Back to Archived Journals » International Journal of Interferon, Cytokine and Mediator Research » Volume 6
Role of CXCL10 in central nervous system inflammation
Authors Michlmayr D, McKimmie C
Received 4 November 2013
Accepted for publication 16 January 2014
Published 27 February 2014 Volume 2014:6 Pages 1—18
DOI https://doi.org/10.2147/IJICMR.S35953
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Daniela Michlmayr, Clive S McKimmie
Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, Scotland, UK
Abstract: The central nervous system (CNS) is a highly complex tissue that is vital for a large number of life-dependent processes. The neurons of the CNS are post-mitotic and cannot be easily replaced or repaired if lost or damaged. The hallmark of neuroinflammation is an influx of leukocytes through the blood–brain barrier or blood–cerebrospinal fluid barrier. Depending on the disease, the presence of leukocytes can have beneficial or detrimental effects on disease outcome. Importantly, the entry of leukocytes into the CNS is primarily controlled by chemokines, a large family of chemotactic cytokines that coordinate the movement of leukocytes into and within tissues. Expression of inflammatory chemokines is almost absent in the resting CNS, but can be highly upregulated during inflammation. This includes CXCL10, a chemokine that has critical roles in controlling the entry of several important leukocyte subsets into the brain and other tissues. CXCL10 is expressed by neurons, glia, and stromal cells in a number of different CNS diseases and can have either a protective or a detrimental role in facilitating disease progression or resolution. CXCL10 together with CXCL9 and CXCL11 binds to CXCR3, which is predominantly expressed on activated T-cells and natural killer cells. Due to the ability of CXCL10 to mediate leukocyte influx in a variety of inflammatory CNS diseases, research has focused on identifying drugs that block the function of CXCL10 or CXCR3. Many of these drugs are under investigation and are discussed here in detail. In this paper, we review the function of CXCL10 during nervous system inflammation and highlight the latest developments that suggest its involvement in a number of key diseases, including multiple sclerosis, Alzheimer's disease, and viral encephalitis.
Keywords: chemokine, neuroinflammation, neuroimmunology, encephalitis, CXCL10
Introduction
The central nervous system (CNS), comprised of the spinal cord, brain, optic nerves, and retina, constitutes a complex network of specialized cells that includes neurons and glial cells. Neurons are post-mitotic cells and are not easily replaced compared with other cells in the body, although limited neurogenesis has been observed in specific areas of the brain.1,2 Accordingly, to protect these cells from damage, the CNS has evolved a variety of strategies that tightly control the initiation and perpetuation of inflammatory responses. Despite the absence of most recognizable immune components in resting neural tissue, florid and sometimes life-threatening immune responses can result following infection or damage. Indeed, due to the sometimes life-threatening consequence of CNS leukocyte entry, this process is highly regulated to prevent inadvertent immune-mediated neuropathology. Thus, the CNS is considered to be an immune-specialized site and is characterized by the following features:3 low expression of major histocompatibility complex I and major histocompatibility complex II, absence of resident dendritic cells, lack of lymphatic vessels, and maintenance of blood–brain and blood–cerebrospinal fluid barriers that, amongst other functions, regulate the entry of leukocytes into the brain. It has long been proposed that, due to the absence of these immunological hallmarks, the brain can be considered “immunoprivileged”. However, it is now clear that the brain is subject to immunosurveillance, albeit via mechanisms that are distinct from those found in most other tissues, and that florid and life-threatening immune responses can occur.4 Indeed, during infection and inflammation, these barriers can become compromised or dysfunctional.4–6 This allows leukocytes, fluid, and plasma components such as complement and antibodies to enter the brain; a process that is often associated with activation and proliferation of local glial cells. Together, these processes are invariably detrimental in the case of sterile autoimmune diseases such as multiple sclerosis (MS). In contrast, during infection with neurotropic pathogens, CNS inflammation can lead to removal of infectious agents, but often at the cost of neuropathology and neuronal dysfunction.
Chemokines are a large family of small chemotactic cytokines (8–12 kDa) that are pivotal for the regulation of leukocyte migration and function during a wide variety of contexts, including infection, inflammation, hematopoiesis, angiogenesis, embryogenesis, lymphoid development, and metastasis of cancer.7 During neuroinflammation, chemokines play an important role in attracting inflammatory leukocytes into the CNS, where they can have detrimental or beneficial effects depending on the context.6 Based on the position of the first two conserved cysteine residues at the amino terminus of the mature protein, chemokines can be divided into four subfamilies, ie, CC, CXC, XC, and CX3C. Both CC and CXC-chemokine subfamilies contain many members, whereas XC and CX3C chemokines are comprised of two members and one member, respectively, in humans.7 CXCR3 is a key chemokine receptor within the CXC chemokine family that binds CXCL9, CXCL10, and CXCL11 and has important functions in mediating leukocyte entry into the inflamed CNS (Figure 1).8,9 The most studied of these chemokines is CXCL10, primarily due to its pivotal role in mediating the influx of inflammatory leukocytes into the CNS in a number of important diseases, and is the focus of this review.
 | Figure 1 Chemokine receptor CXCR3 and its ligands CXCL9, CXCL10, and CXCL11. CXCR3 is a seven transmembrane G-protein coupled receptor. The DRY-motif is critical for engaging with cytoplasmic G-proteins to initiate signaling. CXCR3 binds three different ligands, ie, CXCL9, CXCL10, and CXCL11. These ligands bind to specific and distinct regions within the receptor. |
CXC chemokines
CXC chemokines are the second largest subfamily of chemokines and contain one amino acid between the first two and adjacent cysteine residues at the N-terminus of the mature protein.7 In humans, there are 16 members within the CXC chemokine subfamily, most of which are found clustered at a specific chromosomal location, which is chromosome 4 in humans and chromosome 5 in mice. The clustering of these chemokines within the chromosome indicates that many CXC chemokines evolved at the same time and their proximal location within the chromosome reflects the functional specialization that developed during evolution.10 Many of the CXC chemokines (CXCL1-3, CXCL5-6, and CXCL8) are important drivers of neutrophil trafficking.6 In addition, some CXC chemokines contain a structural domain, consisting of three amino acid residues (glutamine-leucine-arginine), known as the ELR motif, that plays an important role during angiogenesis.12 Finally, the chemokine CXCL12, which binds to the chemokine receptor CXCR4, has important roles in embryogenesis, homeostasis, and inflammation. Interestingly, CXCR4 is one of only two chemokine receptors for which drugs have been successfully licensed for use in the clinic, which in the case of CXCR4, are used to mobilize stem cells from the bone marrow niche.13
The CXCL cluster containing CXCL9, CXCL10, and CXCL11 is located in a separate minicluster in close proximity to the main CXC chemokine cluster.14 Importantly, CXCL9, CXCL10, and CXCL11 lack the ELR motif present in many of the other CXC chemokines and instead are potent inhibitors of angiogenesis.12 The close proximity of CXCL9, CXCL10, and CXCL11 within the chromosome indicates that these chemokines have evolved together and most likely explains why they share the same receptor, CXCR3. CXCL10 and CXCL11 are found in particularly close proximity within the minicluster and also share more structural similarities when compared with CXCL9. A unifying feature is their ability to be highly upregulated in response to interferon (IFN), a characteristic that facilitated their original identification as IFN-inducible proteins.15
Structure and expression of CXCL10
CXCL10 is a small 8.7 kDa protein that was originally identified in 1985 by Luster et al, who discovered its expression by U937 cells (a histiocytic lymphoma cell line with monocytic characteristics) following treatment with IFN-γ.15,16 Recombinant IFN-γ triggered CXCL10 expression within 30 minutes, with maximal accumulation peaking 5 hours posttreatment, a consequence of the presence of an IFN-stimulated response element and a nuclear factor kappa B1 element in its promoter.17,18 In comparison with CXCL9, which is only induced by IFN-γ, CXCL10 can also be induced by both IFN-γ and type I IFNs (IFN-α and IFN-β).19 IFN-γ expression is restricted to subsets of CD4+ T helper (Th)1 lymphocytes, CD8+ cytotoxic T-cells, and natural killer cells. In contrast, type I IFNs can be expressed by most cells following recognition of pathogens through innate sensors such as Toll-like receptors and RNA helicases including retinoic acid-inducible gene 1 and melanoma differentiation-associated protein 5, which leads to activation of transcription factor interferon-regulatory protein-3 and interferon-regulatory protein-7.20 It has also been reported that the cytokines tumor necrosis factor and interleukin-1β can weakly induce CXCL10 expression and elicit synergistic upregulation in combination with IFNs to induce strong CXCL10 expression.18 Finally, direct activation of CXCL10 expression by the nuclear factor kappa B pathway has been shown in a study of hypoxia-reperfusion injury and in a model of obliterative bronchiolitis.21,22 Thus, CXCL10 is differentially regulated compared with CXCL9, and this results in different temporal and spatial expression of this chemokine.
Outside the CNS, CXCL10 is expressed by a variety of cells including monocytes, macrophages, fibroblasts, osteoblasts, and endothelial cells.16,23,24 In the CNS, both glial cells and neurons can express CXCL10. Astrocytes are particularly effective at expressing this chemokine following exposure to viruses or their mimics, eg, as demonstrated by astrocytes stimulated with the viral analog, poly(I:C).25 In addition, infection with lymphocytic choriomeningitis virus (LCMV), mouse hepatitis virus (MHV), and Theiler’s murine encephalitis virus (TMEV) all induce high astrocytic expression of CXCL10.26–29 Microglia can also express CXCL10, as demonstrated following infection with neurotropic viruses such as Semliki Forest virus and cytomegalovirus (CMV).30,31 Microglia also appear to be a source of CXCL10 in Alzheimer’s disease and MS.26 In comparison, infection of mice with cerebral malaria or West Nile virus (WNV) leads to high expression of CXCL10 in neurons rather than in glia.32,33
CXCL10 is a chemoattractant for CXCR3+ cells, such as activated CD4+ Th1 and CD8+ T-cells, natural killer cells, and plasmacytoid dendritic cells, and putatively subsets of B-cells.34,35 Activated T-cells are predominantly attracted to the site of inflammation in the presence of CXCL10. Intriguingly, CXCL10 can be antagonistic for CCR3 when present at high concentrations. CCR3 is typically expressed on eosinophils, cells that constitute an important part of many skewed Th2 cell immune responses. Thus, during infection with, eg, viruses, CXCL10 may prevent the entry of these cells into infected tissues and in doing so reinforce a Th1 cell-dominant immune response.36 The same effect has been reported for the other CXCR3 ligands, CXCL9 and CXCL11. It should be noted that compared with CXCL9 and CXCL11, CXCL10 has an intermediate affinity for CXCR3 binding.37 The 16 amino acid residues closest to the N-terminal of CXCL10 are necessary for CXCR3 binding. Following ligation of CXCR3 with CXCL10, the receptor-ligand complex is internalized.38 The difference in CXCR3 ligand affinity is most likely due to the use of distinct regions of each ligand for receptor binding.
In addition to its roles in mediating an influx of inflammatory leukocytes into infected or inflamed tissues, CXCL10 has a variety of other functions, not all of which may depend on its chemotactic abilities. First, CXCL10 together with CXCL9 and CXCL11 are potent inhibitors of angiogenesis.39,40 Second, CXCL10 has been shown to have direct antibacterial properties similar to α-defensins, for example against Escherichia coli and Listeria monocytogenes.41 Last and most important, CXCL10 has also been shown to be important for the generation and function of effector T-cells, and this is an area of active research, although a specific role for CXCL9 in recruiting CD8+ T-cell memory responses has also been proposed. In a murine neuroblastoma model, CXCL10 has been shown to be pivotal for generation of tumor-specific T-cells and tumor protective immunity during interleukin-12 therapy.42 More recently, a series of key publications by Luster et al and von Andrian et al have shown that CXCL10 and its receptor CXCR3 are key for coordinating and instigating T-cell responses to antigens in lymph nodes draining infected or inflamed tissues.43,45 Following immunization with the antigen ovalbumin, dendritic cell-derived CXCL10 in the draining lymph nodes optimizes T-cell priming by facilitating both interactions between T-cells and dendritic cells and by guiding intranodal positioning of CD4+ T-cells to interfollicular and medullary zones.43 In a separate study, it was shown that the ability of memory CD8 T-cells to instigate rapid and effective immunity against pathogens upon secondary challenge with the same virus may in part be explained by their rapid relocation within the lymph nodes using a CXCR3-mediated process.43,44 Thus, whilst CXCL10 has previously been characterized as a simple attractor of leukocytes to inflamed tissues, it is in fact a pleiotropic chemokine with complex, overlapping chemotactic and nonchemotactic functions. Importantly, this could have implications when using CXCL10 or CXCR3 as a drug target for the treatment of various diseases.
Gene-deficient mice as models for studying the CXCL10-CXCR3 axis
CXCR3-deficient mice can be used to study the role of the CXCL10-CXCR3 axis in the pathogenesis of neuroinflammation, and many studies of viral encephalitis have used this method to investigate the role of CXCR3. However, one disadvantage of using gene-deficient mice is that blockade of the chemokine receptor is absolute and cannot be modulated or varied during the course of disease. Additionally, it has been demonstrated that CXCR3-/- mice, which are otherwise phenotypically similar to wild-type mice, have a deficiency of natural killer cells in their blood, suggesting that CXCR3 is important for the maintenance of natural killer cells in the body.45,46 As discussed above, CXCR3 may also have functions during T-cell priming and generation of effector cells. Thus, one should be careful to exclude the possibility that these extraneural processes are impacting on what appears to be a CNS-centric phenotype.
Caution has also to be exercised when interpreting results obtained from CXCL9- deficient and CXCL10-deficient mice. C57BL/6 mice contain a point mutation within the CXCL11 gene, rendering this mouse strain CXCL11-deficient.47 However, both CXCL9 gene-deficient and CXCL10 gene-deficient mice have been generated using 129 embryonic stem cells that possess a wild-type CXCL11 allele. Due to the close linkage of all three CXCR3 ligands, the wild-type CXCL11 gene has accidently been reintroduced along with the “knocked out” allele.47 Thus, CXCL9-deficient and CXCL10-deficient mice have regained CXCL11 function and cannot be easily compared with their C57Bl/6 wild-type control counterparts that lack CXCL11. Thus, comparisons of CXCL9-deficient and CXCL10-deficient mice with wild-type C57BL/6 mice have to be carefully analyzed with this caveat in mind.47
Etiology of neuroinflammatory and neurodegenerative diseases
The hallmarks of neuroinflammation include activation of glial cells, particularly microglia and astrocytes, expression of inflammatory cytokines and chemokines, increased blood–brain barrier permeability, and infiltration of leukocytes into the CNS.48 Neuroinflammation can occur acutely during infection of the CNS with neurotropic pathogens, such as viruses, bacteria, and parasites, during stroke, head trauma, and entry of toxins into the brain. Acute neuroinflammation is typically short-lived and self-limited, and the immune response within the CNS is often necessary to clear pathogens from the infected brain or to restore homeostasis after injury and damage. If inflammation of the brain persists, it can become chronically inflamed, often resulting in neuronal dysfunction, tissue damage, and neurodegeneration.4,49 As such, chronic CNS inflammation can be considered a highly pathological state that often results in irreversible changes to the brain.
Neurodegenerative disorders of the CNS include MS, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. The etiological basis of these diseases is still a matter of debate, although it has been hypothesized that, in some cases, virus infection can be one etiological trigger for, eg, MS, although this remains controversial.50–52 In the developed world, these chronic neuroinflammatory or neurodegenerative diseases represent the most common form of CNS inflammatory disease and tend to disproportionally affect adults and the elderly. In contrast, acute inflammatory diseases caused by infectious agents, that predominantly affect the young, are far more common in the developing world. This is due to the high prevalence of neurotropic pathogens, many of which are arthropod-borne. Whilst these diseases are most commonly found in tropical and subtropical climates, due to globalization and climate change there exists an increasing opportunity for the spread of infectious agents, such as neurotropic viruses, to more temperate climates. A recent example of this phenomenon is WNV, a neurotropic virus that emerged in the Americas in 1999 and has since become endemic.53 Medically important pathogens that can infect neural tissue are shown in Table 1 and include parasites such as plasmodium and trypanosomes, viruses such as Japanese encephalitis, rabies, herpes simplex virus (HSV), and WNV, and bacteria such as meningococcal and pneumococcal species.
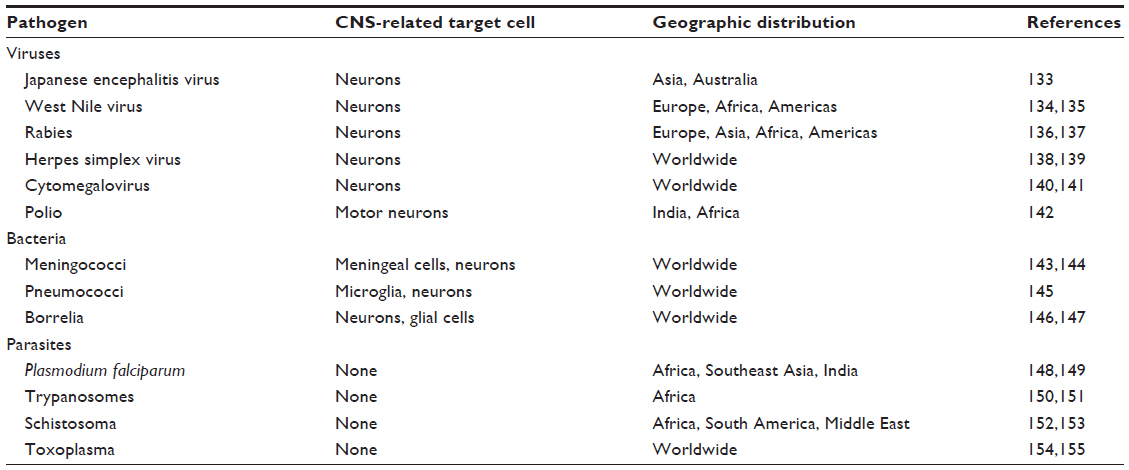 | Table 1 Medically important neurotropic pathogens |
Role of CXCL10 in neurodegenerative diseases
Alzheimer’s disease and prion diseases are both examples of neurodegenerative diseases.54,55 One of the hallmarks of Alzheimer’s disease is loss of neurons and formation of amyloid plaques in the brain, resulting in cognitive impairment and loss of memory. The etiology of Alzheimer’s disease is not yet clear, but genetic, epigenetic, and environmental factors play a pivotal role in the onset of the disease.55 The accumulation of beta amyloid results in aberrant neuronal activity, synaptic depression, and gliosis.56 This occurs concomitantly with an induction of CXCL10 by astrocytes and to a lesser extent in microglia.26,57 Since there is not an obvious influx of inflammatory leukocytes into the brain with Alzheimer’s disease, alternative functions for CXCL10 have been suggested. Fascinatingly, CXCR3 is expressed on neurons in Alzheimer’s disease26 and CXCL10 binding may lead to induction of the extracellular signal-regulated kinase pathway that ultimately results in neuronal dysfunction and apoptosis.58–60 Some have suggested that this may be one mechanism by which loss of memory occurs during Alzheimer’s disease.26,56
Prion diseases or transmissible spongiform encephalopathies are a distinct group of fatal neurodegenerative diseases that can be acquired, inherited, or occur sporadically. They are characterized by accumulation of misfolded prion proteins in the CNS, a process that leads to neuronal malfunction, gliosis, and invariably death.61–63 Infection with the transmissible spongiform encephalopathy agent also leads to induction of cytokines, albeit at low levels, in the brain. This includes CXCL10, along with other cytokines which are primarily expressed by astrocytes and microglia. Similar to Alzheimer’s disease, prion disease does not result in encephalitis or significant leukocyte influx into the brain, suggesting that other factors required for leukocyte influx are missing, or that CXCL10 has alternative functions during prion disease.64 Indeed, the contribution that CXCL10 provides in driving the pathogenesis of prion disease remains to determined, although Riemer et al have shown that CXCR3-deficient mice have a survival benefit over wild-type prion-infected mice, suggesting that signaling through this receptor contributes to the pathogenesis of this disease.65
Role of CXCL10 during neuroinflammation
In many models of neuroinflammation, CXCL10 is highly expressed and performs critical nonredundant roles. This is perhaps best demonstrated following CNS infection with a variety of viruses, such as dengue virus, WNV, or LCMV, in which loss of CXCL10 results in significant disease perturbation that cannot be ameliorated by expression of the other CXCR3 ligands, CXCL9 and CXCL11. This nonredundancy of CXCL10 could be due to spatial and temporal differences in expression compared with the other two CXCR3 ligands, and is elegantly discussed in more detail by Müller et al.66 Diseases in which CXCL10 function or expression has been implicated are shown in Table 2. Fascinatingly, the function of CXCL10 results in processes that can be either protective or detrimental, depending on the etiology and context of neuroinflammation, as summarized in Figure 2. Together, these studies highlight the important role of CXCL10 in recruiting leukocytes and mediating CNS inflammation.10,32,67,68 In this review, we discuss the role of CXCL10 in the context of CNS inflammation in both infectious and autoimmune contexts, with a particular focus on viral encephalitis.
 | Table 2 Role of CXCL10 in autoimmunity and infection of the CNS has been studied using both CXCL10 null mice and a neutralizing antibody |
 | Figure 2 CXCL10 function can be either beneficial or detrimental to the host depending on the disease and context. Diseases of autoimmunity and neurodegeneration tend to be more severe in animals with a functioning CXCL10-CXCR3 axis. In contrast, the role of CXCL10 can be either beneficial or detrimental to the host during CNS infection. For example, during cerebral malaria, CXCL10 promotes a damaging infiltrate of leukocytes that does not appear to aid pathogen clearance. In comparison, CXCL10-driven responses to HSV appear protective by facilitating the entry of leukocytes that enhance pathogen clearance. |
Multiple sclerosis
MS is a cell-mediated autoimmune disease characterized by chronic demyelination of axons and loss of oligodendrocytes, which often results in severe disability with disease progression. In MS, activated CXCR3+ T-cells enter the CNS and can be detected in the cerebrospinal fluid or in the brain parenchyma.11,69–71 These encephalitogenic T-cells are mainly CD4+ Th1 and Th17 lymphocytes that specifically recognize components of myelin, a process that eventually leads to demyelination of neuronal axons.72 The etiology of MS is not properly understood, but is most likely patient-specific and due to a variety of environmental and genetic factors. It has long been suggested that viruses such as Epstein-Barr virus might be one etiological factor that can trigger an autoimmune process in genetically susceptible people. In this scenario, virus infection initiates an otherwise clinically inapparent neuroinflammatory event that facilitates a gradual loss of tolerance to myelin components.51 However, this view remains controversial and will likely be the focus of debate for some time to come.
The expression of both CXCL9 and CXCL10 is highly upregulated during MS. In particular, CXCL10 expression is upregulated early during MS and is mainly released by activated astrocytes.27 In support of this, Sørensen et al have demonstrated in human post mortem brain samples that CXCL10 can be detected in astrocytes, demyelinating plaques, and close to perivascular cell infiltrates within cerebral brain capillaries that are comprised of many CXCR3+ T-cells.29 Another study by Balashov et al showed that CXCL10 protein levels in the cerebrospinal fluid were significantly increased in MS patients, and Franciotta et al demonstrated that CXCL10 levels in the cerebrospinal fluid and serum were higher during acute phases of MS than in patients with stable MS.27,73 However, it should be noted that, despite these pathological observations, CXCL10 levels do not correlate well with clinical signs of MS. In addition to astrocytes, infiltrating macrophages and T-cells are also capable of producing CXCL10. This is in agreement with a previous study of human post mortem brain tissue from MS patients that demonstrated that macrophages express CXCL10 within demyelinating plaques.74 Furthermore, Biddison et al have reported that myelin proteolipid protein-specific CD8+ T-cells in vitro release CXCL10 which attracts myelin-specific CD4+ T-cells.75 In summary, studies using MS tissue have shown that CXCL10 is highly expressed by astrocytes and subsets of leukocytes, although the implications of these findings are yet to be fully determined. Further, human tissue is difficult to obtain for studies and does not allow an indepth analysis of CXCL10, particularly during the early events of disease progression. Importantly, some mechanistic insight has been provided through the use of animal models of MS, such as experimental autoimmune encephalomyelitis (EAE).
Experimental autoimmune encephalomyelitis
MS has been extensively studied using various animal models, the most widely used of which is the mouse model of EAE. EAE is an autoimmune-driven model of CNS inflammation that is most commonly instigated by administration of myelin-specific peptides in combination with highly potent proinflammatory adjuvants, such as complete Freund’s adjuvant and pertussis toxin, to facilitate localization of inflammation to the CNS. Following vaccination, self-reactive CD4+ T-cells proliferate and infiltrate neural tissue, such as the spinal cord and occasionally the brain, resulting in inflammatory demyelination and neurological dysfunction. Alternatively, EAE can be induced by passive transfer of encephalitic myelin-specific T-cells.
By studying the various types of EAE in mice, it has been possible to elucidate mechanisms by which both cell-mediated and humoral-mediated components of the immune system mediate CNS damage.76 However, whilst EAE has been widely used as a model of MS, it has been suggested that the most widely used form of EAE may be more similar to acute human demyelinating diseases such as acute disseminated encephalomyelitis.76,77 However, it should be noted that EAE can be induced by a variety of protocols, that include both active immunization of mice with myelin-specific components or passive transfer of encephalitogenic cells into recipient animals.76 Subsequently, the use of different protocols can lead to variations in disease outcome. Therefore, considering the disease heterogeneity induced by the various EAE regimes, interpretation of results obtained from EAE studies should be treated with some caution.
As in MS, CXCL10 is also highly expressed by astrocytes in EAE,78 is associated with a large influx of inflammatory leukocytes into neural tissue, and correlates with disease severity. Together, this suggests that CXCL10 is involved in the pathogenesis of autoimmune responses in the CNS79,80 by recruiting leukocytes into the brain and putatively by facilitating effector T-cell function. Fife et al used a CXCL10-neutralizing antibody to inhibit leukocyte infiltration into the CNS that critically was associated with concomitant amelioration of clinical signs of EAE.79 Wildbaum et al have shown that CXCL10 drives polarization of CD4+ T-cells into Th1 cells during EAE.81 In this study, a plasmid encoding the CXCL10 gene was vaccinated into mice, resulting in protection against EAE and the development of skewed Th2 immune responses. This further suggests that CXCL10 is involved in T-cell polarization, and that neutralization of CXCL10 can lead to suppression of EAE due to its effects in lymphoid tissues, in addition to its role in mediating a CNS leukocyte influx.79,81
However, it should be noted that there are some studies that suggest CXCL10 has the opposite effect of that suggested above, and instead acts to suppress EAE severity. These apparent contradictory data include a study in which neutralization of CXCL10 in rats leads to exacerbation of EAE symptoms, with increased numbers of CD4+ T-cells.82 Another study by Klein et al has shown that CXCL10 deficient-mice or mice treated with neutralizing CXCL10 antibody did not exhibit an altered clinical course of EAE. Further, CXCL10-/- mice showed an increased susceptibility to EAE following immunization with a decreased dose of myelin components.83 This study suggests that CXCL10 does not play a role in the trafficking of pathogenic T-cells into the CNS but is important in determining susceptibility to EAE. Interestingly, the authors also showed that CXCL10-deficient mice had higher levels of CXCL11 expression in the CNS and decreased CXCL9 levels in the draining lymph nodes, suggesting that compensatory modulation of other CXCR3 ligands may occur in the absence of CXCL10.
In summary, these studies have generated data that suggest divergent roles for CXCL10 during progression of EAE. Indeed, the role of CXCL10 in MS and the most widely used animal model, EAE, remains controversial. However, the use of highly distinct protocols for inducing EAE may go some way towards explaining the apparent conflicting data that indicate involvement of CXCL10. Further, the ability of CXCL9 and CXCL11 to bind CXCR3 may complicate data interpretation. Studies that have focused on these other ligands have been nicely reviewed by Müller et al.66
Infections of the CNS
Mouse hepatitis virus infection
MHV belongs to the family of Coronaviridae and is a well characterized model of viral encephalitis, which leads to acute encephalitis followed by chronic demyelination. Therefore, the chronic stage of MHV infection has similarities to MS and this model can be used to study demyelination. CXCL10 expression in the CNS is upregulated within 24 hours post infection with MHV and has been shown to colocalize with MHV-infected tissue.84 The primary source of CXCL10 in the CNS after infection has been identified as astrocytes, but microglia are also capable of producing this chemokine. The role of CXCL10 during acute MHV infection has been shown to be important for controlling viral replication by mediating both the attraction of CD4+ T-cells to infected tissue and the generation of effector T-cells.85 However, another study by Stiles et al showed that generation of T-cells was not impaired in CXCL10-deficient mice,86 and instead functions to attract T-cells into the CNS. Another study by Liu et al demonstrated that use of CXCL10-neutralizing antibodies during acute MHV infection leads to increased viral loads, higher mortality, and decreased CD4+ and CD8+ T-cell infiltration into the CNS.87 In contrast, during the chronic stages of MHV infection, which are characterized by demyelination, CXCL10 may promote inflammatory responses that are detrimental to host survival. Similar to acute infection, CXCL10 expression levels are also highly elevated in the brain during demyelination. However, unlike during acute infection, neutralization of CXCL10 leads to reduced demyelination, decreased brain CD4+ T-cell numbers, and critically to a clinical improvement of symptoms. Thus, these data demonstrate that expression of CXCL10 is beneficial during the acute stages of MHV infection, but plays a detrimental role during later stages of infection in which demyelination dominates. Studies investigating blockade of CXCR3 during the demyelinating stage of MHV infection produced similar results to those with neutralizing CXCL10.86 However, blockade of CXCR3 did not result in higher viral loads in the CNS or decreased CD8+ T-cell numbers compared with studies using CXCL10-neutralizing antibodies or CXCL10-deficient mice. The reason for this discrepancy is not yet clear.
Lymphocytic choriomeningitis virus infection
LCMV belongs to the family Arenaviridae that can cause fatal meningoencephalitis in mice. In this model, T-cells infiltrate the meninges, choroid plexus, and ependymal and parenchymal sites. Interestingly, this influx of lymphocytes appears to be entirely counterproductive and detrimental to the host, since athymic nude mice, which are deficient for T-cells, are able to survive an otherwise fatal injection with LCMV.88,89 T-cells entering the brain are capable of producing a variety of inflammatory chemokines and cytokines, such as tumor necrosis factor and IFN-γ, which result in permeabilization of the blood–brain barrier and facilitate infiltration of other mononuclear cells into the brain.90 Whilst both CXCL9 and CXCL10 are highly expressed in the LCMV-infected brain, the available evidence suggests that CXCL10 is more influential in controlling the position of CNS T-cells rather than in mediating CNS entry per se. CXCL10 is expressed in subpial astrocytes and expression of this chemokine correlates with the presence of LCMV RNA and leukocyte infiltration.91 In contrast, the kinetics of CXCL9 expression are both much delayed compared with CXCL10 and limited to isolated pockets of activated microglia and perivascular macrophages. Thus, the pattern of CXCL9 expression is in stark contrast with that of CXCL10, which occurs both earlier during infection and in a more extensive fashion.92,93 Importantly, CXCL10-deficient mice are protected from an otherwise lethal LCMV infection and show impaired trafficking of CD8+ T-cells into the CNS.91,94 Further, accumulation of CD8+ T-cells in the CNS during LCMV infection is critical for both permeabilization of the blood-brain barrier95 and for perforin-induced cell lysis of virally infected cells.96 Subsequently, from these data, it appears that CXCL10 is an important and necessary mediator of CD8+ T-cell CNS influx, and that in the context of LCMV infection, this leads to a detrimental series of events that culminate in a lethal panencephalitis.
In comparison, CXCL9 and CXCL11 are not able to compensate for the loss of CXCL10 function during LCMV infection.68 This is perhaps best exemplified in mice treated with a CXCL9-neutralizing antibody in which disease progression was not significantly altered compared with untreated mice.97 As such, LCMV infection represents an interesting example of a noncytolytic virus infection, in which widespread infection of neurons can be relatively well tolerated without significant neurological deficit in the absence of a T-cell influx.
West Nile virus infection
WNV belongs to the family of Flaviviridae and can cause fatal encephalitis and meningitis in humans. During brain infection, leukocytes accumulate in the CNS, and CD8+ T-cells in particular have been shown to be important for viral clearance.98 CXCL10 expression is restricted mainly to neurons and correlates with the presence of WNV antigens, particularly in cerebellar granule neurons.32,83 During the later stages of WNV infection, both CXCL9 and CXCL10 expression can also occasionally be detected in astrocytes and infiltrating macrophages.32 Studies using CXCL10-deficient mice or mice treated with the CXCL10-neutralizing antibody have shown that a lack of functional CXCL10 significantly decreases the number of CD8+ T-cells in the virally infected brain, resulting in persistence of WNV within the CNS and significantly higher mortality and morbidity compared with wild-type mice.32,99 Interestingly, the number of CD4+ T-cells was not significantly lower in infected CXCL10-deficient brains, suggesting that other chemokines mediate their entry to the CNS. Importantly, CXCL9-deficient mice are not more susceptible to WNV infection, suggesting that CXCL10 has a pivotal and nonredundant role in mediating antiviral immune responses to WNV infection of the CNS.99
Theiler’s murine encephalitis virus infection
TMEV belongs to the family of Picornaviridae. TMEV causes acute encephalitis in mice characterized by infection of glial cells, mainly astrocytes, and infiltration of mononuclear cells into the CNS.100 The acute phase is then followed by a chronic phase, characterized by chronic demyelination that leads to paralysis. In both acute and chronic phases of TMEV infection, CXCL10 is highly upregulated in mice regardless of the viral strain used.101–103 Macrophages and T-cells can also be a source of CXCL10 production following infiltration of these cells into the brain.74,75 Interestingly, the levels of CXCL10 expression correlated with the viral antigen and not with the number of T-cells in the CNS, suggesting that infected glial cells are mainly responsible for CXCL10 expression. Despite high CXCL10 expression in the CNS, neutralization of CXCL10 did not have an effect on disease severity, progression, or the pathology of TMEV infection in mice.103 Instead, CXCL9 may have more pivotal nonredundant roles during the pathogenesis of TMEV infection, and as such is distinct from the results obtained using MHV, LCMV, and WNV.104 Thus, it would seem that not all CNS RNA virus infections are alike in their requirement for CXCL10-mediated immune function. This suggests that the role of CXCL10 during neuroinflammation is more complex than might be assumed. The importance and involvement of different leukocyte populations in each of these neuroinflammatory models could account for the differential role that CXCL10 plays during brain inflammation.
Herpes virus infection of the CNS
HSV belongs to the family of Herpesviridae that can induce encephalitis in humans and mice. CXCL9 and CXCL10 are expressed at high levels in the CNS, primarily in microglia.105–108 Following HSV infection, CXCL10-deficient mice show higher viral loads in the CNS, decreased numbers of natural killer cells and CD8+ T-cells, and higher mortality.109 However, it should be noted that another study using CXCR3/CXCL10 double-deficient mice demonstrated that accumulation of CD8+ T-cells into the CNS was not impaired, making firm conclusions about CXCL10 involvement during HSV encephalitis difficult.109 However, other studies have suggested that CXCL10 in collaboration with CXCL9 are pivotal for the attraction of both natural killer cells and CD8+ T-cells into the spinal cord.110 Together, these studies suggest that CXCL9 and CXCL10 can have either synergistic or opposing functions when competing for CXCR3, depending on the context of infection.50
Cerebral malaria
Infection with Plasmodium falciparum, particularly in children, can lead to cerebral malaria, which is a common and severe complication of malarial infection.111 Infection of the brain with malarial parasites can result in severe neurocognitive impairments, coma, and death. This disease has been extensively studied in mice infected with Plasmodium berghei ANKA, which replicates many aspects of human disease, including an influx of mononuclear cells into the infected brain. In particular, CXCR3-expressing natural killer cells and CD8+ T-cells are thought to play important roles in the pathogenesis of this disease.112,113 After infection with P. berghei ANKA , CXCL10 mRNA and protein levels are localized to neurons and occasionally to endothelial cells of the blood–brain barrier,33,114 although detection in astrocytes has also been reported using in situ hybridization.115 In contrast, CXCL9 is predominantly expressed by neurovascular endothelial cells.33 In the absence of significant type I IFN expression, induction of these two chemokines is dependent upon IFN-γ signaling.114 In human studies of children in Ghana suffering cerebral malaria due to P. falciparum infection, CXCL10 levels were higher in the cerebrospinal fluid than in serum,116 and as such CXCL10 could be used as a putative biomarker for stratifying infected patients.
Studies using CXCL10-deficient mice demonstrated that mice were only partially protected from P. berghei ANKA infection, with a mortality rate of 30%–40% compared with 80%–100% in wild-type mice. A similar result was obtained using CXCL9-deficient mice.33 In contrast, CXCR3-deficient mice were completely protected from cerebral malaria.33,115 These data suggest that CXCL9 and CXCL10 play nonredundant roles in cerebral malaria and both chemokines are important for complete attraction of T-cells into the CNS. The differential source of CXCL9 and CXCL10 expression would explain their nonoverlapping roles in cerebral malaria. Together, these studies indicate that blockade of CXCR3 should be explored as a novel treatment for cerebral malaria.33
Trypanosome infection
Trypanosomes are unicellular parasitic protozoa that can infect a variety of animals and humans. Human African trypanosomiasis, also known as sleeping sickness, occurs in sub-Saharan Africa and is usually caused by subspecies of Trypanosoma brucei.117 After initial infection, the parasite remains in the blood and can then migrate into lymph and into the CNS where it can cause severe meningoencephalitis characterized by a high mortality rate due to a lack of efficient drugs available to treat trypanosomiasis.118 Similar to malarial infection of the brain, CXCL10 is consistently and highly upregulated in the cerebrospinal fluid of patients and also in mice, and this expression correlates with disease severity.119 Expression of CXCL10 occurs predominantly in astrocytes and is dependent on IFN-γ.93,115,120 Accordingly, CXCL10 levels in cerebrospinal fluid could be used as a biomarker for late-stage trypanosome infection.
Following infection, CXCL10-deficient mice show reduced morbidity, decreased infiltration of lymphocytes into the CNS, and lower mortality. However, the levels of parasitemia are similar in CXCL10-deficient and wild-type mice, suggesting that CXCL10 exerts its role by affecting neuroimmune processes and not by modulating parasite load. Further, histological analyses revealed that perivascular cuffing of trypanosomes and T-cells around cerebral capillaries were reduced in CXCL10-deficient mice compared with wild-type mice and fewer parasites could generally be detected in the brain parenchyma.119 Importantly, this implies that CXCL10-mediated inflammation facilitates not only the entry of leukocytes, but also further enhances the entry of parasites into the brain. Together, these data suggest that CXCL10 plays an important role in the pathogenesis of both malaria and trypanosome-induced encephalitis, has potential as a future biomarker for late-stage disease, and putatively may constitute a new drug target.121,122
Chemokine redundancy and targeting of CXCL10 during neuroinflammatory diseases
Chemokines belong to a complex system in which highly promiscuous ligands can potentially bind multiple receptors and in which receptors can bind multiple ligands. In the face of this complexity and apparent overlapping function, many have described the chemokine system as highly redundant. In support of this, the targeting of a specific chemokine-receptor axis for therapeutic purposes has proven to be particularly challenging. However, this viewpoint has been increasingly challenged, particularly in the context of drug discovery. This viewpoint suggests that redundancy is not the main feature of this system during many diseases.123 Instead, it has been suggested that many drug trials to date that have targeted chemokines or their receptors may have selected an inappropriate target, timed the intervention inappropriately, or administered ineffective doses of the drug. Thus, it is not surprising that most of these trials were unsuccessful in developing successful chemokine-based drugs. Arguably, once we are better informed about the hierarchies of chemokine function in specific disease contexts, it should be possible to develop specific chemokine-based therapies, and improved strategies of their application need to be considered.
It may be too much of an oversimplification to ask whether CXCL10 is a friend or foe during neuroinflammation. It is clear from the studies reported above that CXCL10 has an important nonredundant role in a number of CNS inflammatory contexts. However, the impact of modulating the function of CXCL10 is likely to be highly disease-specific and context-specific (Figure 2). Studies using MHV, WNV, and HSV as models of viral encephalitis have demonstrated a beneficial role of CXCL10 during CNS infection with regard to facilitating the elimination of virus-infected cells.32,85–87,110 In contrast, CXCL10-driven inflammation appears to be detrimental in encephalitis induced by LCMV, malaria, and trypanosomes.33,91,119 Further, CXCL10 is likely to play a role in the pathogenesis of MS, although the jury is still out on whether it is a pivotal player in this disease, and further studies are required to clarify this matter.81,82 Similarly, data on TMEV have also not yet shown a convincing role for CXCL10 in affecting the disease course.100 However, in summary, the consensus finding from studies on viral encephalitis indicates that if direct pathogen-induced damage can be reasonably tolerated without loss of neurological function, CXCL10-mediated inflammation is best suppressed to avoid unnecessary immune-driven pathology. This interpretation is likely to be highly pathogen-specific.
CXCL10 can be targeted either directly or through targeting of its receptor, CXCR3. Targeting CXCR3 has clear benefits in that it will also abrogate the effects of all three CXCR3 ligands.124 This is of particular relevance to cerebral malaria in which blockade of CXCL9 or CXCL10 alone leads to partial protection only, whereas blockade of CXCR3 confers full protection in mice.33 Therefore, in this review, we consider the use of drugs that target both CXCL10 and CXCR3.
There are various possibilities for targeting either chemokines or their receptors that include use of neutralizing antibodies, modified chemokines as agonists, and small molecule antagonists.124 Examples include a neutralizing antibody that binds to CXCL10, known as MDX1100, which has entered a Phase II clinical trial for the treatment of ulcerative colitis and rheumatoid arthritis.124 In addition, a CXCR3-blocking antibody has also been developed and used in a rat model of arthritic joint inflammation, where the antibody was capable of reducing leukocyte infiltration into the joint and prevented weight loss.125 To our knowledge, whilst CXCL10-neutralizing antibodies have been used experimentally to modulate CNS disease, they have yet to be used in clinical trials for the treatment of neuroinflammatory diseases.
Small molecule antagonists for CXCR3
CXCR3, like all chemokine receptors, are seven membrane-spanning G-protein coupled receptors. Due to the large number of small molecule antagonists and agonists available that can bind this class of receptor, there exists a realistic possibility that among these compounds there exists one that can successfully modulate receptor function. Indeed, since 2001, several small molecule antagonists against CXCR3 have emerged, primarily via high throughput screenings. Whether these compounds are able to cross the blood-brain barrier is not yet known; however, all drugs showed good bioavailability by entering the blood.
One of the best characterized CXCR3 antagonists is TAK-779, a nonpeptidergic antagonist that is capable of binding CCR2, CCR5, and CXCR3.126 However, upon further study it is now apparent that, whilst it is a potent CCR5 blocker, the CXCR3 binding profile of TAK-779 is limited and its use as a CXCR3 blocker has not been continued. Thus, the use of TAK-779 seems to be more appropriate in CCR5-dependent diseases such as human immunodeficiency virus infection.127
Amgen (Thousand Oaks, CA, USA) has identified several CXCR3-interacting compounds, including antagonists based on azaquinazolinone. Initially, most compounds had unacceptable pharmacokinetic properties and were not used for further studies. However, compound 12, also known as AMG 487 and the more active 4-F, and 3CF3 analog 13 (known as NBI-74330), have been studied extensively in both in vitro and animal models.128,129 Both compounds bind noncompetitively to CXCR3 and block the activity of this receptor. In a bleomycin-induced lung inflammation model, AMG 487 significantly reduced the infiltration of leukocytes into the lung. These preclinical studies finally resulted in the initiation of a Phase I clinical trial in 2003.124 This trial showed that the tolerance of AMG 487 was good in patients with psoriasis but unfortunately the drug did not show a significant therapeutic effect.130 In subsequent steps, Amgen replaced the azaquinazolinone core with an imidazole group and through several modification steps they obtained compound 21.131 This small molecule antagonist has been shown to bind specifically to CXCR3 and effectively inhibits binding of CXCL9, CXCL10, and CXCL11 to their receptors, and to significantly reduce bleomycin-induced lung inflammation.131 This exciting new compound has yet to be tested in clinical trials, although it should have wide applicability for diseases in which CXCR3 is known to be a dominant driver of inflammation. The original AMG 487 compound has since been further optimized to generate another potent CXCR3 inhibitor, known as compound 25.132 Like compound 21, compound 25 was tested in several in vitro and in vivo experiments, and both showed good oral bioavailability in animal models and as such are promising candidates for the development of CXCR3 blockers.
Schering-Plough (Kenilworth, NJ, USA) has filed a patent for a piperazinyl-piperidine-based CXCR3 antagonist, which shows exceptionally high affinity for CXCR3.124 However, no functional or in vivo data have been published to date. Other CXCR3 antagonists include compounds based on 1-aryl-3-piperidin-4-yl-urea (developed by Union Chimique Belge, Brussels, Belgium), 4-N-aryl-diazepanylurea (US Pharmacopeia), 2-iminobenzimidazoles (Abbott Laboratories, Abbott Park, IL, USA), bispiperidines (Janssen Pharmaceuticals, Inc., Titusville, NJ, USA) and ergolines (Hoffmann-La Roche, Basel, Switzerland).124 In summary, compounds showing high affinity for CXCR3 have arisen from a variety of different structural classes. A summary of CXCR3 small molecule antagonists is provided in Table 3. Next, it will be critical to determine whether these compounds have clear efficacy in modulating inflammation in vivo, and in which diseases CXCR3 function is detrimental. Thus, a better understanding of the exact role of CXCL10 and its receptor in mediating neuroinflammation will greatly aid in determining the feasibility of using these CXCR3 blockers to ameliorate disease.
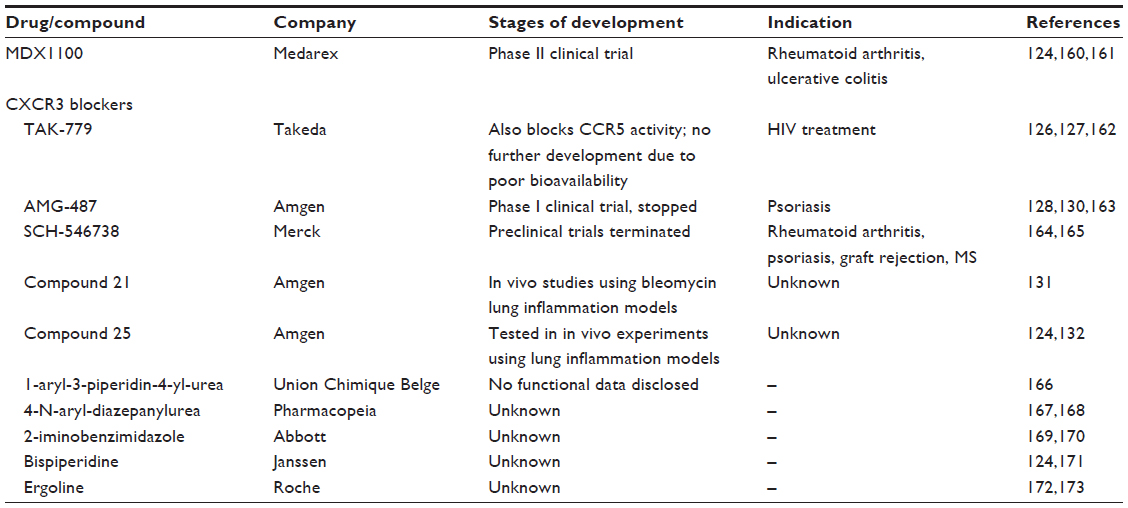 | Table 3 Summary of small molecule inhibitors that have been used to modulate CXCL10 function |
Conclusion
In this review we have highlighted the function of CXCL10 during many neuroinflammatory diseases. Interestingly, CXCL10 is highly expressed in a wide variety of these diseases. Reports that have studied both human clinical samples and animal models have revealed that CXCL10 is a potent and sometimes nonredundant chemoattractant of both T-cells and natural killer cells into the CNS. The ability of CXCL10 to mediate either beneficial or detrimental processes during neuroinflammation appears to be highly context-specific and disease-specific. In some situations, CXCL10 appears to have a highly beneficial role, eg, in WNV infection, in which CXCR3-expressing effector T-cells enter the CNS and clear the virus. Alternatively, in other diseases, CXCL10-mediated inflammation can result in an inflammatory infiltrate that triggers a damaging and sometimes counterproductive response, eg, during trypanosomiasis. These apparent contradictory functions remain to be fully clarified. However, at least in the case of CNS infections, the ability of CXCL10 to mediate processes beneficial to the host is most likely dependent on how well the CNS can tolerate infection without obvious loss of function, eg, in LCMV infection where T-cell entry is associated with a worse outcome for the host. In the case of sterile neuroinflammation, the net result of CXCL10 function is likely to be more damaging than protective, although it is not yet clear whether, in these noninfectious diseases, the CXCL10-CXCR3 chemotactic axis is the most relevant target for modulating CNS leukocyte influx, compared with other chemokine axes. Nonetheless, despite these uncertainties, it is clear that CXCL10 plays a central role in mediating the migration of several important leukocyte subsets. Accordingly, pharmaceutical companies have developed several promising small molecule antagonists that effectively bind and block the function of CXCL10 or its receptor CXCR3. Further research that clarifies the positioning of CXCL10 in the hierarchy of CNS chemokine function will ultimately determine the validity of targeting this important chemotactic axis in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
Eriksson P, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4(11):1313–1317. | |
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002; 415(6875):1030–1034. | |
Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28(1):12–18. | |
Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12(9):623–635. | |
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200(6):629–638. | |
Ransohoff RM. The chemokine system in neuroinflammation: an update. J Infect Dis. 2002;186 Suppl 2:S152–S156. | |
Zlotnik A, Yoshie O. Chemokines: a new classification review system and their role in immunity. Immunity. 2000;12:121–127. | |
Vanguri P. Interferon-gamma-inducible genes in primary glial cells of the central nervous system: comparisons of astrocytes with microglia and Lewis with brown Norway rats. J Neuroimmunol. 1995;56(1):35–43. | |
Cole KE, Strick CA, Paradis TJ, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187(12):2009–2021. | |
Nomiyama H, Osada N, Yoshie O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 2010;21(4):253–262. | |
Patel L, Charlton SJ, Chambers JK, Macphee CH. Expression and functional analysis of chemokine receptors in human peripheral blood leukocyte populations. Cytokine. 2001;14(1):27–36. | |
Belperio JA, Keane MP, Arenberg DA, et al. CXC chemokines in angiogenesis. J Leukoc Biol. 2000;68(1):1–8. | |
Doitsidou M, Reichman-Fried M, Stebler J, et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 2002;111(5):647–659. | |
Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7(12):243. | |
Luster AD, Jhanwar SC, Chaganti RS, Kersey JH, Ravetch JV. Interferon-inducible gene maps to a chromosomal band associated with a (4;11) translocation in acute leukemia cells. Proc Natl Acad Sci U S A. 1987;84(9):2868–2871. | |
Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315(6021):672–676. | |
Majumder SS, Zhou LZ, Chaturvedi PP, Babcock GG, Aras SS, Ransohoff RM. p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J Immunol. 1998;161(9):4736–4744. | |
Ohmori Y, Hamilton TA. Cooperative interaction between interferon (IFN) stimulus response element and kappa B sequence motifs controls IFN gamma-and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J Biol Chem. 1993;268(9):6677–6688. | |
Farber JM. Mig and IP-10: CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61(3):246–257. | |
Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86(6):2900–2910. | |
Medoff BD, Wain JC, Seung E, et al. CXCR3 and its ligands in a murine model of obliterative bronchiolitis: regulation and function. J Immunol. 2006;176(11):7087–7095. | |
Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J Exp Med. 2001;193(8):975–980. | |
Gasper NA, Petty CC, Schrum LW, Marriott I, Bost KL. Bacterium-induced CXCL10 secretion by osteoblasts can be mediated in part through toll-like receptor 4. Infect Immun. 2002;70(8):4075–4082. | |
Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J Exp Med. 1987;166(4):1084–1097. | |
McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun. 2010;394(4):1006–1011. | |
Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108(1–2):227–235. | |
Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96(12):6873–6878. | |
Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol. 2013;87(6):3382–3392. | |
Sørensen TL, Trebst C, Kivisäkk P, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. 2002;127(1–2):59–68. | |
McKimmie CS, Roy D, Forster T, Fazakerley JK. Innate immune response gene expression profiles of N9 microglia are pathogen-type specific. J Neuroimmunol. 2006;175(1–2):128–141. | |
Cheeran MC, Hu S, Sheng WS, Peterson PK, Lokensgard JR. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J Virol. 2003;77(8):4502–4515. | |
Klein RS, Lin E, Zhang B, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79(17):11457–11466. | |
Campanella GSV, Tager AM, El Khoury JK, et al. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A. 2008;105(12):4814–4819. | |
Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28(11):3696–3705. | |
Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746–754. | |
Loetscher P, Pellegrino A, Gong JH, et al. The ligands of CXC chemokine receptor 3, I-TAC, Mig, and IP10, are natural antagonists for CCR3. J Biol Chem. 2001;276(5):2986–2991. | |
Cox MA, Jenh CH, Gonsiorek W, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59(4):707–715. | |
Colvin RA, Campanella GSV, Manice LA, Luster AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26(15):5838–5849. | |
Strieter RM, Kunkel SL, Arenberg DA, Burdick MD, Polverini PJ. Interferon gamma-inducible protein 10 (IP-10), a member of the C-X-C chemokine family, is an inhibitor of angiogenesis. Biochem Biophys Res Commun. 1995;210(1):51–57. | |
Angiolillo AL, Sgadari C, Taub DD, et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182:155–162. | |
Cole AM, Ganz TT, Liese AM, Burdick MD, Liu LL, Strieter RM. Cutting edge: IFN-inducible ELR- CXC chemokines display defensin-like antimicrobial activity. J Immunol. 2001;167(2):623–627. | |
Pertl U, Luster AD, Varki NM, et al. IFN-gamma-inducible protein-10 is essential for the generation of a protective tumor-specific CD8 T cell response induced by single-chain IL-12 gene therapy. J Immunol. 2001;166(11):6944–6951. | |
Groom JR, Richmond J, Murooka TT, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37(6):1091–1103. | |
Sung JH, Zhang H, Moseman EA, et al. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell. 2012;150(6):1249–1263. | |
Christensen JE, Nansen A, Moos T, et al. Efficient T-cell surveillance of the CNS requires expression of the CXC chemokine receptor 3. J Neurosci. 2004;24(20):4849–4858. | |
Yates CC, Whaley D, Kulasekeran P, et al. Delayed and deficient dermal maturation in mice lacking the CXCR3 ELR-negative CXC chemokine receptor. Am J Pathol. 2007;171(2):484–495. | |
Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207–215. | |
Jacobs AH, Tavitian B. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32(7):1393–1415. | |
Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–439. | |
Sotelo J, Corona T. Varicella zoster virus and relapsing remitting multiple sclerosis. Mult Scler Int. 2011;2011(3):1–5. | |
Lunemann JD, Kamradt T, Martin R, Munz C. Epstein-Barr virus: environmental trigger of multiple sclerosis? J Virol. 2007;81(13):6777–6784. | |
Simmons A. Herpesvirus and multiple sclerosis. Herpes. 2001;8(3):60–63. | |
Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358(9278):261–264. | |
Gomez-Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491–4500. | |
Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. | |
Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812–818. | |
Saruhan-Direskeneli G, Yentur SP, Akman-Demir G, Isik N, Serdaroglu P. Cytokines and chemokines in neuro-Behcet’s disease compared with multiple sclerosis and other neurological diseases. J Neuroimmunol. 2003;145(1–2):127–134. | |
Sui Y, Stehno-Bittel L, Li S, et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23(4):957–964. | |
Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207(1–2):92–100. | |
Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 2004;156(1–2):74–87. | |
Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. | |
Head MW, Bunn TJR, Bishop MT, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991–2002. Ann Neurol. 2004;55(6):851–859. | |
Brown AR, Rebus S, McKimmie CS, Robertson K, Williams A, Fazakerley JK. Gene expression profiling of the preclinical scrapie-infected hippocampus. Biochem Biophys Res Commun. 2005;334(1):86–95. | |
Tribouillard-Tanvier D, Striebel JF, Peterson KE, Chesebro B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J Virol. 2009;83(21):11244–11253. | |
Riemer C, Schultz J, Burwinkel M, et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3-/- mice. J Virol. 2008;82(24):12464–12471. | |
Müller M, Carter S, Hofer MJ, Campbell IL. Review: the chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity – a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36(5):368–387. | |
Hsieh M-F, Lai S-L, Chen J-P, et al. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J Immunol. 2006;177(3):1855–1863. | |
Christensen JE, de Lemos C, Moos T, Christensen JP, Thomsen AR. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol. 2006;176(7):4235–4243. | |
Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol. 2004;25(3):132–137. | |
Sorensen TL, Tani M, Jensen J, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103(6):807–815. | |
Kivisakk P, Trebst C, Liu Z, et al. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin Exp Immunol. 2002;129(3):510–518. | |
El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5(2):189–197. | |
Franciotta D, Martino G, Zardini E, et al. Serum and CSF levels of MCP-1 and IP-10 in multiple sclerosis patients with acute and stable disease and undergoing immunomodulatory therapies. J Neuroimmunol. 2001;115(1–2):192–198. | |
Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of the interferon-gamma-inducible chemokines IP-10 and Mig and their receptor, CXCR3, in multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2000;26(2):133–142. | |
Biddison WE, Cruikshank WW, Center DM, Pelfrey CM, Taub DD, Turner RV. CD8+ myelin peptide-specific T cells can chemoattract CD4+ myelin peptide-specific T cells: importance of IFN-inducible protein 10. J Immunol. 1998;160(1):444–448. | |
Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129(8):1953–1971. | |
Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol. 2005;58(6):939–945. | |
Ransohoff RM, Hamilton TA, Tani M, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7(6):592–600. | |
Fife BT, Kennedy KJ, Paniagua MC, et al. CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2001;166(12):7617–7624. | |
Glabinski AR, Tani M, Tuohy VK, Tuthill RJ, Ransohoff RM. Central nervous system chemokine mRNA accumulation follows initial leukocyte entry at the onset of acute murine experimental autoimmune encephalomyelitis. Brain Behav Immun. 1995;9(4):315–330. | |
Wildbaum G, Netzer N, Karin N. Plasmid DNA encoding IFN-gamma-inducible protein 10 redirects antigen-specific T cell polarization and suppresses experimental autoimmune encephalomyelitis. J Immunol. 2002;168(11):5885–5892. | |
Narumi S, Kaburaki T, Yoneyama H, Iwamura H, Kobayashi Y, Matsushima K. Neutralization of IFN-inducible protein 10/CXCL10 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2002;32(6):1784–1791. | |
Klein RS, Izikson L, Means T, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172(1):550–559. | |
Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, Buchmeier MJ. Dynamic regulation of α-and β-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol. 1998;160(2):970–978. | |
Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168(7):3195–3204. | |
Stiles LN, Hosking MP, Edwards RA, Strieter RM, Lane TE. Differential roles for CXCR3 in CD4+ and CD8+ T cell trafficking following viral infection of the CNS. Eur J Immunol. 2006;36(3):613–622. | |
Liu MT, Armstrong DD, Hamilton TA, Lane TE. Expression of Mig (monokine induced by interferon-gamma) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J Immunol. 2001;166(3):1790–1795. | |
Christoffersen PJ, Volkert M, Rygaard J. Immunological unresponsiveness of nude mice to LCM virus infection. Acta Pathol Microbiol Scand C. 1976;84C(6):520–523. | |
Leist TP, Cobbold SP, Waldmann H, Aguet M, Zinkernagel RM. Functional analysis of T lymphocyte subsets in antiviral host defense. J Immunol. 1987;138(7):2278–2281. | |
Kim JV, Kang SS, Dustin ML, McGavern DB. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2008;457(7226):191–195. | |
Christensen JE, Simonsen S, Fenger C, et al. Fulminant lymphocytic choriomeningitis virus-induced inflammation of the CNS involves a cytokine-chemokine-cytokine-chemokine cascade. J Immunol. 2009;182(2):1079–1087. | |
Asensio VC, Campbell IL. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71(10):7832–7840. | |
Hofer MJ, Carter SL, Müller M, Campbell IL. Unaltered neurological disease and mortality in CXCR3-deficient mice infected intracranially with lymphocytic choriomeningitis Virus-Armstrong. Viral Immunol. 2008;21(4):425–433. | |
Fung-Leung WP, Kündig TM, Zinkernagel RM, Mak TW. Immune response against lymphocytic choriomeningitis virus infection in mice without CD8 expression. J Exp Med. 1991;174(6):1425–1429. | |
Andersen IH, Marker O, Thomsen AR. Breakdown of blood-brain barrier function in the murine lymphocytic choriomeningitis virus infection mediated by virus-specific CD8+ T cells. J Neuroimmunol. 1991;31(2):155–163. | |
Storm P, Bartholdy C, Sørensen MR, Christensen JP, Thomsen AR. Perforin-deficient CD8+ T cells mediate fatal lymphocytic choriomeningitis despite impaired cytokine production. J Virol. 2006;80(3):1222–1230. | |
Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol. 2003;171(12):6838–6845. | |
Wang Y, Lobigs M, Lee E, Müllbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003;77(24):13323–13334. | |
Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180(4):2641–2649. | |
Tsunoda I, Lane TE, Blackett J, Fujinami RS. Distinct roles for IP-10/CXCL10 in three animal models, Theiler’s virus infection, EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP-10. Mult Scler. 2004;10(1):26–34. | |
Theil DJ, Tsunoda I, Libbey JE, Derfuss TJ, Fujinami RS. Alterations in cytokine but not chemokine mRNA expression during three distinct Theiler’s virus infections. J Neuroimmunol. 2000;104(1):22–30. | |
Hoffman LM, Fife BT, Begolka WS, Miller SD, Karpus WJ. Central nervous system chemokine expression during Theiler’s virus-induced demyelinating disease. J Neurovirol. 1999;5(6):635–642. | |
Ransohoff RM, Wei T, Pavelko KD, Lee JC, Murray PD, Rodriguez M. Chemokine expression in the central nervous system of mice with a viral disease resembling multiple sclerosis: roles of CD4+ and CD8+ T cells and viral persistence. J Virol. 2002;76(5):2217–2224. | |
Ure DR, Lane TE, Liu MT, Rodriguez M. Neutralization of chemokines RANTES and MIG increases virus antigen expression and spinal cord pathology during Theiler’s virus infection. Int Immunol. 2005;17(5):569–579. | |
Wickham S, Lu B, Ash J, Carr DJ. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol. 2005;162(1–2):51–59. | |
Carr DJ, Tomanek L. Herpes simplex virus and the chemokines that mediate the inflammation. Curr Top Microbiol Immunol. 2006;303: 47–65. | |
Lokensgard JR, Hu S, Sheng W, et al. Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol. 2001;7(3):208–219. | |
Sui Y, Potula R, Dhillon N, et al. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol. 2004;164(5):1557–1566. | |
Wuest TR, Carr DJ. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol. 2008;181(11):7985–7993. | |
Thapa M, Welner RS, Pelayo R, Carr DJ. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J Immunol. 2008;180(2):1098–1106. | |
Hunt NH, Golenser J, Chan-Ling T, et al. Immunopathogenesis of cerebral malaria. Int J Parasitol. 2006;36(5):569–582. | |
Nitcheu J, Bonduelle O, Combadiere C, et al. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol. 2003;170(4):2221–2228. | |
Hansen DS, Bernard NJ, Nie CQ, Schofield L. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol. 2007;178(9):5779–5788. | |
Van den Steen PE, Deroost K, Van Aelst I, et al. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur J Immunol. 2008;38(4):1082–1095. | |
Miu J, Mitchell AJ, Müller M, et al. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J Immunol. 2008;180(2):1217–1230. | |
Armah HB, Wilson NO, Sarfo BY, et al. Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J. 2007;6:147. | |
Uzureau P, Uzureau S, Lecordier L, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. 2013;501(7467):430–434. | |
Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113(4):496–504. | |
Amin DN, Rottenberg ME, Thomsen AR, et al. Expression and role of CXCL10 during the encephalitic stage of experimental and clinical African trypanosomiasis. J Infect Dis. 2009;200(10):1556–1565. | |
Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes forCxcl9 andCxcl10 is dependent on IFN-γ but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microgliain vitro. Glia. 2007;55(16):1728–1739. | |
Tiberti N, Matovu E, Hainard A, et al. New biomarkers for stage determination in Trypanosoma brucei rhodesiense sleeping sickness patients. Clin Transl Med. 2013;2(1):1. | |
Hainard A, Tiberti N, Robin X, et al. A combined CXCL10, CXCL8 and H-FABP panel for the staging of human African trypanosomiasis patients. PLoS Negl Trop Dis. 2009;3(6):e459. | |
Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11(5):355–363. | |
Wijtmans M, Verzijl D, Leurs R, de Esch IJ, Smit MJ. Towards small-molecule CXCR3 ligands with clinical potential. Chem Med Chem. 2008;3(6):861–872. | |
Mohan K, Issekutz TB. Blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol. 2007;179(12):8463–8469. | |
Gilliam BL, Riedel DJ, Redfield RR. Clinical use of CCR5 inhibitors in HIV and beyond. J Transl Med. 2011;9 Suppl 1:S9. | |
Gao P, Zhou X-Y, Yashiro-Ohtani Y, et al. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol. 2003;73(2):273–280. | |
Tonn GR, Wong SG, Wong SC, et al. An inhibitory metabolite leads to dose- and time-dependent pharmacokinetics of (R)-N- (3-(4-Ethoxy-phenyl)-4-oxo-3,4-dihydro-pyrido (2,3-d)pyrimidin-2-yl)-ethyl)-N-pyridin-3-yl-methyl-2-(4-trifluoromethoxy-phenyl)-acetamide (AMG 487) in human subjects after multiple dosing. Drug Metab Dispos. 2009;37(3):502–513. | |
Jopling LA, Watt GF, Fisher S, Birch H, Coggon S, Christie MI. Analysis of the pharmacokinetic/pharmacodynamic relationship of a small molecule CXCR3 antagonist, NBI-74330, using a murine CXCR3 internalization assay. Br J Pharmacol. 2007;152(8):1260–1271. | |
Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8(1):23–33. | |
Du X, Gustin DJ, Chen X, et al. Imidazo-pyrazine derivatives as potent CXCR3 antagonists. Bioorg Med Chem Lett. 2009;19(17):5200–5204. | |
Chen X, Mihalic J, Deignan J, et al. Discovery of potent and specific CXCR3 antagonists. Bioorg Med Chem Lett. 2012;22(1):357–362. | |
Campbell GL, Hills SL, Fischer M, et al. Estimated global incidence of Japanese encephalitis: a systematic review. Bull World Health Organ. 2011;89(10):766–774, 774A–774E. | |
Centers for Disease Control. West Nile virus disease and other arboviral diseases – United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61(27):510–514. | |
Samuel MA, Diamond MS. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80(19):9349–9360. | |
Schnell MJ, McGettigan JP, Wirblich C, Papaneri A. The cell biology of rabies virus: using stealth to reach the brain. Nat Rev Micro. 2010;8(1):51–61. | |
Warrell M. Rabies and African bat lyssavirus encephalitis and its prevention. Int J Antimicrob Agents. 2010;36:S47–S52. | |
Sabah M, Mulcahy J, Zeman A. Herpes simplex encephalitis. BMJ. 2012;344:e3166. | |
Banatvala JE. Herpes simplex encephalitis. Lancet Infect Dis. 2011; 11(2):80–81. | |
Jain M, Duggal S, Chugh TD. Cytomegalovirus infection in non-immunosuppressed critically ill patients. J Infect Dev Ctries. 2011;5(8):571–579. | |
Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010;20(4):202–213. | |
Obregon R, Chitnis K, Morry C, et al. Achieving polio eradication: a review of health communication evidence and lessons learned in India and Pakistan. Bull World Health Organ. 2009;87(8):624–630. | |
Gammelgaard LK, Colding H, Hartzen SH, Penkowa M. Meningococcal disease and future drug targets. CNS Neurol Disord Drug Targets. 2011;10(1):140–145. | |
Tzeng YL, Stephens DS. Epidemiology and pathogenesis of Neisseria meningitidis. Microbes Infect. 2000;2(6):687–700. | |
Fitch MT, van de Beek D. Emergency diagnosis and treatment of adult meningitis. Lancet Infect Dis. 2007;7(3):191–200. | |
Halperin JJ. Nervous system Lyme disease: is there a controversy? Semin Neurol. 2011;31(3):317–324. | |
O’Connell S. Lyme borreliosis: current issues in diagnosis and management. Curr Opin Infect Dis. 2010;23(3):231–235. | |
Idro R, Jenkins NE, Newton CR. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 2005;4(12):827–840. | |
Shikani HJ, Freeman BD, Lisanti MP, Weiss LM, Tanowitz HB, Desruisseaux MS. Cerebral malaria: we have come a long way. Am J Pathol. 2012;181(5):1484–1492. | |
Kennedy PG. Diagnostic and neuropathogenesis issues in human African trypanosomiasis. Int J Parasitol. 2006;36(5):505–512. | |
Keita M, Bouteille B, Enanga B, Vallat JM, Dumas M. Trypanosoma brucei brucei: a long-term model of human African trypanosomiasis in mice, meningo-encephalitis, astrocytosis, and neurological disorders. Exp Parasitol. 1997;85(2):183–192. | |
Ross AG, McManus DP, Farrar J, Hunstman RJ, Gray DJ, Li Y-S. Neuroschistosomiasis. J Neurol. 2012;259(1):22–32. | |
Finsterer J, Auer H. Parasitoses of the human central nervous system. J Helminthol. 2013;87(3):257–270. | |
Feustel SM, Meissner M, Liesenfeld O. Toxoplasma gondii and the blood-brain barrier. Virulence. 2012;3(2):182–192. | |
Yan J, Huang B, Liu G, et al. Meta-analysis of prevention and treatment of toxoplasmic encephalitis in HIV-infected patients. Acta Trop. 2013;127(3):236–244. | |
Nie CQ, Bernard NJ, Norman MU, et al. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog. 2009;5(4):e1000369. | |
Khan IA, MacLean JA, Lee FS, et al. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12(5):483–494. | |
Lepej SZ, Rode OD, Jeren T, Vince A, Remenar A, Barsic B. Increased expression of CXCR3 and CCR5 on memory CD4+ T-cells migrating into the cerebrospinal fluid of patients with neuroborreliosis: the role of CXCL10 and CXCL11. J Neuroimmunol. 2005;163(1–2):128–134. | |
Giunti D, Borsellino G, Benelli R, et al. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J Leukoc Biol. 2003;73(5):584–590. | |
Suzuki K, Kawauchi Y, Palaniyandi SS, et al. Blockade of interferon-gamma-inducible protein-10 attenuates chronic experimental colitis by blocking cellular trafficking and protecting intestinal epithelial cells. Pathol Int. 2007;57(7):413–420. | |
Yellin M, Paliienko I, Balanescu A, et al. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1730–1739. | |
Baba M, Nishimura O, Kanzaki N, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 1999;96(10):5698–5703. | |
Johnson M, Li A-R, Liu J, et al. Discovery and optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett. 2007;17(12):3339–3343. | |
Jenh C-H, Cox MA, Cui L, et al. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC Immunol. 2012;13(1):2. | |
Kim SH, Anilkumar GN, Zawacki LG, et al. III. Identification of novel CXCR3 chemokine receptor antagonists with a pyrazinyl-piperazinyl-piperidine scaffold. Bioorg Med Chem Lett. 2011;21(23):6982–6986. | |
Allen DR, Bolt A, Chapman GA, et al. Identification and structure-activity relationships of 1-aryl-3-piperidin-4-yl-urea derivatives as CXCR3 receptor antagonists. Bioorg Med Chem Lett. 2007;17(3):697–701. | |
Cole AG, Stroke IL, Brescia M-R, et al. Identification and initial evaluation of 4-N-aryl-[1,4]diazepane ureas as potent CXCR3 antagonists. Bioorg Med Chem Lett. 2006;16(1):200–203. | |
Stroke IL, Cole AG, Simhadri S, et al. Identification of CXCR3 receptor agonists in combinatorial small-molecule libraries. Biochem Biophys Res Commun. 2006;349(1):221–228. | |
Hayes ME, Wallace GA, Grongsaard P, et al. Discovery of small molecule benzimidazole antagonists of the chemokine receptor CXCR3. Bioorg Med Chem Lett. 2008;18(5):1573–1576. | |
Hayes ME, Breinlinger EC, Wallace GA, et al. Lead identification of 2-iminobenzimidazole antagonists of the chemokine receptor CXCR3. Bioorg Med Chem Lett. 2008;18(7):2414–2419. | |
Bongartz J-P, Buntinx M, Coesemans E, Hermans B, Lommen GV, Wauwe JV. Synthesis and structure-activity relationship of benzetimide derivatives as human CXCR3 antagonists. Bioorg Med Chem Lett. 2008;18(21):5819–5823. | |
Thoma G, Baenteli R, Lewis I, et al. Special ergolines efficiently inhibit the chemokine receptor CXCR3 in blood. Bioorg Med Chem Lett. 2011;21(16):4745–4749. | |
Thoma G, Baenteli R, Lewis I, et al. Special ergolines are highly selective, potent antagonists of the chemokine receptor CXCR3: discovery, characterization and preliminary SAR of a promising lead. Bioorg Med Chem Lett. 2009;19(21):6185–6188. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.