Back to Journals » International Journal of General Medicine » Volume 14
ROHHAD (Rapid-onset Obesity with Hypoventilation, Hypothalamic Dysfunction, Autonomic Dysregulation) Syndrome—What Every Pediatrician Should Know About the Etiopathogenesis, Diagnosis and Treatment: A Review
Authors Lazea C ![]() , Sur L, Florea M
, Sur L, Florea M ![]()
Received 22 November 2020
Accepted for publication 31 December 2020
Published 29 January 2021 Volume 2021:14 Pages 319—326
DOI https://doi.org/10.2147/IJGM.S293377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Cecilia Lazea,1 Lucia Sur,1 Mira Florea2
1Department Pediatrics I, Emergency Pediatric Hospital, University of Medicine and Pharmacy “Iuliu Hatieganu”, Cluj-Napoca, Romania; 2Community Medicine Department, University of Medicine and Pharmacy “Iuliu Hatieganu”, Cluj-Napoca, Romania
Correspondence: Cecilia Lazea
Department Pediatrics I, Emergency Pediatric Hospital, University of Medicine and Pharmacy “Iuliu Hatieganu”, 68, Motilor Street, Cluj-Napoca 400370, Romania
Tel +40744353764
Fax +40264402539
Email [email protected]
Abstract: Rapid-onset obesity with hypoventilation, hypothalamic dysfunction, autonomic dysregulation (ROHHAD) syndrome is a rare disease with unknown and debated etiology, characterized by precipitous obesity in young children, hypoventilation and autonomic dysregulation with various endocrine abnormalities. Neuroendocrine tumors can be associated in more than half of the cases. This rare condition has a severe outcome because of high morbidity and mortality. We provide a comprehensive description of the etiopathogenetic theories of the disease, clinical presentation, diagnostic workup and treatment possibilities.
Keywords: obesity, hypoventilation, hypothalamic dysfunction, autonomic dysregulation
Introduction
Rapid onset obesity with hypoventilation, hypothalamic dysfunction and autonomic dysregulation (ROHHAD) syndrome is a rare disease first described by Fishman et al1 and renamed ROHHAD by Ize-Ludlow et al in 2007.2 The acronym ROHHAD describes the typical sequence of symptoms. This disease is characterized by early and rapid onset of obesity associated with hypoventilation, autonomic dysregulation and endocrine abnormalities. The association with tumors with neural crest origin has included the termination NET into the acronym (ROHHADNET).3–5
The clinical manifestations of ROHHAD syndrome overlap with those of congenital central hypoventilation syndrome (CCHS) and late-onset central hypoventilation syndrome (LO-CHS). Clear delineation from these entities is provided on genetic basis (mutation of the paired-like homeobox 2B gene PHOX2B which is present in patients with CCHS) and the absence of hypothalamic dysfunction in LO-CHS.6 The diagnosis of ROHHAD syndrome is challenging due to unknown etiology, absence of confirmatory tests and is made based on clinical presentation. The condition is characterized by high morbidity and mortality rates.4,5
Because of global epidemic of childhood obesity, it is very important for every pediatrician to recognize this condition early in order to avoid the complications and ensure a good quality of life for these patients.
Incidence
ROHHAD syndrome is a very rare disorder, about 100 cases being reported to date and it is considered a relatively new disease.4,5 Because of the explosion of the exogenous obesity in children worldwide, ROHHAD syndrome must be considered for differential diagnosis in obese children.
Etiopathogenesis
Three main etiopathogenetic hypothesis have been postulated: genetic, epigenetic, and autoimmune.
Genetic Theory
Genetic basis of ROHHAD syndrome has been extensively investigated and various studies ruled out the mutations in several candidate genes, including PHOX2B, the gene causative for congenital central hypoventilation syndrome and genes responsible for development and function of the hypothalamic, neuroendocrine and autonomic systems. Candidate genes studied and their role in the pathogenesis of ROHHAD syndrome are presented in Table 1.
 |
Table 1 Candidate Genes in ROHHAD Syndrome |
PHOX2B encodes a transcription factor that has an important role in the regulation of neural crest migration and development of the autonomic nervous system and is considered the disease-defining gene for CCHS. Similarly with ROHHAD syndrome, the CCHS phenotype includes autonomic nervous system dysregulation and endocrine abnormalities, which makes the clinical differentiation very difficult, but consideration of PHOX2B as a disease-defining gene for CCHS allows the genetic distinction between the two entities.2,6–9
Mutations in the gene of BDNF with roles in the neuronal development and the impairment of activation of its receptor TrkB were analyzed in patients with ROHHAD syndrome and obesity and no significant correlation was found.2,10,11
Variations of the genes with role in the development of the hypothalamus and autonomic nervous system such as 5-hydroxytryptamine receptor 1A (HTR1A), orthopedia (OTP), pituitary adenylate cyclase activating polypeptide (PACAP) were analyzed but they were not significantly correlated with ROHHAD syndrome.12 The absence of hypocretin-1 in the cerebrospinal fluid of a patient with ROHHAD syndrome and narcolepsy was reported, suggesting a causative relation, but other studies demonstrated the absence of mutations in HCRT, HCRTR1 and HCRTR2 genes in patients with ROHHAD.13–15 Mutations in NECDIN gene with role in respiratory rhythmogenesis and hypothalamic insufficiency and in ASCLI gene, required for the generation of ventral neuroendocrine neurons which acts as potential modifier gene of PHOX2B were not correlated with ROHHAD syndrome.16
A nonsense mutation was reported in the retinoic acid-induced 1 (RAI1) gene known to cause Smith–Magenis syndrome (SMS), in a patient with morbid obesity and clinical diagnosis of ROHHAD syndrome, suggesting a potential overlap with SMS.17
Epigenetic Theory
Epigenetic hypothesis is supported by report of discordant presentation of ROHHAD syndrome in monozygotic twins.15,22 Patwari et al reported a pair of monozygotic twin girls with concordant growth and development until eight years of age and characteristic features of ROHHAD syndrome appearance in the affected twin after this age.22 The authors highlighted the possibility of variation in the epigenomes of identical twins leading to discordance in phenotypes of twins. Barclay et al did not identify coding differences in the same pair of discordant monozygotic twins.15
Immunologic Theory
The immune-mediated pathogenesis has been suggested by several authors who reported patients with clinical presentation consisted to ROHHAD in whose cerebrospinal fluid analysis disclosed an intrathecal synthesis of oligoclonal bands and antihypothalamus and antipituitary antibodies were detected.23,24 Association with celiac disease, may suggest further evidence for immune-mediated etiology.25 Autoimmune-mediated process has been illustrated by the positive effect of the intensive immunosuppressive treatment (cyclophosphamide, rituximab, immunoglobulin and corticoids) for the neuropsychological function in four patients with ROHHAD syndrome and ganglioneuroblastoma. In these patients the autoimmune process was considered plausible, given that neuroblastoma is associated with autoimmune-mediated paraneoplastic syndromes, as opsoclonus-myoclonus syndrome.15,26,27 In another 15-old-year patient with ROHHAD syndrome with focal inflammation in the periaqueductal grey matter and hypothalamus, corticosteroids, immunoglobulins and mycophenolate mofetil long-term administration had a beneficial effect for the neuropsychological function and autonomic disorders.28 Autopsy findings in six children with ROHHAD syndrome revealed features of encephalitis characterized by lymphocytic infiltrate mainly perivascular, various distributing in the brain, suggesting also the immune mediated pathogenesis.29
Clinical Presentation
The onset of this disease ranges from 0 to 9 years, but the most common onset is in early childhood, at ages between two and four years, with hyperphagia and rapid weight gain. Children with ROHHAD syndrome usually have normal growth and development and good general health prior to onset of symptoms. Clinical presentation of these patients is variable in severity and timing.2,4,5 The clinical presentation is heterogenous, there are cases with marked endocrine involvement, while others exhibit marked behavioral disturbances.
Rapid Obesity
Rapid obesity in early childhood is often the first recognizable sign of the disease.
Hypothalamic Dysfunction
Hypothalamic dysfunction may manifest as growth hormone deficiency, diabetes insipidus, transient syndrome of inappropriate antidiuretic hormone secretion (SIADH), hypodipsia, central precocious puberty, premature adrenarche, amenorrhea, hypogonadotropic hypogonadism, hyperprolactinemia, hypothyroidism, corticotrophin deficiency, low or normal IGF1 level. Dysnatremia (hypernatremia and hyponatremia) may be present and is linked with impaired water balancing such as polydipsia or diabetes insipidus.4,5,30,31,34 These manifestations appear from months to years following the rapid-onset obesity.
Autonomic Dysregulation
Autonomic dysregulation may present as ophthalmologic abnormality, such as blurred vision, altered pupil response to light, strabismus, ptosis, altered perception of pain, gastrointestinal dysmotility with chronic constipation or diarrhea, bradycardia, neurogenic bladder, excessive sweating, thermal dysregulation (hypothermia, hyperthermia), cold hands and feet, livedo reticularis, pseudo Raynaud’s phenomenon, syncope, urinary incontinence, dysarthria.4,5,32–34
Behavioral Disorders
Behavioral change is the most common form of cognitive dysfunction and the symptoms include mood changes such as irritability and aggression, hyperactivity, fatigue, social withdrawal, poor school performance, intellectual disability, flat affect, hallucination, major depressive disorder, anxiety, attention deficit disorder, self-injurious behavior, obsessive-compulsive disorder, oppositional-defiant disorder, bipolar disorder, and psychosis.4,5,26–28,34
Neurologic Abnormalities
Neurologic abnormalities consist of seizure, blurring of consciousness, sleep disturbance, hypersomnolence, narcolepsy, developmental delay, developmental regression, gait disturbance, nystagmus, general weakness. Seizures may be related to episodes of hypoxemia due to inadequate ventilator support. Enlargement of the pituitary gland and generalized brain atrophy were also reported.4,5,34,35
Hypoventilation
Hypoventilation is the most life-threatening feature of ROHHAD syndrome because it can lead to cardiorespiratory arrest. Most of children with ROHHAD syndrome have obstructive sleep apnea, hypoxemia and hypercapnia at early ages, but in more severe cases hypoventilation can also occur while awake. The spectrum of sleep disorders breathing is completed by central sleep apnea, abnormal ventilatory response to carbon dioxide, nocturnal hypoventilation, and cyanotic episodes. Early recognition of respiratory abnormalities raises the index of suspicion of ROHHAD syndrome.36,37
Hypothyroidism, one of the most common associated endocrine disorders can influence the central ventilatory control based on decrease of oxygen consumption.2,4,5
Tumors of Neural Crest Origin
Approximately 40–56% of the patients with ROHHAD syndrome develop tumors of neural crest origin such as ganglioneuroma and ganglioneuroblastoma.3–5 These tumors are localized in the chest, abdomen or along the sympathetic nervous system chain. Hamartomatous masses with neural elements were also reported in one case.4 Most of the children recorded a short period of time (approximatively two years) between the onset of obesity and the diagnosis of neural crest origin tumor.5
Dysmorphic Features
Dysmorphic features as depressed nasal bridge, macrocephaly, anteverted nares and hypertelorism were also described in ROHHAD patients.32
Metabolic Disorders
Insulin resistance, impaired glucose intolerance, diabetes mellitus, hypertriglyceridemia and progressive fatty liver disease were reported in several cases.2,5,38
Other Clinical Manifestations
Other clinical manifestations may be fever, rash, enuresis, headache, edema, pulmonary hypertension, cough, renal failure, rectal prolapse secondary to dysregulation of the digestive system, scoliosis.4,5,33 Recurrent upper respiratory tract infections are reported in these children.39
Diagnosis
The diagnosis of ROHHAD syndrome is based on clinical presentation and clinical course and involves a cooperative consultation by specialists in the fields of pediatrics, pneumology, endocrinology, oncology, psychiatry, otorhinolaryngology, cardiology, surgery, nutrition, and psychology. There is no genetic testing available to diagnose this disorder.
The diagnosis is made based on the presence of following features: (1) rapid-onset obesity starting in early childhood and alveolar hypoventilation during sleep; (2) signs and symptoms of hypothalamic dysfunction and autonomic disturbances; and (3) exclusion of other condition causing similar features, such as congenital central hypoventilation syndrome. Rapid onset obesity and the most common endocrine disorders such as precocious puberty and hypothyroidism are very often the early recognizable signs. Sequential comprehensive evaluation is recommended for children with ROHHAD syndrome as the clinical presentation is very variable (Table 2).
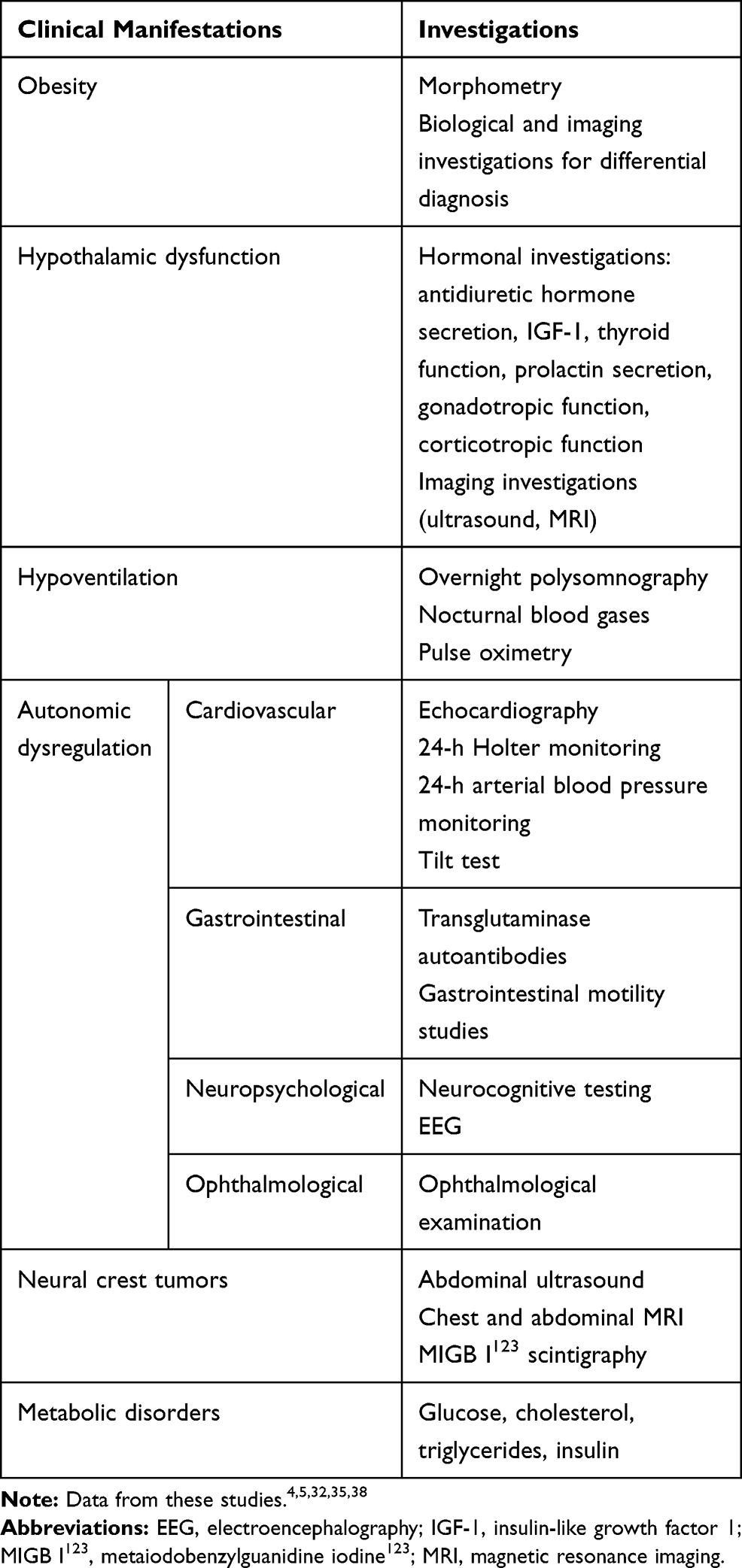 |
Table 2 Evaluation Workup in ROHHAD Syndrome |
Differential Diagnosis
Because of phenotypic similarities, the differential diagnosis of patients with ROHHAD syndrome, typically involves consideration of another disorder marked by early childhood obesity (Prader–Willi syndrome) and a rare disorder with breathing abnormalities and variable features of autonomic nervous system dysregulation (congenital central hypoventilation syndrome). Divergent and similar clinical features of the ROHHAD syndrome, Prader–Willi syndrome (PWS) and congenital central hypoventilation syndrome (CCHS) are presented in Table 3.
 |
Table 3 Clinic and Genetic Diagnostic Criteria in ROHHAD Syndrome, PWS and CCHS |
Treatment
Multidisciplinary care is crucial for the management of these patients, to optimize the quality of life. Another very active and important member of this team is the family. Early diagnosis and adequate conservative intervention are critical for optimizing the quality of life and neurocognitive outcome.
The treatment of ROHHAD syndrome is based on the clinical features (Table 4).
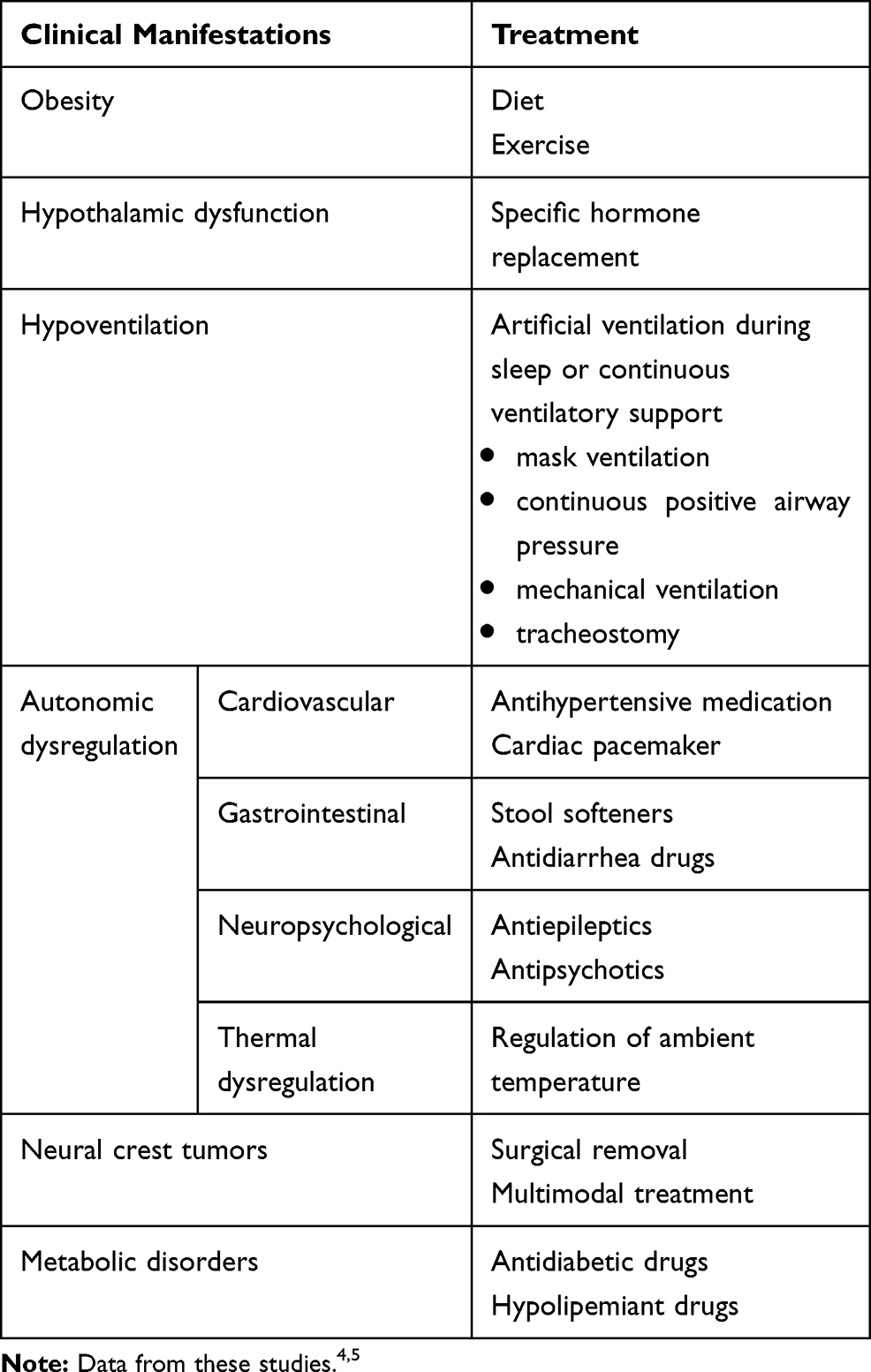 |
Table 4 Therapeutic Options in ROHHAD Syndrome |
The obesity control based on the strict caloric intake is difficult and requires the intervention of a nutritionist.
Moderate exertion is recommended and pulse oximetry monitoring is required during exercise.
Hypothalamic dysfunction is variable and the treatment may include a strict fluid intake regimen and specific hormone replacement.
Hypoventilation may need artificial ventilation during sleep in the first years of evolution with progressive need for continuous ventilatory support. These procedures, available at home, may improve the quality of life and prevent sudden death.4,5,36,37 Early recognition of sleep disorders breathing and targeted therapeutic interventions will limit morbidity and mortality associated with ROHHAD syndrome.36,37
Autonomic dysregulation may need various therapeutical interventions according to specific symptoms.
Neural crest tumors require surgical removal and multimodal treatment.4,5
Complications and Evolution
Insulin resistance, diabetes mellitus, hypertriglyceridemia, progressive fatty liver disease, metabolic syndrome, bradycardia, cor pulmonale, right ventricular hypertrophy, pulmonary hypertension, heart failure and scoliosis have been described in patients with ROHHAD syndrome.38,39,43,44
Mortality rate is high at 50–60%, due to hypoventilation, cardiopulmonary failure and cardiopulmonary arrest.4,5,36
Conclusions and Key Points
- ROHHAD syndrome is a relatively new disease with multisystemic involvement, with potentially severe evolution. Rapid-onset obesity associated with hypothalamic dysfunction and central hypoventilation are the clinical markers of the disease.
- The etiology of ROHHAD syndrome is still obscure, although genetic, epigenetic and immune-modulated etiopathogenetic theories were formulated.
- As there are not specific laboratory findings, the diagnosis is only supported by clinical criteria and the exclusion of CCHS based on the absence of PHOX2B gene mutation.
- Therapeutic options addressed to each clinical disturbance are supportive and involve a multidisciplinary team. Careful monitoring of these children is essential to limit morbidity and mortality associated with ROHHAD syndrome.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fishman LS, Samson JH, Sperling DR. Primary alveolar hypoventilation syndrome (Ondine’s curse). Am J Dis Child. 1965;110(2):155–161. doi:10.1001/archpedi.1965.02090030165011
2. Ize-Ludlow D, Gray JA, Sperling MA, et al. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation presenting in childhood. Pediatrics. 2007;120(1):e179–e188. doi:10.1542/peds.2006-3324
3. Bougneres P, Pantalone L, Linglart A, Rothenbuhler A, Le Stunff C. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab. 2008;93(10):3971–3980. doi:10.1210/jc.2008-0238
4. Lee J, Shin J, Kim S, et al. Rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation and neuroendocrine tumors (ROHHADNET) syndrome: a systematic review. Bio Med Res Int. 2018. doi:10.1155/2018/1250721.
5. Harvengt J, Gernay C, Mastouri M, Farhat N, Lebrethon M-C, Seghaye M-C M-C. ROHHAD(NET) syndrome: systematic review of the clinical timeline and recommendations for diagnosis and prognosis. J Clin Endocrinol Metab. 2020;105(7):1–13. doi:10.1210/clinem/dgaa247.
6. Cielo C, Marcus C. Central hypoventilation syndromes. Sleep Med Clin. 2014;9(1):105–118. doi:10.1016/j.jsmc.2013.10.005.
7. Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A. 2003;123A(3):267–278. doi:10.1002/ajmg.a.20527
8. Sasaki A, Kanai M, Kijima K, et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet. 2003;114(1):22–26. doi:10.1007/s00439-003-1036-z.
9. Patwari P, Carroll M, Rand C, Kumar R, Harper R, Weese-Mayer DE. Congenital central hypoventilation syndrome and the PHOX2B gene: A model of respiratory and autonomic dysregulation. Respir Physiol Neurobiol. 2010;173(3):322–335. doi:10.1016/j.resp.2010.06.013.
10. Sandrini L, Di Minno A, Amadio P, Ieraci A, Tremoli E, Barbieri S. Association between obesity and circulating brain-derived neurotrophic factor (BDNF) levels: systematic review of the literature and meta-analysis. Int J Mol Sci. 2018;19(8):2281. doi:10.3390/ijms19082281.
11. Pandit M, Behl T, Sachdeva M, Arora S. Role of brain neurotropic factor in obesity. Obes Med. 2020;17:100109. doi:10.1016/j.obmed.2020.100189.
12. Rand CM, Patwari PP, Rodikova EA, et al. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation: analysis of hypothalamic and autonomic candidate genes. Pediatr Res. 2011;70(4):375–378. doi:10.1203/PDR.0b013e318229474d
13. Dhondt K, Verloo P, Verlhest H, Van Coster R, Overeem S. Hypocretin- deficiency in a girl with ROHHAD syndrome. Pediatrics. 2013;132(3):e788–92. doi:10.1542/peds.2012-3225.
14. Barclay S, Rand C, Gray P, et al. Absence of mutations in HCRT, HCRTR1 and HCRTR2 in patients with ROHHAD. Resp Phys Neurobiol. 2015;221:59–63. doi:10.1016/j.resp.2015.11.002
15. Barclay S, Rand C, Borch L, et al. Rapid-Onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD): exome sequencing of trios, monozygotic twins and tumours. Orphanet J Rare Dis. 2015;10(1):103. doi:10.1186/s13023-015-0314-x.
16. De Pontual L, Trochet D, Caillat-Zucman S, et al. Delineation of late onset hypoventilation associated with hypothalamic dysfunction syndrome. Pediatr Res. 2008;64(6):689–694. doi:10.1203/PDR.0b013e318187dd0e
17. Thaker V, Esteves K, Towne M, et al. Whole Exome Sequencing Identifies RAI1 Mutation in a Morbidly Obese Child Diagnosed With ROHHAD Syndrome. J Clin Endocrinol Metab. 2015;100(5):1723–1730. doi:10.1210/jc.2014-4215
18. Larkin E, Patel S, Goodloe R, et al. A candidate gene study of obstructive sleep apnea in European Americans and African Americans. Am J Respir Crit Care Med. 2010;182(7):947–953. doi:10.1164/rccm.201002-0192OC
19. Tennese A, Gee C, Wevrick R. Loss of the Prader-Willi syndrome protein necdin causes defective migration, axonal outgrowth, and survival of embryonic sympathetic neurons. Dev Dyn. 2008;237(7):1935–1943. doi:10.1002/dvdy.21615.
20. Han JC. Rare Syndromes and Common Variants of the Brain-Derived Neurotrophic Factor Gene in Human Obesity. In: Tao Y-X, editor. Genetics of Monogenic and Syndromic Obesity. Vol. 140. Chennai: Academic Press; 2016:75–95.
21. Yeo G, Connie Hung -C-C, Rochford J, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7(11):1187–1189. doi:10.1038/nn1336.
22. Patwari P, Rand C, Berry-Kravis E, Ize-Ludlow D, Weese-Mayer D. Monozygotic twins discordant for ROHHAD phenotype.. Pediatrics. 2011;128(3):e711–e715. doi:10.1542/peds.2011-0155.
23. Sartori S, Priante E, Pettenazzo A, et al. Intrathecal synthesis of oligoclonal bands in rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome: new evidence supporting immunological pathogenesis. J Child Neurol. 2014;29(3):421–425. doi:10.1177/0883073812469050.
24. Giacomozzi C, Guaraldi F, Cambiaso P, et al. Anti-Hypothalamus and Anti-Pituitary Auto-antibodies in ROHHAD Syndrome: additional Evidence Supporting an Autoimmune Etiopathogenesis. Horm Res Paediatr. 2019;92(2):124–132. doi:10.1159/000499163.
25. Cemeroglu AP, Eng DS, Most LA, Stalsonburg C, Kleis L. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome and celiac disease in a 13-year-old girl: further evidence for autoimmunity? J Pediatr Endocrinol Metab. 2016;29(1):97–101. doi:10.1515/jpem-2015-0129.
26. Paz-Priel I, Cooke D, Chen A. Cyclophosphamide for Rapid-Onset Obesity, Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation Syndrome. J Pediatr. 2011;158(2):337–339. doi:10.1016/j.jpeds.2010.07.006.
27. Jacobson L, Rane S, McReynolds L, Steppan D, Chen A, Paz-Priel I. Improved Behavior and Neuropsychological Function in Children With ROHHAD After High-Dose Cyclophosphamide. Pediatrics. 2016;138(1):e20151080. doi:10.1542/peds.2015-1080.
28. Chow C, Fortier MV, Das L, et al. Rapid-Onset Obesity With Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD) Syndrome May Have a Hypothalamus–Periaqueductal Gray Localization. Pediatr Neurol. 2015;52(5):521–525. doi:10.1016/j.pediatrneurol.2014.11.019.
29. Gharial J, Ganesh A, Curtis C, et al. Neuroimaging and Pathology Findings Associated With Rapid Onset Obesity, Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD) Syndrome.. J Pediatr Hematol Oncol. 2020;11. doi:10.1097/MPH.0000000000001927.
30. Valea A, Silaghi CA, Ghervan CMV, et al. Morbid child obesity with possible ROHHADNET-ROHHAD syndrome. Case report. Acta Endocrinol. 2014;10:515–524. doi:10.4183/aeb.2014.515/.
31. Al-Harbi A, Al-Shamrani A, Al-Shawwa B. Rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysregulation in Saudi Arabia. Saudi Med J. 2016;37(11):1258–1260. doi:10.15537/smj.2016.11.15578.
32. Kot K, Moszczynska E, Lecka-Ambroziak A, Migdal M, Szalecki M. Zespół ROHHAD u 9-letniego chłopca — opis przypadku. Endokrynologia Polska. 2016;67(2):226–231. doi:10.5603/EP.a2016.0037.
33. Şiraz ÜG, Ökdemir D, Direk G, et al. ROHHAD syndrome, a rare cause of hypothalamic obesity: report of two cases. Journal of Clinical Research in Pediatric Endocrinology. 2018;10(4):382–386. doi:10.4274/jcrpe.0027.
34. Filippidou M, Petropoulou T, Botsa E, et al. ROHHAD syndrome – A still unrecognized cause of childhood obesity: report of three cases. J Pediatr Endocrinol Metab. 2020;33(10):1341–1348. doi:10.1515/jpem-2020-0111.
35. Aljabban L, Kassab L, Bakoura NA, Alsalka MF, Maksoud I. Rapid-onset obesity, hypoventilation, hypothalamic dysfunction, autonomic dysregulation and neuroendocrine tumor syndrome with a homogenous enlargement of the pituitary gland: a case report. J Med Case Rep. 2016;10(1):328. doi:10.1186/s13256-016-1116-z.
36. Reppucci D, Hamilton J, Yeh A, Katz S, Al-Saleh S, Narang I. ROHHAD syndrome and evolution of sleep disordered breathing. Orphanet J Rare Dis. 2016;11(1):106. doi:10.1186/s13023-016-0484-1.
37. Trang H, Samuels M, Ceccherini I, et al. Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet J Rare Dis. 2020;15(1):252. doi:10.1186/s13023-020-01460-2.
38. Eldin JAW, Tombayoglu D, Butz L, et al. Natural history of ROHHAD syndrome: development of severe insulin resistance and fatty liver disease over time. Clinical Diabetes and Endocrinology. 2019;5(1):9. doi:10.1186/s40842-019-0082-y.
39. Chew HB, Ngu LH, Keng WT. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation and autonomic dysregulation (ROHHAD): a case with additional features and review of the literature. BMJ Case Rep. 2011;2011(jan20 1):1–6. doi:10.1136/bcr.02.2010.2706
40. Barclay S, Rand C, Nguyen L, et al. ROHHAD and Prader-Willi syndrome (PWS): clinical and genetic comparison. Orphanet J Rare Dis. 2018;13(1):124. doi:10.1186/s13023-018-0860-0.
41. Patwari P, Wolfe L. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation: review and update. Curr Opin Pediatr. 2014;26(4):487–492. doi:10.1097/MOP.0000000000000118.
42. Charnay A, Antisdel-Lomaglio J, Zelko FA. Congenital central hypoventilation syndrome: neurocognition already reduced in preschool-aged children. Chest. 2016;149(3):809–815. doi:10.1378/chest.15-0402.
43. Stowe R, Afolabi‐Brown O. Pulmonary hypertension and chronic hypoventilation in ROHHAD syndrome treated with average-volume assured pressure support. Pediatric Invest. 2019;3(4):253–256. doi:10.1002/ped4.12168
44. Kocaay P, Siklar Z, Camtosun E, Kendirli T, Berberoğlu M. ROHHAD syndrome: reasons for diagnostic difficulties in obesity. J Clin Res Pediatric Endocrinol. 2014;6(4):254–257. doi:10.4274/jcrpe.1432
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.