy that has adequately sampled lung tissue and shows a UIP histopathologic pattern.4 A multidisciplinary discussion (clinicians, radiologists, and pathologists with adequate experience with ILD and IPF) provides an optimal opportunity to reach a consensus diagnosis of IPF versus other forms of fibrosing ILD.31,32
Pirfenidone in clinical trials
Although mechanisms involved in the development of IPF are not fully understood, a complex and interconnected process occurs, which includes damage to alveolar epithelium and disruption of alveolar basement membranes, release of proinflammatory and profibrotic cytokines, myofibroblast activation and proliferation, and deposition of extracellular matrix proteins by mesenchymal cells that leads to progressive destruction of normal lung architecture and irreversible fibrosis with loss of functional lung units as aberrant lung remodeling occurs.11,12 Transforming growth factor-beta (TGF-β) and other profibrotic growth factors have been implicated as playing a key role,33,34 and biomechanical characteristics of the matrix environment that lead to increased lung stiffness may also promote fibroblast responses and progression of fibrosis.35 Pirfenidone is an orally bioavailable pyridone derivative that exhibits both anti-inflammatory and anti-fibrotic activities. It has been shown to ameliorate bleomycin-induced lung injury in hamsters,36,37 and pirfenidone suppresses TGF-β-induced myofibroblast differentiation and fibrogenic activity of human lung fibroblasts.38,39
Despite the lack of efficacy for many drugs that showed promise for the treatment of IPF, primary end points were met for both pirfenidone17,40 and nintedanib41 in Phase III, placebo-controlled, randomized clinical trials (RCTs). These results led to approval by the FDA in 2014 for both drugs to be prescribed for patients with IPF in the US (Table 2). Pirfenidone was also shown to be efficacious in the second Japanese Phase III RCT,42 which demonstrated a significant reduction in the primary end point of FVC decline and in a secondary end point of progression-free survival. Although the multinational CAPACITY Phase III RCTs40 that were completed with results published after the Japanese study showed a significant reduction in the primary end point of FVC decline in one (CAPACITY 004) but not the other (CAPACITY 006), pirfenidone was approved for treatment in many countries outside of the US (eg, Japan, Canada, India, and various European countries) on the basis of results from the Phase III Japanese and CAPACITY 004 studies. However, because the US FDA required an additional RCT that showed efficacy before pirfenidone could be approved in the US, the ASCEND Phase III RCT was performed (2,403 mg/d of pirfenidone vs placebo) and enrolled 555 subjects.17 The end point of the ASCEND RCT was met with pirfenidone, demonstrating a relative risk reduction of 47.9% for FVC decline ≥10%; additionally, two secondary end points of a significant attenuation of decline in 6-minute walk test (6-MWT) distance and improved progression-free survival were also achieved for the pirfenidone-treated cohort. Finally, analysis of pooled data from both the CAPACITY and the ASCEND RCTs showed a significant reduction in death due to any cause at 52 weeks as well as death from IPF progression for the treatment cohort.40,43
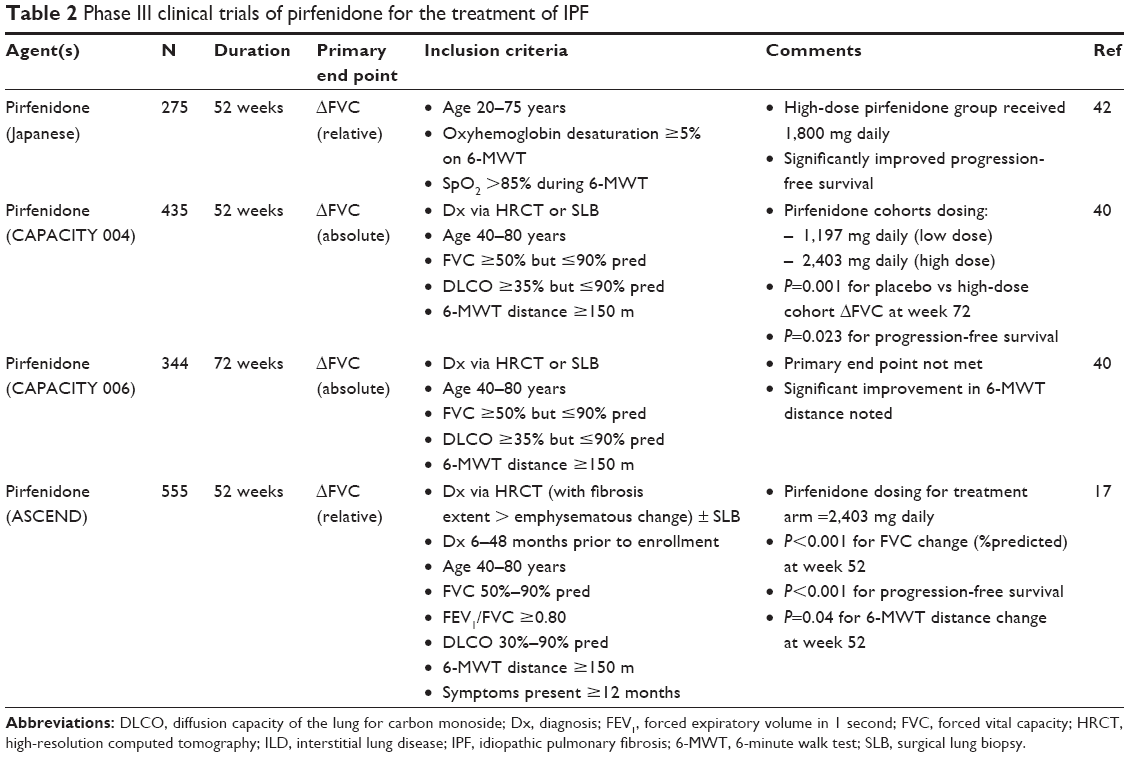 | Table 2 Phase III clinical trials of pirfenidone for the treatment of IPF |
A more recent analysis of pooled CAPACITY and ASCEND RCT data43 also showed that various confounders (US vs non-US patients, age, gender, race, various measures of degree of lung function impairment, use of supplemental oxygen, smoking status, or time since diagnosis) did not appear to alter treatment effects. Because pirfenidone had already been approved for prescription in many countries outside of the US prior to FDA approval, “real world” experiences with pirfenidone in other countries have appeared in the literature and report that the drug is relatively well-tolerated and appears to benefit a substantial number of patients.44,45 Recent publications also support safety and efficacy of pirfenidone for the treatment of IPF. An open-label extension study (RECAP) of patients enrolled in the CAPACITY RCTs showed similar effects for patients who had been in the placebo arm but were allowed to be newly treated with pirfenidone, and the safety profile was similar to that for the pirfenidone-treated cohort.46 Additionally, pooled data from five RCTs (CAPACITY 002, 004, 006, ASCEND, and RECAP) with 1,299 patients analyzed for safety outcomes and prospectively followed for up to 9.9 years support previous observations that long-term treatment with pirfenidone is well tolerated by the majority of patients and generally safe.47 Furthermore, although longitudinal data from patients in the CAPACITY and ASCEND RCTs showed considerable intrasubject variability in FVC measurements, sustained treatment beyond the initial 6-month treatment period appeared to continue to reduce risk of both FVC decline and death for pirfenidone-treated subjects versus patients receiving placebo.48
Although N-acetylcysteine (NAC) in the NAC-only arm of the PANTHER trial did not appear to have any efficacy in preventing FVC decline in IPF,49 whether NAC in combination with pirfenidone may enhance tolerability of pirfenidone or have an additive effect on delaying FVC decline was assessed by a Phase II RCT (PANORAMA) that was recently published.50 Despite enrolling 123 patients in this multicenter, multinational RCT, an exploratory analysis did not support any benefit of combination therapy (NAC plus pirfenidone) on FVC decline. Indeed, a possible harmful effect was suggested with a significantly greater decline in FVC reported for those receiving combination therapy versus those receiving pirfenidone monotherapy. This observation should give pause to clinicians who may consider giving NAC as an adjunctive therapy while treating patients with pirfenidone, as such therapy may not only lack benefit but could also potentially cause harm.
Clinical use of pirfenidone to treat IPF
Although pirfenidone was relatively well tolerated in the Phase III RCTs and follow-up safety data show it to be relatively safe, significant side effects can occur (Table 3). Pirfenidone is metabolized primarily in the liver via the cytochrome system (CYP1A2 enzyme), and inhibitors that can reduce pirfenidone metabolism (eg, amiodarone, ciprofloxacin, and fluvoxamine) should be avoided or, if needed, reduced dosage of pirfenidone should be considered.25,51,52 CYP1A2 inducers (eg, omeprazole and tobacco smoking) may reduce bioavailability of pirfenidone and should also be avoided. Significant increases in liver function test values may occur, and monitoring should occur monthly for the first 6 months of therapy and every 3 months thereafter to detect hepatotoxicity should it occur. If pirfenidone is administered to patients with mild to moderate hepatic dysfunction, liver function should be carefully monitored. Pirfenidone should not be prescribed for patients with severe hepatic dysfunction or patients with advanced renal dysfunction (glomerular filtration rate <30 mL/min). Common side effects of pirfenidone consist of gastrointestinal intolerance (eg, nausea, emesis, abdominal discomfort, dyspepsia, and diarrhea) or skin reactions (photosensitivity and rash). Avoiding direct exposure to sunlight, using sunscreen, and wearing protective clothing can reduce the risk of photosensitivity skin reactions. Gastrointestinal side effects can be reduced by gradually escalating the dose of pirfenidone when initially starting therapy, taking the drug with meals, and using a proton-pump inhibitor. Although patients with advanced disease with severely reduced FVC and gas transfer values were excluded from enrollment in Phase III RCTs, pirfenidone may be offered to any patient regardless of disease severity, but patients should adequately understand the risks and benefits of this therapy.
 | Table 3 Pirfenidone pharmacologic characteristics, precautions, and monitoring |
Prior to initiating treatment with pirfenidone, a thorough dialog with the patient, which includes an educational overview of the benefits, risks, and limitations of therapy, is recommended (Figure 2). Discussions should include educating patients concerning the nature of their disease so that patients can more fully understand the rationale for taking pirfenidone to delay progression of IPF. The decision whether to begin therapy should address logistical information that includes the required prior authorization approval process (for patients in the US), identifying out-of-pocket patient responsibility after prescription benefit review of coverage, review of patient assistance programs available to offset significant out-of-pocket expenses, specialty pharmacy dispensing processes and procedures, registration with an identified specialty pharmacy, coordination of regular communication regarding monthly refills, and a plan for patient–provider follow-up. Clinical information should also be adequately discussed with the patient, including mechanism of action, review of common and serious adverse drug reactions (ADRs), the process for ADR reporting, a planned schedule for necessary baseline and on-going laboratory work for monitoring purposes, a review of strategies to maximize medication adherence, and the plan for periodic follow-up visits to the clinic. These discussions can require a significant time commitment, as medication-related logistical and clinical information is fairly complex and easily misunderstood, particularly in elderly patients with IPF.
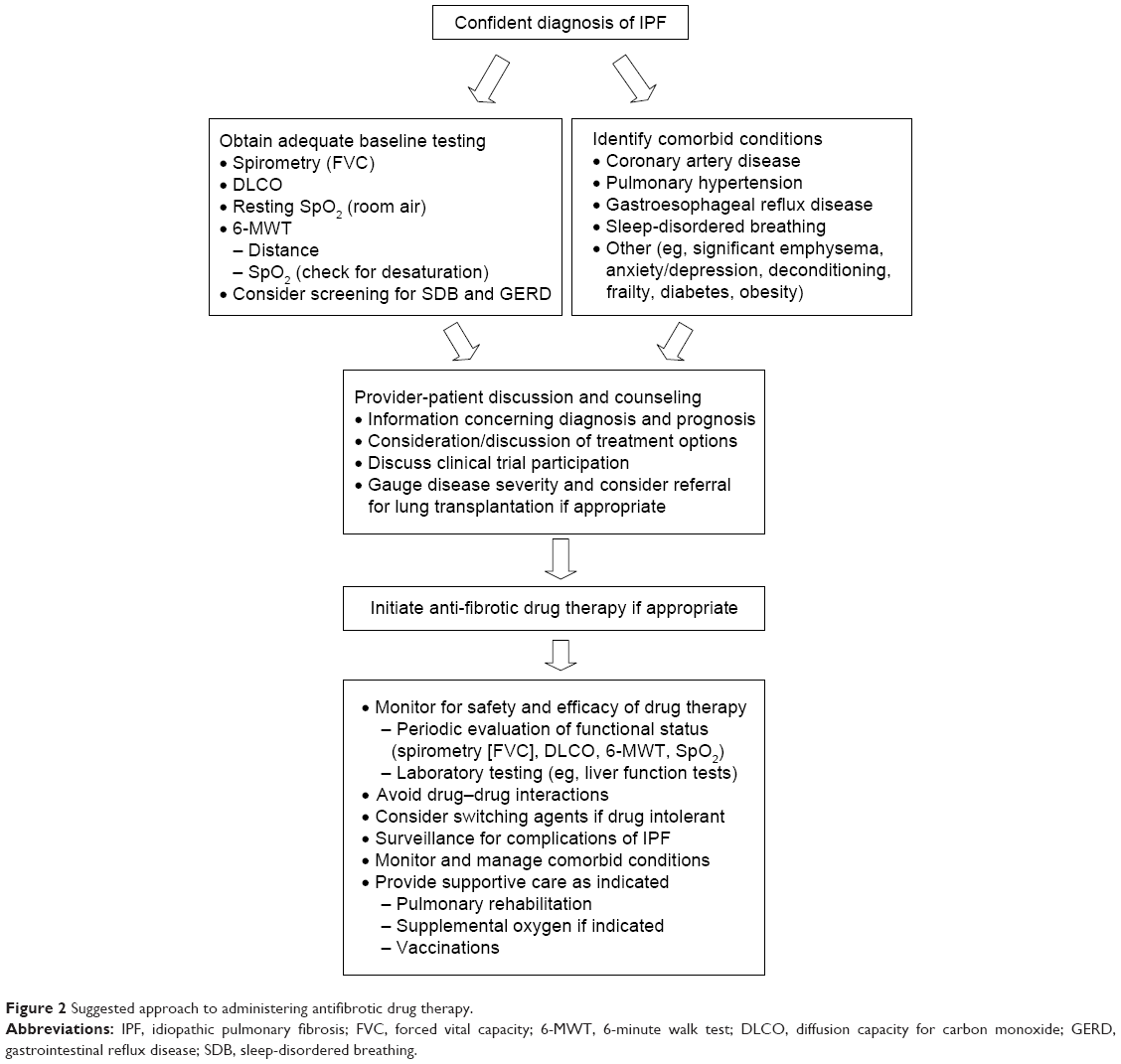 | Figure 2 Suggested approach to administering antifibrotic drug therapy. |
Each pirfenidone capsule contains 267 mg of active drug, and the dose should be gradually increased to a target of 801 mg three times daily over a period of 3 weeks. During the first week of therapy, patients are instructed to take one capsule of pirfenidone orally three times daily, then two capsules (534 mg) three times daily during week 2, and then escalate the dose to 801 mg (three capsules) three times daily during week 3. This dose escalation represents a significant adherence challenge if side effects such as gastrointestinal upset are experienced, and the maintenance dose introduces a significant pill burden. Despite a decrease in maximal and area under the curve concentrations (Cmax and AUC) when pirfenidone is taken with food, it remains advisable to take each dose of pirfenidone with a full meal to decrease the likelihood of dizziness, nausea, or gastrointestinal upset. Additionally, avoidance of grapefruit and grapefruit juice has been recommended,53 and weight loss and anorexia have been reported. Because skin rashes, eruptions, irritations, and photosensitivity are well documented, patients should be counseled to wear sunscreen with SPF of at least ≥50 when exposed to sunlight. Wearing a wide brimmed hat and using specially designed sun protection clothing can also be considered. Increased fatigue and dizziness have also been associated with pirfenidone use; these side effects range from mild to severe and may impair physical or mental abilities and have been linked to therapy discontinuation in some patients. Indeed, fatigue has been reported as a significant adverse effect in both RCTs54 and in “real-life” follow-up assessments55,56 of patients receiving pirfenidone therapy and may lead to discontinuation of pirfenidone. Additionally, tobacco smoking can interfere with the bioavailability of pirfenidone and may abrogate its antifibrotic effects; patients should be counseled to avoid tobacco usage, which could negate any beneficial effect of pirfenidone.
When deciding to prescribe antifibrotic therapy with either pirfenidone or nintedanib, currently available data do not support superiority of one agent over the other. However, analysis of pooled data for pirfenidone supported a reduction in all-cause and IPF-related mortality in addition to its effects on FVC.40 The evidence-based update to the 2011 clinical practice guideline for the treatment of IPF57 provided a conditional recommendation for use of either pirfenidone or nintedanib to treat patients with IPF (Table 4). The recommendation for both drugs was termed conditional because the available evidence for treatment with either agent was based upon limited evidence (potential impact on disease progression and/or mortality, cost of treatment, risk of possible adverse events). This was perceived by the expert panel as not having enough certainty to merit assigning a strong recommendation for clinical use, although the majority of patients are likely to choose such therapy when adequately informed of the balance of desirable and undesirable consequences associated with these drugs. Because IPF is highly prevalent in elderly patients, many patients will have significant comorbid conditions.25 These should be identified, and appropriate management of such comorbidities may improve survival and quality of life above and beyond therapeutic intervention with an antifibrotic agent. Finally, patients should be made aware of the existence of clinical trials and the need for RCT participation to identify new and potentially better agents to treat IPF; new trials generally allow patients to enroll when on stable doses of pirfenidone or nintedanib.
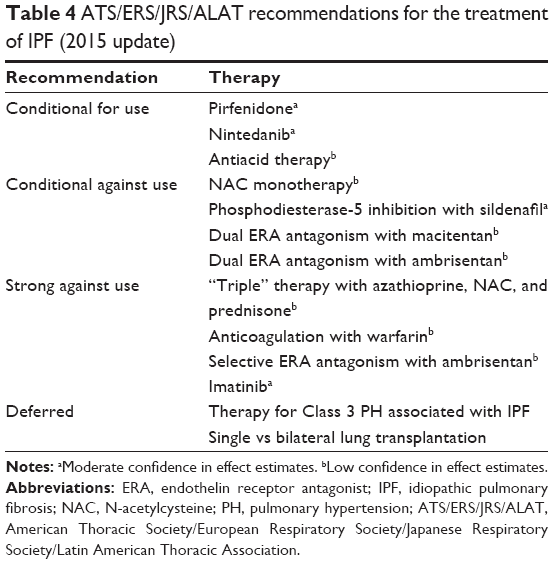 | Table 4 ATS/ERS/JRS/ALAT recommendations for the treatment of IPF (2015 update) |
Whether the benefit of very early treatment with pirfenidone (eg, when UIP is detected as an incidental finding in an asymptomatic patient) outweighs potential risks is not known. Also, whether patients with advanced disease, such as patients with FVC <50% of predicted normal value, a DLCO <30% of predicted, or those requiring use of supplemental oxygen, are likely to experience significant benefit from pirfenidone therapy is also unknown, and patients should not forego lung transplant evaluation and potential listing if they have progressive disease and are potential candidates who wish to be a lung transplant recipient if they meet criteria for listing.58 Whether progressive respiratory impairment that continues to worsen while on therapy (eg, ≥10% decline in FVC) should trigger a switch to an alternative agent (nintedanib) or whether dual therapy (simultaneous administration of both pirfenidone and nintedanib) could be considered represent two additional unanswered questions. The mechanism of action of nintedanib differs significantly from that of pirfenidone (disruption of TGF-β signaling) in that nintedanib inhibits multiple intracellular kinases that are involved in signaling and stimulation of matrix deposition via a number of growth factor (vascular endothelial growth factor, platelet-derived growth factor, and fibroblast growth factor) receptors,41 and combination therapy may have an additive or synergistic effect but needs to be evaluated in an adequately powered and rigorously performed RCT.
An AEIPF event can occur at any time during the course of IPF, and an AEIPF episode is estimated to occur in 5%–10% of patients per year.59 Treatment of such events consists of providing supportive measures (supplemental oxygen, relief of dyspnea, and psychological support), using broad-spectrum anti-infectives as clinically indicated, and judiciously administering corticosteroids. Although the use of immunosuppressive medications when such an event occurs is not considered appropriate, established treatment with an antifibrotic drug can be continued.60
The potential role of pirfenidone in treating other fibrotic disorders
Many other forms of fibrosing ILD, such as chronic HP, CTD-ILD, non-IPF IIP, or drug-induced PF, can progress despite immunomodulatory/anti-inflammatory therapies. Although these patients are often younger and may have less comorbidities than older patients with IPF and be more eligible for possible lung transplantation, antifibrotic agents may have a significant impact on disease progression. RCTs that are generally early phase have recently been initiated for some forms of CTD-ILD (scleroderma and rheumatoid arthritis and HP, Table 5), and the possibility of synergy when pirfenidone is combined with nintedanib for the treatment of IPF is also being explored. The open-label Phase II study of pirfenidone in patients with scleroderma-associated ILD (LOTUSS) showed an acceptable tolerability and safety profile, even when coadministered with mycophenolate mofetil.61 Because some of these forms of PF may respond to immunomodulatory therapies, an approach that combines traditional therapies with an antifibrotic agent may prove to have enhanced potential to have a significant impact on disease progression. Pirfenidone may also have an significant impact on disorders other than fibrotic ILD, such as progressive airway or parenchymal fibrosis that characterizes chronic lung allograft dysfunction following lung transplantation or obliterative bronchiolitis complicating hematopoietic stem cell transplantation.62
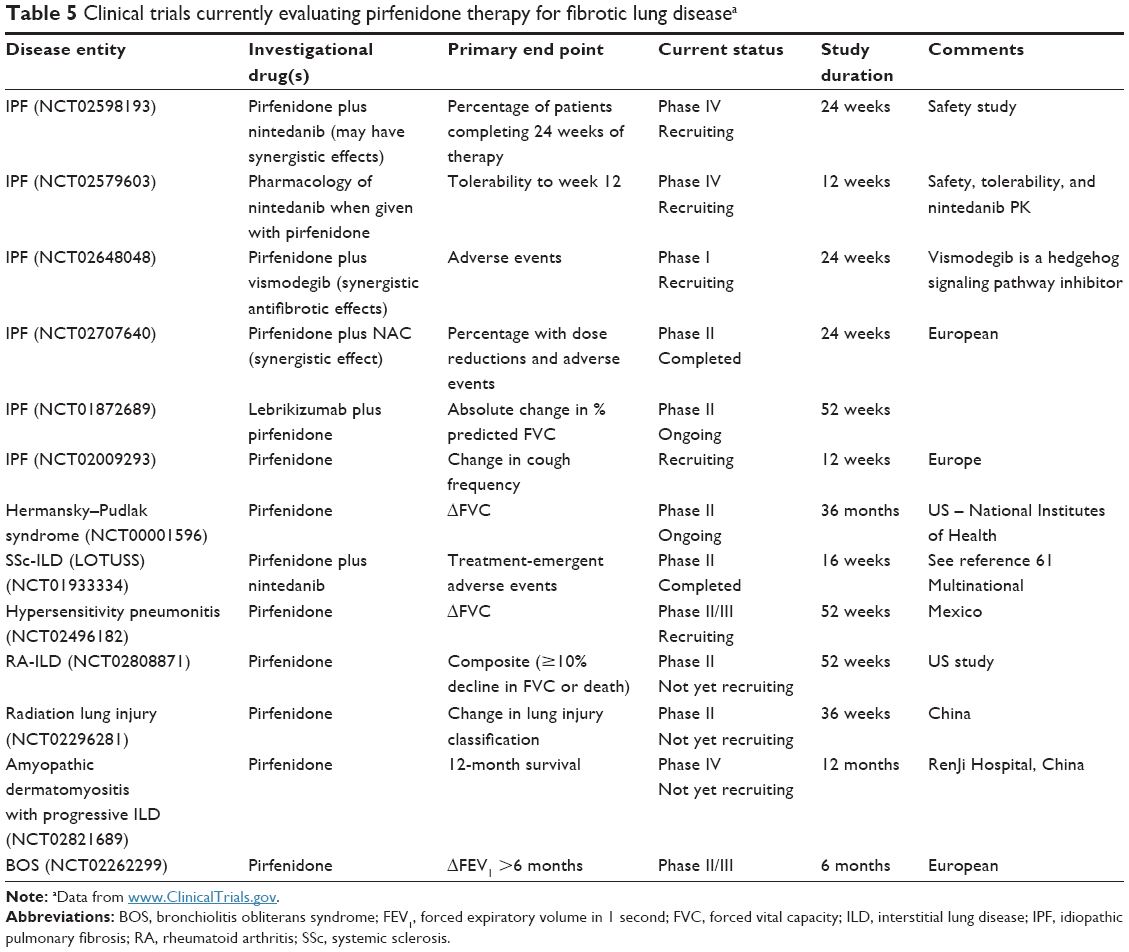 | Table 5 Clinical trials currently evaluating pirfenidone therapy for fibrotic lung diseasea |
Precision medicine has the goal of identifying and harnessing true biological explanations for drug efficacy in individual patients,63 and IPF and other forms of PF are clearly diseases with considerable heterogeneity in patient characteristics, disease course, and responses to therapeutic interventions. Precision medicine seeks to maximize therapeutic efficacy by exploiting knowledge of genetic, molecular, environmental, and lifestyle differences to identify the best preventive and treatment approaches for individual patients. Ideally, one could use specific genetic characteristics or molecular biomarkers to identify patients with IPF for whom a specific pharmacologic intervention or combination therapies represent the best choice to arrest the disease process and minimize potential harms, but considerable research must be done before this can become a reality for a heterogeneous disease such as IPF.64 Clearly, not all patients with IPF show a response to treatment with pirfenidone or nintedanib, and the number of patients needed to treat with pirfenidone to have a significant impact on FVC decline for one patient appears to be approximately eight, and combined data suggest that number needed to treat is in the range of 30 for the outcome of enhanced survival. However, a recent report of a “real-life” experience with pirfenidone suggested that individuals with more rapidly declining lung function prior to beginning pirfenidone treatment are more likely to experience benefit (attenuation of FVC decline) than patients with more stable lung function prior to treatment onset.65 Advances in our understanding of the genetic underpinnings of IPF and other forms of PF and the role of epigenetic factors in shaping the differences that determine disease heterogeneity will facilitate advances in precision medicine that may allow identification of patients who will have a meaningful response to pirfenidone or other antifibrotic therapies versus those patients who are unlikely to benefit and may be harmed.
Summary and conclusion
Pirfenidone has been shown to have a significant effect on FVC decline and progression-free survival in Phase III clinical trials that enrolled patients with IPF with mild to moderately severe respiratory impairment. Treatment with antifibrotic agents has now entered the clinical arena, and treatment of IPF patients with pirfenidone is being perceived as a standard-of-care approach to disease management. Although many non-IPF forms of ILD can also lead to loss of lung function due to progressive fibrosis, there has been little clinical research to establish a potential role for antifibrotic agents in non-IPF ILD. Additionally, diagnosis of IPF and differentiation from other forms of fibrotic ILD can be difficult due to similar radiological and histopathologic characteristics. Nonetheless, antifibrotic agents such as pirfenidone may also have a meaningful impact on other forms of pulmonary fibrosis, and pirfenidone is reasonably well tolerated by a majority of patients with IPF. However, monitoring for adverse effects including hepatic dysfunction and potential drug–drug interactions is required, and dose manipulation and/or lifestyle changes can allow patients to achieve tolerance of side effects such as gastrointestinal upset. Clinical trials are needed to determine if pirfenidone can benefit patients with other forms of fibrosing ILD such as chronic HP or CTD-associated ILD.
Acknowledgment
This study was supported in part by the George and Julie Mosher Pulmonary Research Fund.
Disclosure
Dr Meyer has been an investigator in clinical trials sponsored by Boehringer-Ingelheim, Bristol Myers Squibb, Fibrogen, Genentech, Gilead, InterMune, Promedior, and Roche. All authors do not report any other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Leslie KO. My approach to interstitial lung disease using clinical, radiological and histopathological patterns. J Clin Pathol. 2009;62(5):387–401. | ||
Tabaj GC, Fernandez CF, Sabbagh E, Leslie KO. Histopathology of the idiopathic interstitial pneumonias (IIP): a review. Respirology. 2015;20(6):873–883. | ||
American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304. | ||
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Katzenstein AL, Mukhopadhyay S, Myers JL. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol. 2008;39(9):1275–1294. | ||
Swigris JJ, Brown KK, Flaherty KR. The idiopathic interstitial pneumonias and connective tissue disease-associated interstitial lung disease. Curr Rheum Rev. 2010;6:91–98. | ||
Larsen BT, Smith ML, Elicker BM, et al. Diagnostic approach to advanced fibrotic interstitial lung disease: bringing together clinical, radiologic, and histologic clues. Arch Pathol Lab Med. Epub 2016 Sep 15. | ||
Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4(7):557–565. | ||
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2(7):566–572. | ||
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;11(2 pt 1):646–664. | ||
Sandbo N. Mechanisms of fibrosis in IPF. In: Meyer KC, Nathan SD, editors. Idiopathic Pulmonary Fibrosis. New York, NY: Humana Press, Springer; 2014:161–205. | ||
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. | ||
Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710–1720. | ||
Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. | ||
Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med. 2015;21(5):454–462. | ||
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. | ||
Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Tourin O, Swigris JJ, Olson AL. Idiopathic pulmonary fibrosis: the epidemiology and natural history of disease. In: Meyer KC, Nathan SD, editors. Idiopathic Pulmonary Fibrosis: A Comprehensive Clinical Guide. New York, NY: Humana Press, Springer; 2014:9–34. | ||
Caminati A, Madotto F, Cesana G, Conti S, Harari S. Epidemiological studies in idiopathic pulmonary fibrosis: pitfalls in methodologies and data interpretation. Eur Respir Rev. 2015;24(137):436–444. | ||
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. | ||
Nathan SD, Shlobin OA, Weir N, et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest. 2011;140(1):221–229. | ||
Nathan SD. Idiopathic pulmonary fibrosis phenotypes. In: Meyer KC, Nathan SD, editors. Idiopathic Pulmonary Fibrosis: A Comprehensive Clinical Guide. New York, NY: Humana Press, Springer; 2014:221–238. | ||
King C, Nathan SD. Identification and treatment of comorbidities in idiopathic pulmonary fibrosis and other fibrotic lung diseases. Curr Opin Pulm Med. 2013;19:466–473. | ||
Meyer KC, Danoff SK, Lancaster LH, Nathan SD. Management of idiopathic pulmonary fibrosis in the elderly patient: addressing key questions. Chest. 2015;148(1):242–252. | ||
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. | ||
Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–728. | ||
Ley B, Bradford WZ, Weycker D, Vittinghoff E, du Bois RM, Collard HR. Unified baseline and longitudinal mortality prediction in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1374–1381. | ||
Wadhwa A, Flaherty KR. The keys to making a confident diagnosis of IPF. In: Meyer KC, Nathan SD, editors. Idiopathic Pulmonary Fibrosis: A Comprehensive Clinical Guide. New York, NY: Humana Press, Springer; 2014:77–101. | ||
Meyer KC. Diagnosis and management of interstitial lung disease. Transl Respir Med. 2014;2:4. eCollection 2014. | ||
Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170:904–910. | ||
Meyer KC. Multidisciplinary discussions and interstitial lung disease diagnosis: how useful is a meeting of the minds? Lancet Respir Med. 2016;4:529–531. | ||
Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L525–L534. | ||
Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. 2012;9(3):111–116. | ||
Froese AR, Shimbori C, Bellaye PS, et al. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:84–96. | ||
Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125(6):779–785. | ||
Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291(1):367–373. | ||
Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008;82(3–4):210–217. | ||
Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. | ||
Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. | ||
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. | ||
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–253. | ||
Ogura T, Azuma A, Inoue Y, et al. All-case post-marketing surveillance of 1371 patients treated with pirfenidone for idiopathic pulmonary fibrosis. Respir Investig. 2015;53(5):232–241. | ||
Harari S, Caminati A. Idiopathic pulmonary fibrosis: from clinical trials to real-life experiences. Eur Respir Rev. 2015;24(137):420–427. | ||
Costabel U, Albera C, Bradford WZ, et al. Analysis of lung function and survival in RECAP: an open-label extension study of pirfenidone in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(3):198–205. | ||
Lancaster L, Albera C, Bradford WZ, et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir Res. 2016;3(1):e000105. eCollection 2016. | ||
Nathan SD, Albera C, Bradford WZ, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–435. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez FJ, de Andrade JA, Anstrom KJ, et al. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2093–2101. | ||
Behr J, Bendstrup E, Crestani B, et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med. 2016;4(6):445–453. | ||
King CS, Nathan SD. Practical considerations in the pharmacologic treatment of idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2015;21(5):479–489. | ||
Spagnolo P, Maher TM, Richeldi L. Idiopathic pulmonary fibrosis: recent advances on pharmacological therapy. Pharmacol Ther. 2015;152:18–27. | ||
Esbriet (pirfenidone) [prescribing information]. South San Francisco, CA: Genentech USA; 2015. | ||
Jiang C, Huang H, Liu J, Wang Y, Lu Z, Xu Z. Adverse events of pirfenidone for the treatment of pulmonary fibrosis: a meta-analysis of randomized controlled trials. PLoS One. 2012;7(10):e47024. | ||
Oltmanns U, Kahn N, Palmowski K, et al. Pirfenidone in idiopathic pulmonary fibrosis: real-life experience from a German tertiary referral center for interstitial lung diseases. Respiration. 2014;88(3):199–207. | ||
Milger K, Kneidinger N, Neurohr C, Reichenberger F, Behr J. Switching to nintedanib after discontinuation of pirfenidone due to adverse events in IPF. Eur Respir J. 2015;46(4):1217–1221. | ||
Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An Official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19. | ||
Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014 – an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015;34(1):1–15. | ||
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbations of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2007;2016(194):265–275. | ||
Maher TM, Whyte MK, Hoyles RK, et al. Development of a consensus statement for the definition, diagnosis, and treatment of acute exacerbations of idiopathic pulmonary fibrosis using the Delphi technique. Adv Ther. 2015;32(10):929–943. | ||
Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. 2016;43(9):1672–1679. | ||
Meyer KC. Diagnosis and management of bronchiolitis obliterans syndrome following lung or hematopoietic cell transplantation. Expert Rev Respir Med. 2016;10:599–602. | ||
Hey SP, Kesselheim AS. Countering imprecision in precision medicine. Science. 2016;353:448–449. | ||
Brownell R, Kaminski N, Woodruff PG, et al. Precision medicine: the new frontier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193(11):1213–1218. | ||
Loeh B, Drakopanagiotakis F, Bandelli GP, et al. Intraindividual response to treatment with pirfenidone in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;191(1):110–113. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.