")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Role of lipegfilgrastim in the management of chemotherapy-induced neutropenia
Authors Hoggatt J, Tate T, Pelus L
Received 13 January 2015
Accepted for publication 5 March 2015
Published 1 April 2015 Volume 2015:10(1) Pages 2647—2652
DOI https://doi.org/10.2147/IJN.S55796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Jonathan Hoggatt,1 Tiffany A Tate,1 Louis M Pelus2
1Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, MA, USA; 2Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, USA
Abstract: Chemotherapy, irradiation, and other agents are widely used to target the process of cell division in neoplastic cells. However, while these therapies are effective against most cancers, the high proliferative rate of the cells of the hematopoietic system that produce billions of blood cells needed daily throughout life is extremely sensitive to these agents, resulting in loss of blood cell populations, which can be life threatening. Neutropenia is the most serious hematologic toxicity of chemotherapy, which can result in patient morbidity and mortality due to opportunistic infection and often is the limiting factor in dose escalation or duration of chemotherapeutic administration. Neutropenic patients often require hospitalization and incur substantial medical costs associated with anti-infective therapy. Treatment of iatrogenic and congenic neutropenia was changed in the early 1990s with the introduction of filgrastim (Neupogen®) and pegfilgrastim (Neulasta®). With the expiration of patent lives of both of these drugs, biosimilars have begun to emerge. In this review, we will summarize the chemical characteristics, pharmacokinetics, safety and efficacy of lipegfilgrastim (Lonquex®), the first long-acting biosimilar filgrastim to receive regulatory approval and enter the marketplace.
Keywords: granulocyte-colony stimulating factor, biosimilar, filgrastim, neutrophil, XM22, cancer
Corrigendum for this paper has been published.
Introduction
The use of chemotherapeutics to treat malignancies is rooted in the use of mustard gas during the World Wars. Sinking of the USS John Harvey carrying a cargo of mustard bombs in the port city of Bari, Italy, in 1943 released the toxic payload and exposed survivors and residents to nitrogen mustard gas. Within 3–4 days, many of those exposed had severe drops in their white blood cell counts.1 Hypothesizing that the actions of the mustard gas on white blood cells could potentially be used to treat hematologic diseases, several clinicians studied the effectiveness of intravenously administered nitrogen mustards, tris(β-chloroethyl)amine and methyl-bis(β-chloroethyl)amine, and published landmark findings in 1946 that nitrogen mustards were effective in treating various forms of leukemia and lymphoma.2 Their conclusion heralded in the era of cancer chemotherapeutic discovery:
Chemicals discovered to be therapeutically active in neoplastic disease deserve close study […]. From this point of view the heuristic aspects of the actions of the β-chloroethylamines here reported may eventually prove of greater importance than the chemical results obtained to date.2
Cancer is traditionally thought of as unregulated cell replication, where tumor cells acquire mutations allowing them to bypass normal cell cycle checkpoints and apoptosis regulatory pathways. Nitrogen mustards work by alkylating DNA, preventing cell division, and causing cell cycle checkpoint-mediated apoptosis. Many chemotherapeutic agents similarly target cell division. Unfortunately, this mechanism of action is not specific to malignant cells, resulting in chemotherapy-related toxicities to healthy tissue. The nonspecific effects of most chemotherapeutic agents are the primary limiting factor in the treatment of malignancies. Methods to reduce these toxicities therefore allow for higher dosage regimens of chemotherapeutics and enhance clinical outcomes in many forms of cancer.
The blood-producing hematopoietic system is particularly susceptible to the toxicities of chemotherapeutics given the production demand of approximately 1 trillion blood cells daily.3 The most abundant white blood cells in the blood are neutrophils, which are essential members of the innate immune system and provide protection from a wide variety of bacterial and fungal pathogens. Given the abundance of neutrophils and their short half-life in the blood of 6–8 hours, the production rate from hematopoietic progenitor cells in the bone marrow is 5×1010 to 10×1010 neutrophils per day.4 The notable amount of cell divisions required to meet this high cellular demand makes the myeloid compartment of the bone marrow hematopoietic system particularly susceptible to chemotherapy.
Chemotherapy-induced neutropenia
Neutropenia is the most serious hematologic toxicity of chemotherapy and often is the limiting factor in dose escalation or duration of chemotherapeutic administration.5 The National Cancer Institute’s Common Toxicity Criteria rates the severity of neutropenia into five classifications: Grade 1 is <2.0×109–1.5×109 cells/L; Grade 2 is <1.5×109–1.0×109 cells/L; Grade 3 <1.0×109–0.5×109 cells/L; Grade 4 is <0.5×109 cells/L; and Grade 5 is death.6 Neutrophils are the primary cellular mediators of the innate immune system and the first line of defense to numerous pathogens. Neutropenic patients are susceptible to infections from the respiratory tract (35%–40% of cases), bloodstream (15%–35%), urinary tract (5%–15%), skin (5%–10%), gastrointestinal tract (5%–10%), and other sites (5%–10%).7 Many of the symptoms manifested in response to bacterial infection are a consequence of the inflammatory immune response to the pathogen. In neutropenic patients, because they lack neutrophils that are mounting the normal response, they lack many of the normal symptoms of infection and hence it can go unnoticed. Oftentimes, the only clinical sign of infection early on is a fever, which defines a serious clinical outcome of chemotherapy known as “febrile neutropenia”, having Grade 4 neutropenia with a fever greater than 38.3°C. More than 60,000 patients are hospitalized annually as a result of neutropenia, and these hospitalizations are estimated to have resulted in $13,400 in medical costs.8 The average costs for febrile neutropenia encounters were reported to be $22,086, with patients who had febrile neutropenia incurring a mean cost difference of $1,149 per patient, per month.9 Similarly, another study reported that mean hospitalization costs per patient were $18,042 for patients with neutropenia, $22,839 for patients with neutropenia plus infection or fever, and $27,587 for patients with neutropenia plus infection.10 Therefore, boosting the bone marrow production of neutrophils is imperative to successful chemotherapy treatment and reduction of medical costs.
The landscape of febrile neutropenia was changed dramatically in the mid-1980s with the molecular cloning of granulocyte colony–stimulating factor (G-CSF).11–14 Initially believed to be a multipotent hematopoietic growth factor as well an inducer of granulocyte differentiation, G-CSF was found to stimulate proliferation15,16 and differentiation13 of several types of myeloid progenitor cells alone and in combination with other growth factors,17–19 and this stimulation of progenitor cells leads to a significant increase in mature neutrophil output. The first clinical trials were performed in cancer patients receiving chemotherapy20–23 leading to US Food and Drug Administration approval in 1991 of Neupogen® (filgrastim). Filgrastim is approved to decrease the incidence of neutropenia in patients with nonmyeloid malignancies receiving chemotherapeutics associated with a significant incidence of severe neutropenia with fever; to reduce incidence and duration of neutropenia in patients with congenital, cyclic, or idiopathic neutropenia; for reducing the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of adults with acute myeloid leukemia (AML); and to reduce the duration of neutropenia and neutropenia-related clinical sequelae, eg, febrile neutropenia, in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by hematopoietic transplantation.
While successful, the burden and associated pain of daily injections of filgrastim can result in patient discomfort and/or noncompliance, resulting in missed doses and reduced efficacy. Improved dosage forms that reduce the daily injection requirement can thus lead to enhanced patient care. A longer lasting monomethoxy polyethylene glycol (PEG)–conjugated filgrastim (pegfilgrastim; Neulasta®) having the same mechanism of action24 and clinical effects compared to filgrastim25 was subsequently introduced, but requires only single administration per cycle of chemotherapy rather than multiple daily injections of filgrastim.26 Since the introduction of filgrastim and pegfilgrastim to clinical practice, the management of neutropenia has been enhanced, and several biosimilars have recently entered the marketplace and others are in development (reviewed in Hoggatt and Pelus27). To date, only a single long-acting biosimilar filgrastim has been introduced. This review will focus on lipegfilgrastim (Lonquex®) approved by the European Medicines Agency (EMA) for the treatment of neutropenia.
Lipegfilgrastim
Lipegfilgrastim is a long-acting, site-specific glycol-pegylated r-metHu G-CSF produced by conjugation of a single 20-kDa PEG to the natural O-glycosylation site of G-CSF (threonine 134), using a novel glycoPEGylation technology (Figure 1). Because the recombinant G-CSF is produced in Escherichia coli, the glycosylation site is empty. Addition of the O-glycan was achieved by enzymatic activity of a truncated N-acetylgalactosaminyltransferase isoform 2 fused with maltose-binding protein at the threonine residue site. A 20-kDa PEG-sialic acid derivative was enzymatically transferred to the O-glycan with a sialyltransferase. In contrast, pegfilgrastim (Neulasta®) is a recombinant methionyl human G-CSF with a methoxy-polyethylene glycol propionaldehyde 20-kDa PEG covalently conjugated to its N-terminus.28,29 The novel pegylation process used in lipegfilgrastim results in different pharmacokinetic and pharmacodynamic profiles than pegfilgrastim, as discussed later.
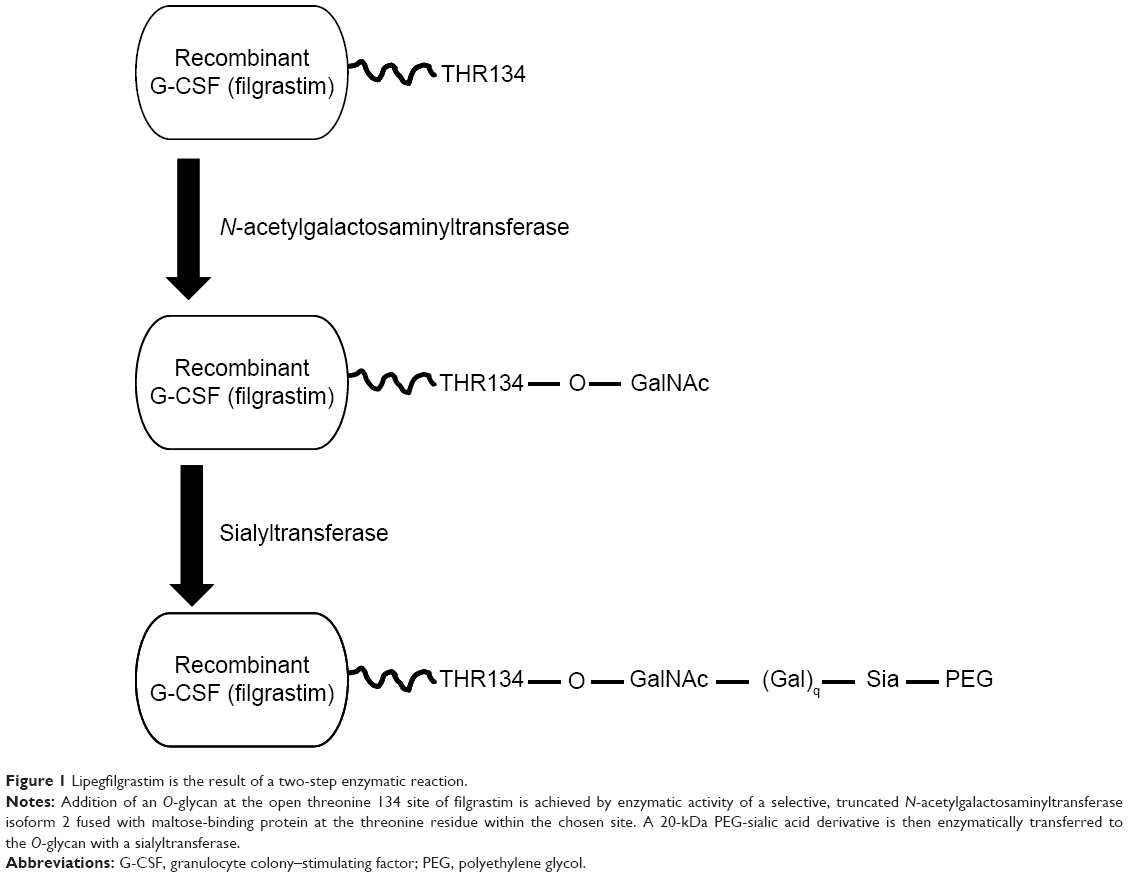 | Figure 1 Lipegfilgrastim is the result of a two-step enzymatic reaction. |
In preclinical studies, lipegfilgrastim and pegfilgrastim binding to the G-CSF receptor was evaluated using an NFS-60 cell-based [125I]-G-CSF competitive G-CSF receptor-binding assay. In the cell-based [125I]-G-CSF competitive G-CSF receptor-binding assay, G-CSF receptor binding was equivalent between lipegfilgrastim and pegfilgrastim as indicated by the inhibition of [125I]-G-CSF binding (0.70±0.09 nM IC50 versus 0.72±0.18 nM IC50 [mean ± standard deviation]).28
Pharmacokinetics
Lipegfilgrastim was evaluated in three separate healthy volunteer studies.30,31 In a limited healthy volunteer study, three separate subcutaneous injection sites were evaluated; upper arm, abdomen, and the thigh. Bioavailability was found to be lower after injection in the thigh compared to the upper arm or abdomen. However, increases in neutrophil counts were similar across the three injection sites and the recommended administration of lipegfilgrastim is for any of these three sites.30 In additional clinical studies, lipegfilgrastim was administered as a single subcutaneous injection and in some analyses compared to pegfilgrastim. In the dose escalation phase, administration of lipegfilgrastim was not continued beyond 100 μg/kg as two subjects experienced ANCMAX (maximum absolute neutrophil count) levels greater than 70 neutrophils/nL, the limit for hyperleukocytosis in healthy subjects.31 The maximal blood concentration was reached after 30–36 hours postadministration. The terminal half-life ranged from approximately 32 to 62 hours, which was 7–10 hours longer for lipegfilgrastim 100 μg/kg compared with pegfilgrastim 100 μg/kg.
Lipegfilgrastim is mainly internalized and eliminated by neutrophils and can be metabolized via intra- or extracellular degradation by proteolytic enzymes, which becomes saturated at higher doses. In vitro data suggest there is little to no effects on CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4/5 activity, suggesting lipegfilgrastim does not affect human cytochrome P450 metabolism.30
Safety
In conventional safety and pharmacology studies in animal models, no special hazard for humans was found. In all healthy volunteer studies, the overall safety and tolerability was good, and no serious adverse events were reported.31 While there have been reports of PEGylation-inducing anti-PEG antibodies in patients,32,33 no subject tested positive for antibodies against filgrastim, pegfilgrastim, or lipegfilgrastim demonstrating no significant immunogenicity of the compound. In Phase III trials in breast cancer patients, the safety of lipegfilgrastim was similar to pegfilgrastim.34,35 The most commonly reported adverse events in both treatment groups were alopecia, nausea, asthenia, bone pain, diarrhea, fatigue, anorexia, vomiting, headache, and myalgia.
Efficacy
Lipegfilgrastim was compared to pegfilgrastim in two separate clinical trials. The first trial was a randomized, double-blind Phase II study, conducted between June and November of 2008.35 This study included screening of 229 patients and ultimately enrollment of 208 patients at 37 centers in the Czech Republic, Germany, Hungary, Romania, Russia, and Ukraine. A total of 202 patients (97.1%) completed the study, with three patients withdrawing due to adverse events, two withdrew consent, and one was lost to follow-up. Male and female patients (only three male patients were in the study) ≥18 years of age with high-risk stage II, III, or IV breast cancer were eligible if they had an Eastern Cooperative Oncology Group performance status ≤2; absolute neutrophil count ≥1.5×109/L; platelet count ≥100×109/L; and adequate cardiac, hepatic, and renal function. Patients had to be chemotherapy-naive and eligible for doxorubicin/docetaxel chemotherapy. Patients received four 3-week chemotherapy cycles and were randomized 1:1:1:1 to receive 3.0-, 4.5-, or 6.0-mg lipegfilgrastim or 6.0-mg pegfilgrastim subcutaneously on chemotherapy day 2. Chemotherapy consisted of doxorubicin 60 mg/m2 given as an intravenous bolus injection followed 1 hour later by a 1-hour intravenous infusion of docetaxel 75 mg/m2.
No significant difference in the duration of severe neutropenia for the first chemotherapy cycle among the three lipegfilgrastim doses and pegfilgrastim was observed. The percentage of patients who did not experience severe neutropenia was higher in the 6.0-mg lipegfilgrastim dosage group (62.0%) compared with the 3.0- and 4.5-mg dosage groups (43.4% and 49.0%, respectively). This percentage of patients was also higher than the 6.0-mg pegfilgrastim dosage group (46.3%). For chemotherapy cycles 2–4, the mean duration of severe neutropenia was significantly shorter for the 6.0-mg lipegfilgrastim dosage group versus the pegfilgrastim dosage group. This study established 6.0-mg lipegfilgrastim as the optimal dose for breast cancer patients receiving chemotherapy and established noninferiority to pegfilgrastim in reducing cycle 1 duration of severe neutropenia.
Following this study, a similar experimental approach was taken in a randomized, multicenter Phase III study.34 Breast cancer patients receiving chemotherapy were randomized 1:1 to receive either the 6.0-mg lipegfilgrastim dose (n=101) or 6.0-mg pegfilgrastim dose (n=101). Lipegfilgrastim was found to be noninferior to pegfilgrastim in the duration of severe neutropenia following cycle 1 of chemotherapy, with a 95% confidence interval of -0.498%, 0.062% days (P=0.1260). Similarly, there was no significant difference in the duration of severe neutropenia between the two groups following cycles 2–4. Intriguingly, the absolute neutrophil count nadir after cycles 2–4 was higher in the lipegfilgrastim group compared to the pegfilgrastim group (2.6 vs 2.0, 2.5 vs 2.0, and 2.7 vs 2.3×109/L; P=0.0189, P=0.0353, and P=0.1122, respectively). Similarly, the time to neutrophil recovery was ~1.5 days shorter for the lipegfilgrastim group compared to the pegfilgrastim group (P<0.05). The results of this study further demonstrated noninferiority of 6.0-mg lipegfilgrastim compared to 6.0-mg pegfilgrastim and suggest a possible enhancement over pegfilgrastim in neutrophil recovery following chemotherapy.
Recently, the results of a double-blind, randomized, placebo-controlled study in elderly patients with non–small-cell lung cancer receiving cisplatin/etoposide chemotherapy were reported at the 12th Meeting of the International Society of Geriatric Oncology.36 Patients received 6.0-mg lipegfilgrastim or placebo on day 4 of the chemotherapy cycle, and the incidence of febrile neutropenia was assessed. Fewer elderly patients receiving lipegfilgrastim (0/53; 0.00%) experienced febrile neutropenia compared to the placebo group (4/30; 13.33%) (P=0.0064). There was no significant difference in incidence severity or type of adverse events between the treatment and placebo groups.
Conclusion
Lipegfilgrastim, in the setting of breast cancer chemotherapy–induced neutropenia is at least as equivalent to pegfilgrastim in reducing neutropenic complications. Lipegfilgrastim, Lonquex®, represents the first long-acting biosimilar filgrastim to reach the market in Germany and has received EMA approval and will likely be marketed throughout Europe.
As the Neupogen® and Neulasta® patent lives have ended, and given the large economic market of G-CSF therapies in reducing the complications associated with chemotherapy, there has been a large effort in biosimilar development. To date, several filgrastim biosimilars have received US Food and Drug Administration and/or EMA approval and have entered the marketplace (recently reviewed in Hoggatt and Pelus27). While lipegfilgrastim is the first long-acting biosimilar filgrastim to reach the market, additional long-acting biosimilars, some with unique modifications to increase half-life, are currently in late-stage clinical development (reviewed in Hoggatt and Pelus27). To date, no long-acting biosimilar filgrastim has been approved by the US Food and Drug Administration. Additionally, dosage forms that eliminate the need for injection altogether, such as the topical filgrastim product Nupen, may enhance patient compliance and resulting care in some clinical applications, although further clinical studies are required.
Overall, the use of biosimilar filgrastim and pegfilgrastim products will be dictated by market prices and individual center preferences until further enhancements in efficacy or ease of administration, eg, oral, are identified and developed. In the shorter term, it is expected that biosimilar G-CSF agonists may reduce the significant medical costs of neutropenia treatment and facilitate market expansion.
Disclosure
The authors report no conflicts of interest in this work.
References
Hirsch J. An anniversary for cancer chemotherapy. JAMA. 2006;296(12):1518–1520. | ||
Goodman LS, Wintrobe MM. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. JAMA. 1946;132:126–132. | ||
Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. 1993;81(11):2844–2853. | ||
Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31(8):318–324. | ||
Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia: risks, consequences, and new directions for its management. Cancer. 2004;100(2):228–237. | ||
National Cancer Institute Common Terminology Criteria for Adverse Advents v3.0 (CTCAE) [Internet]; 2015. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed August 9, 2006. | ||
Nesher L, Rolston KV. The current spectrum of infection in cancer patients with chemotherapy related neutropenia. Infection. 2014;42(1):5–13. | ||
Caggiano V, Weiss RV, Rickert TS, Linde-Zwirble WT. Incidence, cost, and mortality of neutropenia hospitalization associated with chemotherapy. Cancer. 2005;103(9):1916–1924. | ||
Michels SL, Barron RL, Reynolds MW, Smoyer Tomic K, Yu J, Lyman GH. Costs associated with febrile neutropenia in the US. Pharmacoeconomics. 2012;30(9):809–823. | ||
Schilling MB, Parks C, Deeter RG. Costs and outcomes associated with hospitalized cancer patients with neutropenic complications: a retrospective study. Exp Ther Med. 2011;2(5):859–866. | ||
Nagata S, Tsuchiya M, Asano S, et al. The chromosomal gene structure and two mRNAs for human granulocyte colony-stimulating factor. EMBO J. 1986;5(3):575–581. | ||
Platzer E, Oez S, Welte K, et al. Human pluripotent hemopoietic colony stimulating factor: activities on human and murine cells. Immunobiology. 1986;172(3–5):185–193. | ||
Souza LM, Boone TC, Gabrilove J, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232(4746):61–65. | ||
Welte K, Platzer E, Lu L, et al. Purification and biochemical characterization of human pluripotent hematopoietic colony-stimulating factor. Proc Natl Acad Sci U S A. 1985;82(5):1526–1530. | ||
Dührsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72(6):2074–2081. | ||
Welte K, Bonilla MA, Gillio AP, et al. Recombinant human granulocyte colony-stimulating factor. Effects on hematopoiesis in normal and cyclophosphamide-treated primates. J Exp Med. 1987;165(4):941–948. | ||
Ikebuchi K, Ihle JN, Hirai Y, Wong GG, Clark SC, Ogawa M. Synergistic factors for stem cell proliferation: further studies of the target stem cells and the mechanism of stimulation by interleukin-1, interleukin-6, and granulocyte colony-stimulating factor. Blood. 1988;72(6):2007–2014. | ||
Ikebuchi K, Clark SC, Ihle JN, Souza LM, Ogawa M. Granulocyte colony-stimulating factor enhances interleukin 3-dependent proliferation of multipotential hemopoietic progenitors. Proc Natl Acad Sci U S A. 1988;85(10):3445–3449. | ||
Metcalf D, Nicola NA. Proliferative effects of purified granulocyte colony-stimulating factor (G-CSF) on normal mouse hemopoietic cells. J Cell Physiol. 1983;116(2):198–206. | ||
Bronchud MH, Scarffe JH, Thatcher N, et al. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer. 1987;56(6):809–813. | ||
Gabrilove JL, Jakubowski A, Scher H, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. N Engl J Med. 1988;318(22):1414–1422. | ||
Gabrilove JL, Jakubowski A, Fain K, et al. Phase I study of granulocyte colony-stimulating factor in patients with transitional cell carcinoma of the urothelium. J Clin Invest. 1988;82(4):1454–1461. | ||
Morstyn G, Campbell L, Souza LM, et al. Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet. 1988;1(8587):667–672. | ||
Lord BI, Woolford LB, Molineux G. Kinetics of neutrophil production in normal and neutropenic animals during the response to filgrastim (r-metHu G-CSF) or filgrastim SD/01 (PEG-r-metHu G-CSF). Clin Cancer Res. 2001;7(7):2085–2090. | ||
Johnston E, Crawford J, Blackwell S, et al. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol. 2000;18(13):2522–2528. | ||
Green MD, Koelbl H, Baselga J, et al; International Pegfilgrastim 749 Study Group. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol. 2003;14(1):29–35. | ||
Hoggatt J, Pelus LM. New G-CSF agonists for neutropenia therapy. Expert Opin Investig Drugs. 2014;23(1):21–35. | ||
Abdolzade-Bavil A, Cooksey B, Scheckermann C, et al. Pegylated versus glycopegylated G-CSFs and their biochemical and physiological properties [abstract]. Blood. 2013;122:4851. | ||
Mahlert F, Schmidt K, Allgaier H, Liu P, Müller U, David Shen W. Rational development of lipegfilgrastim, a novel long-acting granulocyte colony-stimulating factor, using glycopegylation technology [abstract]. Blood. 2013;122:4853. | ||
European Medicines Agency Product Information: Lonquex [Internet]; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002556/WC500148380.pdf | ||
Buchner A, Lammerich A, Abdolzade-Bavil A, Muller U, Bias P. Lipegfilgrastim: pharmacodynamics and pharmacokinetics for body-weight-adjusted and 6 mg fixed doses in two randomized studies in healthy volunteers. Curr Med Res Opin. 2014;30(12):2523–2533. | ||
Armstrong JK, Hempel G, Koling S, et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110(1):103–111. | ||
Richter AW, Akerblom E. Polyethylene glycol reactive antibodies in man: titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int Arch Allergy Appl Immunol. 1984;74(1):36–39. | ||
Bondarenko I, Gladkov OA, Elsaesser R, Buchner A, Bias P. Efficacy and safety of lipegfilgrastim versus pegfilgrastim: a randomized, multicenter, active-control phase 3 trial in patients with breast cancer receiving doxorubicin/docetaxel chemotherapy. BMC Cancer. 2013;13:386. | ||
Buchner A, Elsasser R, Bias P. A randomized, double-blind, active control, multicenter, dose-finding study of lipegfilgrastim (XM22) in breast cancer patients receiving myelosuppressive therapy. Breast Cancer Res Treat. 2014;148(1):107–116. | ||
Volovat C, Bondarenko IM, Gladkov OA, et al. Efficacy and safety of lipegfilgrastim in elderly patients with non-small-cell lung cancer receiving cisplatin/etoposide chemotherapy. J Geraitr Oncol. 2012;3(Sup 1):S97–S98. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.