Back to Journals » Drug Design, Development and Therapy » Volume 15
Roflumilast Attenuates Doxorubicin-Induced Cardiotoxicity by Targeting Inflammation and Cellular Senescence in Cardiomyocytes Mediated by SIRT1
Authors Zhang S, Wu P, Liu J, Du Y, Yang Z
Received 28 July 2020
Accepted for publication 19 November 2020
Published 11 January 2021 Volume 2021:15 Pages 87—97
DOI https://doi.org/10.2147/DDDT.S269029
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Sheng Zhang,1,2 Peng Wu,1 Jiabao Liu,1 Yingqiang Du,1 Zhijian Yang1
1Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, People’s Republic of China; 2Department of Cardiology, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu 213004, People’s Republic of China
Correspondence: Zhijian Yang
Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, No. 300, Guangzhou Road, Gulou District, Nanjing, Jiangsu, 210029, People’s Republic of China
Tel/Fax +86-25-52271000
Email [email protected]
Background and Purpose: Cardiotoxicity is an important side effect of the treatment of a malignant tumor with Doxorubicin. Currently, decreasing the dosage of Doxorubicin to alleviate the side effects on cardiac function is the common method to deal with the cardiotoxicity induced by Doxorubicin. The present study aims to investigate the therapeutic effects of Roflumilast on Doxorubicin-induced inflammation and cellular senescence, as well as the potential mechanism in H9c2 myocardial cells.
Methods: The injured cardiac cell model was established by incubation with 5 μmol/L Doxorubicin. MTT was used to evaluate the cell viability of treated H9c2 cardiac cells. The expression of 4-HNE was determined using an immunofluorescence assay. The gene expression levels of IL-17, IL-6, TNF-α, IL-4, PAI-1, p21, and SIRT1 were evaluated using qRT-PCR and the protein levels of Gpx4, PAI-1, p21, and SIRT1 were determined using Western blot analysis. Secretions of IL-17, IL-6, TNF-α, IL-4, CK-MB, and cTnI were measured using ELISA. Cellular senescence was assessed using SA-β-Gal staining. Si-RNA technology was used to knockdown the expression of SIRT1 in H9c2 cardiac cells.
Results: Cell viability of H9c2 cardiac cells was significantly inhibited by Doxorubicin but rescued by Roflumilast. The upregulated 4-HNE and downregulated Gpx4 were reversed by Roflumilast. The secretions of IL-6 and IL-17 were promoted by Doxorubicin and suppressed by Roflumilast. The increased SA-β-Gal staining induced by Doxorubicin was inhibited by Roflumilast. P21 and PAI-1 were significantly upregulated and SIRT1 was greatly downregulated by Doxorubicin, all of which were reversed by Roflumilast. The anti-senescent effect of Roflumilast was abolished by knocking down SIRT1.
Conclusion: Roflumilast might attenuate Doxorubicin-induced inflammation and cellular senescence in cardiomyocytes by upregulating SIRT1.
Keywords: roflumilast, doxorubicin, SIRT1, cellular senescence, chronic heart failure
Introduction
In recent years, cancer therapeutics have made great improvements. As chemotherapy continues to improve, the adverse side effects associated with anticancer treatments have increasingly been recognized as an obvious problem. We are now aware that chemotherapy-induced cardiomyopathy can cause heart problems for cancer patients. A clinical study shows that chemotherapy can cause cardiotoxicity years after the treatment, resulting in hypertension, heart failure, and left ventricular dysfunction.1 The study estimates cancer patients have an average 2~6-fold high risk of mortality due to cardiovascular diseases.2 Therefore, chemotherapy treatment strategies for cancer patients must consider the risk of cardiotoxicity and also monitor the patients’ responses.3 Researchers have been seeking the early-stage biomarkers of cardiotoxicity and exploring the mechanisms of chemotherapeutic agents. Anthracyclines are a class of chemotherapy drugs, which have been used in treating different types of cancer for many years. Doxorubicin is one of the most used anthracyclines. Since the late 1960s, Doxorubicin has been widely used as the chemotherapeutic for the treatment of malignant tumors. In the history of chemotherapy, Doxorubicin is regarded as a powerful anti-cancer agent by a separate dose or combination dose with other agents.4,5 However, with the broad application of Doxorubicin, several acute or chronic side effects are reported, which significantly impact the treatment of patients and the therapeutic effects of the agent. Bone marrow suppression, nausea, emesis, and arrhythmia are reported to be the main acute side effects of Doxorubicin. Chronic heart failure (CHF) is one of the chronic side effects induced by Doxorubicin and is mainly caused by irreversible cardiac damage.6,7 It is reported that excessive inflammation, oxidative stress, and cellular senescence are involved in the pathological mechanism underlying the side effects of Doxorubicin on heart function.8,9 Cellular senescence is one of the most important factors that induces cardiotoxicity and is reported to be activated by Doxorubicin through upregulating PAI-1, and p21, or downregulating SIRT1.10–12 Further investigation of the pathological mechanism is profitable for the treatment of cardiotoxicity induced by Doxorubicin clinically.
Roflumilast is a selective phosphodiesterase 4 (PDE-4) inhibitor, by which cellular cAMP could be degraded and inflammation could be alleviated.13,14 Roflumilast was approved by the US Food and Drug Administration (FDA) for treatment of severe chronic obstructive pulmonary disease (COPD) due to its promising effects on alleviating the airway edema induced by antigens,15,16 downregulating the expression of Mucin 5AC in the airway epithelial cells and reducing the production of the reactive oxygen species (ROS) in the neutrophils, pulmonary artery vascular smooth muscle cells, and airway epithelial cells.17,18 Previous studies have shown that cardiomyocyte apoptosis in CHF is caused by hypoxic injury, ischemia-reperfusion injury, and oxidative stress.19 cAMP has been reported to play an important role in regulating myocyte apoptosis. Both in vivo and in vitro studies have shown that elevation of intracellular cAMP mitigates apoptosis in cardiomyocytes and reduces mortality in acute myocardial infarction.20 Phosphodiesterase-4 (PDE4) inhibitors are able to block the hydrolysis of cAMP via inhibition of PDE4. PDE4 inhibitors have been considered as attractive approaches for novel anti-inflammatory drugs. Interestingly, it has been demonstrated that Roflumilast exerts a protective effect against nitric oxide (NO)-induced apoptosis in both of H9c2 cells and neonatal rat cardiomyocytes via both cAMP–PKA/CREB and Epac/Akt-dependent pathways.21 In the present study, the therapeutic effects of Roflumilast on damaged myocardial cells induced by Doxorubicin will be investigated to explore a potential therapeutic method for the clinical treatment of cardiotoxicity caused by Doxorubicin.
Materials and Methods
Cell Culture and Treatment
H9c2 myocardial cells were purchased from American Type Culture Collection (ATCC, USA). The myocardial cells were cultured in a DMEM medium supplied with 10% fetal bovine serum. All the cell cultures were maintained at 37°C with 5% CO2. The treatment reagent Doxorubicin and Roflumilast were purchased from Sigma (St. Louis, USA). H9c2 cells growing at 90% confluence were treated with 5 μmol/L Doxorubicin in the presence or absence of Roflumilast (1, 2.5, 5 μM) for 24 hours.
Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from the cells using an RNA Extraction Kit (Thermo Fisher Scientific, Waltham, USA). The extraction methods were performed according to the manufacturers’ instructions. The purified RNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, USA). A mixture of oligo(dT)s primer was used to reverse-transcribe the complementary DNA. For real-time PCR, SYBR Premix Ex TaqTM (Thermo Fisher Scientific, Waltham, USA) were mixed with target gene primers on an Applied Bio-Rad CFX96 Sequence Detection system (Genscript, Nanjing, China). The mRNA expression levels of IL-17, IL-6, PAI-1, p21, and SIRT1 were determined using the threshold cycle (Ct), and relative expression levels were calculated using the 2−ΔΔCt method. The expression levels of GAPDH were used as internal control. Three independent replicates were performed.
Western Blotting Assay
Total protein lysates were obtained from H9c2 cells using the Nuclear and Cytoplasmic Protein Extraction Kit (Thermo Fisher Scientific, Waltham, USA). Approximately 40 μg of proteins was separated on 12% SDS-polyacrylamide gel (SDS-PAGE), and the separated proteins in the gel were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, MA, USA). The blot was blocked with 5% non-fat dry milk in TBST (Tris-buffered saline/0.1% Tween-20, pH 7.4) for 1 hour at room temperature and incubated overnight with primary antibodies against GPX4 (1:1000) PAI-1 (1:1000), p21 (1:1000), SIRT1 (1:1000) and GAPDH (1:1000) (Abcam, Cambridge, MA). A horseradish peroxidase-conjugated antibody against rabbit IgG (1:5000, Abcam, Cambridge, MA) was used as a secondary antibody. Blots were reacted with the ECL reagents (Amersham Pharmacia Biotech, Inc, USA) and exposed to Tanon 5200-multi chemiluminescent imaging system to detect protein expression. Three independent assays were performed for each group.
Immunofluorescence
Cells were washed three times with a phosphate buffer solution (PBS). The cells were then incubated with primary rabbit anti-4-HNE (1:1000, Abcam, Cambridge, MA) antibody overnight at 4°C. Following three-time washes with PBS, cells were incubated with secondary Alexa Fluor488-conjugated anti-rabbit IgG (1:200, Abcam, Cambridge, MA) for 30 minutes at room temperature. The DAPI was added to counterstain the nucleus for 5 minutes and 50% glycerinum was used to block the medium. Stained cells were photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
SA-β-gal staining was performed according to the manufacturer’s protocol (Cell Signaling Technology, Boston, USA). Briefly, cells were washed with PBS and fixed using the fixative solution for 0.5 hours at room temperature and then incubated at 37°C overnight with the SA-β-gal staining solution. The stained senescent cells were photographed. The percentage of positively stained cells was calculated from five different view fields of each sample in three independent experiments.
siRNA Transfection Experiment
24 hours prior to the transfection, the cells were seeded in a 6-well culture plate. SIRT1 siRNAs were transfected into H9c2 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) or X-treme GENE HP DNA Transfection Reagents (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. 24 hours or 48 hours later, the cells were collected and subjected to further analysis. The assays were performed in triplicate samples.
Cellular Injury Assessment
H9c2 cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. The levels of cTnI and CK-MB released into the cell culture medium were measured using commercial ELISA kits from the Cusabio Technology LLC: rat cardiac troponin (cTnI) (#CSB-E08594r) and rat creatine kinase-muscle/brain (CK-MB)(#CSB-E14403r). Results were shown as the fold change of the vehicle group.
Statistical Analysis
Mean ± standard deviation (SD) was used to measure the variability in the data. GraphPad prism 6.0 (GraphPad Inc.) was used to analyze the data. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was utilized to compare the difference among different groups. P<0.05 was determined as a statistically significant difference between groups.
Results
Roflumilast Ameliorates Doxorubicin-Induced Cell Injury in H9c2 Cells
To investigate the effects of Roflumilast on the injured H9c2 cardiac cells induced by Doxorubicin, MTT assay was used to evaluate the proliferation ability of cells treated with different strategies. Firstly, different concentrations of Roflumilast (1, 2.5, 5 μM) were used for incubation with H9c2 cardiac cells to check if the impact of Roflumilast on the proliferation of H9c2 cardiac cells could be observed. As shown in Figure 1A, compared to the control, no significant difference in cell viability was observed on the H9c2 cardiac cells incubated with other concentrations of Roflumilast (1, 2.5, 5 μM). We then measured the effects of Roflumilast on the production of cAMP in H9c2 cells. It was shown that the administration of Roflumilast for 15 minutes increased intracellular cAMP concentrations in a dose-dependent manner (Figure 1B).
 |
Figure 1 Roflumilast increases concentrations of cyclic adenosine monophosphate (cAMP) in H9c2 cardiac cells. (A) Cells were treated with Roflumilast (1, 2.5, 5 μM) for 24 hours. Cell viability was measured using MTT assay; (B) Cells were treated with Roflumilast (1, 2.5, 5 μM) for 15 minutes. 200 µM dibutyryl cyclic adenosine monophosphate (db-cAMP) was used as a positive control. Concentrations of cAMP have been measured (*, **, ***, ****P<0.05, 0.01, 0.001, 0.0001 vs vehicle group). |
Subsequently, the cell viability and LDH release were determined after the H9c2 cardiac cells were incubated with 5 μmol/L Doxorubicin in the presence or absence of Roflumilast (1, 2.5, 5 μM) for 24 hours. As shown in Figure 2A and B, the cell viability was significantly inhibited by the introduction of Doxorubicin but was greatly promoted by Roflumilast at the concentrations of 2.5 and 5 μM. LDH release was promoted by Doxorubicin and was suppressed by Roflumilast at concentrations of 2.5 and 5 μM.
 |
Figure 2 Roflumilast ameliorates Doxorubicin-induced cell death in H9c2 cardiac cells. Cells were treated with 5 μmol/L Doxorubicin in the presence or absence of Roflumilast (1, 2.5, 5 μM) for 24 hours. (A) Cell viability and (B) Lactate dehydrogenase (LDH) release was assayed (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
To further examine the effects of Roflumilast against Doxorubicin-induced cell injury in H9c2 cardiac cells, the release of CK-MB and cTnI, two important biomarkers of cardiomyoblast injury, were measured. Results indicate that Doxorubicin treatment significantly increased the release of both CK-MB (Figure 3A) and cTnI (Figure 3B) but there were remarkably reduced by Roflumilast in a dose-dependent manner.
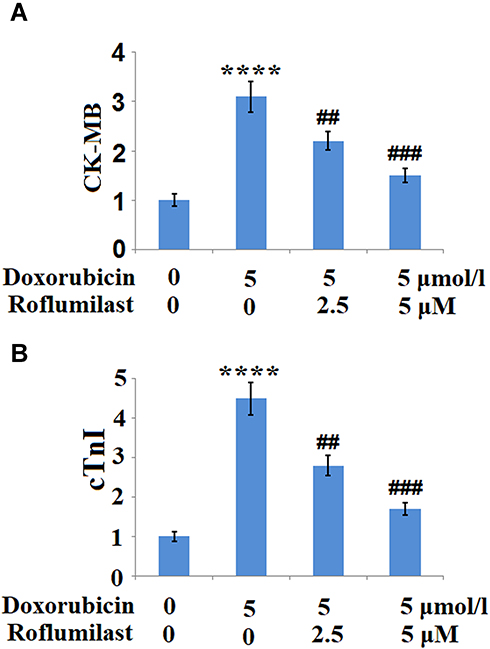 |
Figure 3 Roflumilast reduces the Doxorubicin-induced release of creatine kinase-muscle/brain (CK-MB) and cardiac troponin (cTnI). Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. (A) The release of CK-MB; (B) The release of cTnI (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
Roflumilast Alleviates Doxorubicin-Induced Oxidative Stress
H9c2 cardiac cells were treated with 5 μM Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours and the expressions of 4-HNE and GPX4 were evaluated to illustrate the effects of Roflumilast on the activated oxidative stress. As shown in Figure 4A, 4-HNE in the H9c2 cardiac cells was upregulated by the introduction of Doxorubicin but was significantly downregulated by Roflumilast. The inhibited expression level of GPX4 by Doxorubicin was significantly rescued by Roflumilast (Figure 4B).
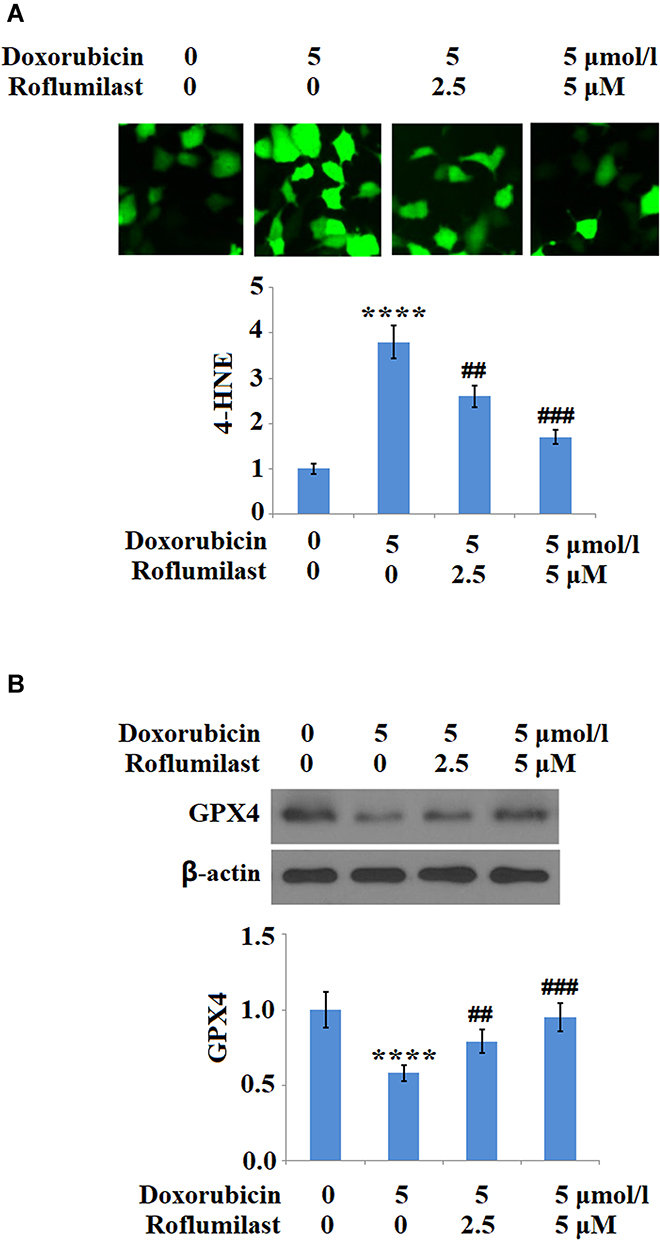 |
Figure 4 Roflumilast attenuates Doxorubicin-induced oxidative stress in H9c2 cardiac cells. Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. (A) Immunostaining of 4-Hydroxynonenal (4-HNE); (B) Expression of glutathione peroxidase 4 (GPX4) was measured by Western blot analysis (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
Roflumilast Inhibits Doxorubicin-Elicited Inflammation
The expressions of the pro-inflammatory cytokines IL-6, IL-17, TNF-α, and the anti-inflammatory cytokine IL-4 were monitored to evaluate the inflammatory response of H9c2 cardiac cells. As shown in Figure 5A–H, the increased concentrations of IL-6, IL-17, and TNF-α induced by Doxorubicin were significantly suppressed by the introduction of Roflumilast. In contrast, Doxorubicin reduced the expression of the anti-inflammatory cytokine IL-4, which was rescued by Roflumilast in a dose-dependent manner. These data indicate that the inflammation in H9c2 cardiac cells was significantly induced by Doxorubicin, and was alleviated by the treatment with Roflumilast.
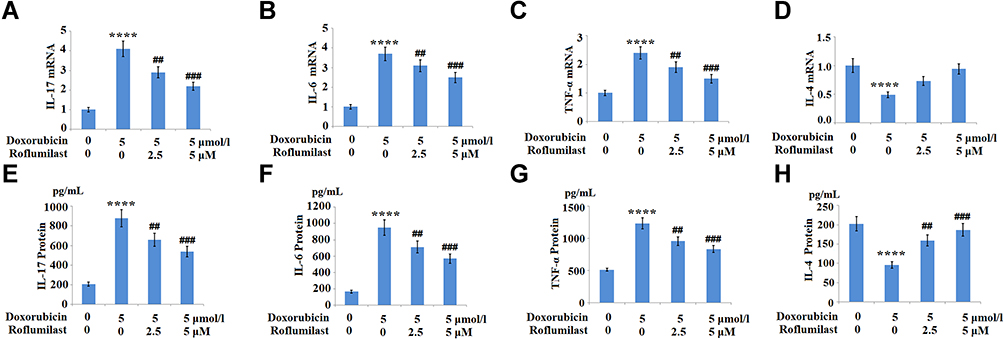 |
Figure 5 The effects of Roflumilast on Doxorubicin-induced pro-inflammatory and anti-inflammatory cytokines production in H9c2 cardiac cells. Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. (A) mRNA of IL-17; (B) mRNA of IL-6; (C) mRNA of TNF-α; (D) mRNA of IL-4; (E) Protein of IL-17; (F) Protein of IL-6; (G) Protein of TNF-α; (H) Protein of IL-4 (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
Roflumilast Ameliorates Doxorubicin Mediated Cellular Senescence
Cellular senescence is one of the pathological features resulting from the administration of Doxorubicin. In the present study, H9c2 cardiac cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours and the cellular senescence was evaluated using SA-β-Gal staining. As shown in Figure 6, we found that the staining of SA-β-Gal solution was significantly enhanced by Doxorubicin, and was suppressed by the introduction of Roflumilast in a dose-dependent manner. These data indicate that the cellular senescence of H9c2 cardiac cells was greatly induced by Doxorubicin and the promoted cellular senescence was significantly inhibited in the presence of Roflumilast.
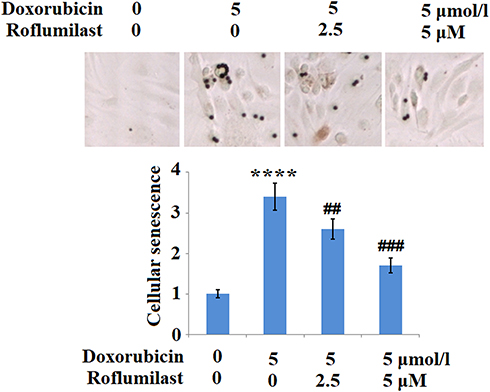 |
Figure 6 Roflumilast prevents Doxorubicin-induced cellular senescence in H9c2 cardiac cells. Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. Cellular senescence was measured using senescence-associated β-galactosidase (SA-β-Gal) staining (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
Roflumilast Suppresses Doxorubicin-Induced Expressions of PAI-1 and p21
To examine the effects of Roflumilast on cellular senescence signaling induced by Doxorubicin, the expression levels of cellular senescence-related proteins, p21 and PAI-1, were determined after the cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours using Western Blot and qRT-PCR. As shown in Figure 7A and B, PAI-1 and p21 in H9c2 cardiac cells were significantly upregulated by the introduction of Doxorubicin and were downregulated in the presence of Roflumilast (2.5, 5 μM).
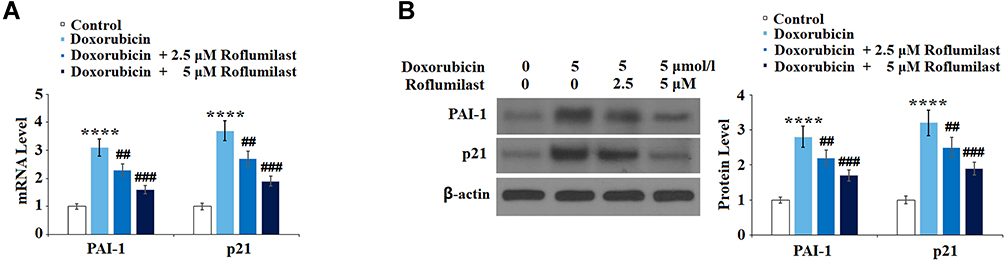 |
Figure 7 Roflumilast prevents Doxorubicin-induced expressions of PAI-1 and p21 in H9c2 cardiac cells. Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. (A) mRNA of PAI-1 and p21; (B) Protein of PAI-1 and p21 (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
Roflumilast Mitigates Doxorubicin-Reduced SIRT1 Expression
The expression level of SIRT1, an anti- cellular senescence protein, was determined after the cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours, using Western Blot and qRT-PCR. As shown in Figure 8A and B, SIRT1 in H9c2 cardiac cells was significantly downregulated by the introduction of Doxorubicin, but upregulated in the presence of Roflumilast (2.5, 5 μM).
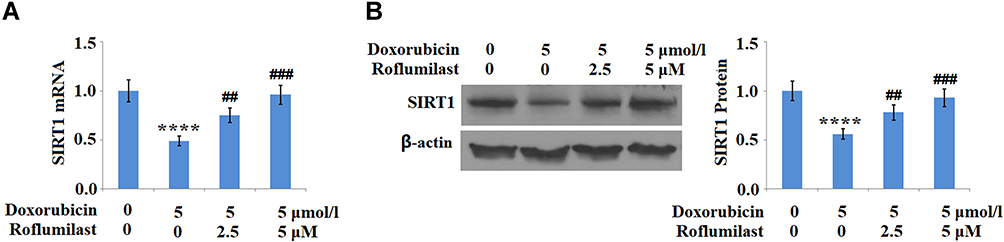 |
Figure 8 Roflumilast restores Doxorubicin-induced reduction of SIRT1 in H9c2 cardiac cells. Cells were treated with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. (A) mRNA of SIRT1; (B) Protein of SIRT1 (****P<0.0001 vs vehicle control; ##, ###P<0.01, 0.001 vs Doxorubicin treatment). |
The Silencing of SIRT1 Abolishes the Inhibitory Effect of Roflumilast on the Cellular Senescence
To investigate the potential mechanism underlying the inhibitory effect of Roflumilast on the cellular senescence in H9c2 cardiac cells, SIRT1 was downregulated in the H9c2 cardiac cells by siRNA technology. As shown in Figure 9A, the result of Western Blot analysis shows that SIRT1 was successfully downregulated in the H9c2 cardiac cells by siRNA technology. The cellular senescence of H9c2 cardiac cells was evaluated using SA-β-Gal staining after incubating the cells with Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 hours. As shown in Figure 9B, knockdown of SIRT1 abolished the protective effects of Roflumilast against cellular senescence induced by Doxorubicin. These data indicate that the inhibitory effect of Roflumilast on the cellular senescence induced by Doxorubicin is associated with its modulation on the expression of SIRT1.
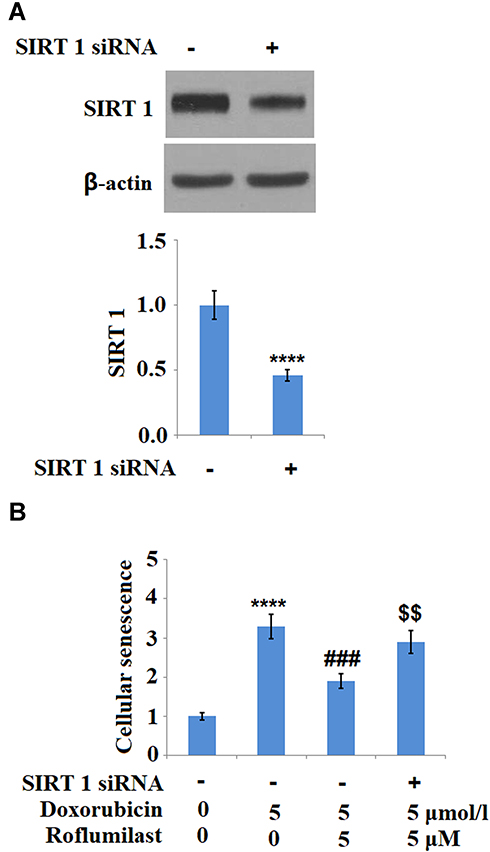 |
Figure 9 Silencing of SIRT 1 abolishes the protective effects of Roflumilast in H9c2 cardiac cells. Cells were transfected with SIRT 1 siRNA, followed by stimulation with 5 μmol/l Doxorubicin in the presence or absence of Roflumilast (2.5, 5 μM) for 24 h. (A) Western Blot analysis revealed successful knockdown of SIRT1; (B) Cellular senescence was measured using SA-β-Gal staining (****P<0.0001 vs vehicle control; ###P<0.001 vs Doxorubicin treatment; $$P<0.01 vs Doxorubicin+ Roflumilast group). |
Discussion
Doxorubicin is a chemotherapeutic drug commonly used in the treatment of malignant tumors in the clinic and is categorized as anthracycline antibiotic. Doxorubicin induces dilated cardiomyopathy and CHF clinically. Therefore, the application of such an effective anti-cancer chemotherapeutic is limited due to the severe side effects of cardiotoxicity and cardiac failure. The proliferation ability and activity of cardiac cells isolated from patients are observed to be significantly suppressed after the treatment with Doxorubicin22 and are aggravated by the continuous administration of Doxorubicin. Currently, decreasing the dosage of Doxorubicin to alleviate the side effects on cardiac function is the common method to deal with the cardiotoxicity that is induced by Doxorubicin. However, the process of CHF cannot be prevented and reversed. Although the pathogenic factor of CHF has been determined, the potential molecular mechanism has not been thoroughly illustrated, which is currently reported to be related to the change in expression levels of some proteins, such as PAI-1, p21, and SIRT1.23 In the present study, the in-vitro CHF model was established by incubating the H9c2 cardiac cells, and was verified by decreased cell viability, promoted LDH release, activated oxidative stress, and induced inflammation. These data indicate that the physiological state of H9c2 cardiac cells was significantly injured by Doxorubicin. In addition, we found that the cellular senescence of H9c2 cardiac cells was significantly induced by Doxorubicin. CHF related proteins, PAI-1 and p21, were upregulated and the anti-CHF protein, SIRT1, was downregulated by incubation with Doxorubicin, which indicates that the characteristic CHF symptoms and the related potential molecular changes were induced by Doxorubicin in the H9c2 cardiac cells. As the application of Doxorubicin in chemotherapy is important, it is necessary to find new therapeutic agents that could alleviate or reverse Doxorubicin-induced cardiotoxicity. In this study, our results demonstrated the protective effects of Roflumilast against Doxorubicin-induced side effects in an in-vitro H9c2 cardiac cell model. It is reasonable to presume that Roflumilast may be beneficial in a patient on Doxorubicin chemotherapy.
As highly differentiated terminal cells, mitosis and proliferation are rarely observed on mature myocardial cells. Initiative or passive death is initiated in myocardial cells under the stimulation of such pathological factors as chronic stress, inflammation, and oxidative stress. The cell death of myocardial cells indicates that cardiac dysfunction will be induced by the running off of myocardial cells. Therefore, to rebuild the injured heart, regulating the cell death of myocardial cells has been paid the most attention in the past.24,25 However, a novel pathological process has been found in obesity or cardiac disease resulting from chemotherapeutics, which is termed cell senescence.26,27 Cell senescence in myocardial cells could be induced by external stimuli, such as oxidative stress, and belongs to “irritable senescence”, which is different from the concept of “replicated senescence”.28 In the concept of replicated senescence, when the cells divide a certain number of times, the proliferation of cells is inhibited and the cell cycle stopped, by which the flat morphology and increased permeability of the cells are observed. This is accompanied by the upregulation of senescence-related proteins, the activation of Senescence-associated D-galactosidase, and the decreased activity of telomerase. Compared to replicated senescence, irritable senescence is not dependent on the activity of telomerase or proliferation times. However, similar changes are also involved in irritable senescence and cardiac dysfunction is directly induced, which decreases the amount of functional myocardial cells and finally induces CHF.29 It is reported that the senescent cells impact the structure and function of organs by secreting such mediators as IL-6, IL-8, IL-17, granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemotactic protein-1 (MCP-1), and matrix metallopeptidase-1 (MMP-1).30 In the present study, cellular senescence in H9c2 cardiac cells was evaluated using SA-β-Gal staining and we found that the cellular senescence was significantly induced by Doxorubicin, and was alleviated by treatment with Roflumilast. These data indicate that the Doxorubicin-activated cellular senescence, which is a classic pathological process of CHF, was greatly suppressed by Roflumilast, illustrating the potential therapeutic effects of Roflumilast against CHF. However, only an in-vitro therapeutic study was performed on the effects of Roflumilast on cellular senescence. A further investigation of the therapeutic effects of Roflumilast on Doxorubicin-induced CHF animal models or even clinical Doxorubicin-induced CHF patients should be explored to verify its potential curative value on CHF.
P21 is reported to be an important regulator of cellular senescence by inducing mitochondrial dysfunction and the production of reactive oxygen species (ROS), and is related to the regulation of the GADD45-MAPK14(p38MAPK)-GRB2-TGFBR2-TGFβ signaling pathway.31 Plasminogen activator inhibitor-1 (PAI-1) is also a mediator paid high attention for regulation of cellular senescence and is reported to be related to the regulation of the TGF-β1/p53 signaling pathway.32 SIRT1 has been widely regarded as a potential anti-senescent agent in the past33 and its regulation effects on PARP1 and NF-κB signaling pathways have been deeply investigated.34 In the present study, to highlight the potential mechanism underlying the therapeutic effects of Roflumilast on Doxorubicin-induced injured cardiac cells, the regulatory effects of Roflumilast on the expression levels of p21, PAI-1, and SIRT1 were taken into consideration. We found that the upregulated expressions of p21 and PAI-1 and the downregulated expression of SIRT1 by Doxorubicin were significantly reversed by Roflumilast, which indicates that the inhibitory effects of Roflumilast on cellular senescence might be related to p21, PAI-1, or SIRT1. For further verification, SIRT1 was knocked down in H9c2 cardiac cells and the inhibitory effects of Roflumilast on cellular senescence were found to be abolished, which verified that the upregulation of SIRT1 might contribute to the effects of Roflumilast on cellular senescence. A representative schematic of the molecular mechanism is shown in Figure 10. However, in future work, the molecular mechanism needs to be further investigated for the relationship between the anti-senescent effects of Roflumilast and the upregulation of SIRT1, such as the TGF-β1/p53 signaling pathway. The sirtuin family proteins (SIR1-7) play critical roles in the diet- and exercise-induced cardiometabolic benefits and aging. A previous study showed that exposure to Doxorubicin of H9c2 cells leads to cellular injury and the reduction of SIRT1.35 Among SIRT proteins, SIRT1 and SIRT3 are highly expressed in cardiomyocytes, and both of them have been shown to be involved in Doxorubicin cardiotoxicity.36 While SIRT1 has been the dominant regulator in Doxorubicin-induced cardiomyocyte injury,37 our data indicate that Roflumilast significantly mitigated Doxorubicin-induced SIRT1 reduction, indicating its protective role in drug-induced cardiotoxicity.
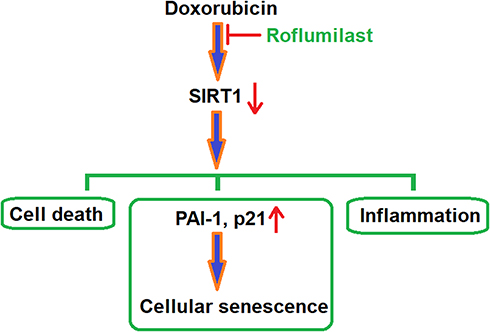 |
Figure 10 A representative schematic of the molecular mechanism. |
There are several limitations to the current study. We tested the protective role of Roflumilast against Doxorubicin-induced cardiotoxicity in rat heart tissue-derived H9c2 cells. The H9c2 cell line has been experimentally validated as a convenient in vitro model to assess Doxorubicin-induced cardiotoxicity.38 However, the cell line cannot be the replacement for primary cultured cardiomyocytes. H9c2 cells are an undifferentiated myoblasts cell line, and the immortalized nature of H9c2 cells with an active cell cycle is different from fully differentiated beating cardiomyocytes in vivo.39 Therefore, the transcriptional response to Roflumilast in H9c2 cells may not be the same as in cardiomyocytes in vivo. Further validation of the effect of Roflumilast in primary rat cardiomyocytes or cardiotoxicity in vivo model is warranted in our future investigation.
PDE family enzymes are the targets of pharmacological drug development. PDE4 is the major form of PDE in rodents, while PDE3 is the highly expressed PDE in the human heart. A previous study suggests that the expression of PDE4 in the human heart plays a protective role in limiting the susceptibility to arrhythmias.40 Therefore, the current notion is that Roflumilast is unlikely to cause cardiac side effects in patients as long as they are not administered concomitantly with PDE3 inhibitors.41 Taken together, our data indicate that the PDE4 inhibitor, Roflumilast, ameliorates Doxorubicin-induced inflammation, and cellular senescence in cardiomyocytes by upregulating SIRT1. Roflumilast is a protective force against Doxorubicin-induced cardiomyocytes injury in vitro. Further investigations of its protective mechanism on chemotherapy-induced cardiotoxicity could lay ground on its potential in therapeutic implication.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81170102 & No.81441011 & No.81670328), the Fourth Period Project “333” of Jiangsu Province (BRA2012207), and the Natural Science of Jiangsu Province Youth Fund (BK-20141020).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Dong J, Chen H. Cardiotoxicity of anticancer therapeutics. Front Cardiovasc Med. 2018;5:9. doi:10.3389/fcvm.2018.00009
2. Herrmann J. From trends to transformation: where cardio-oncology is to make a difference. Eur Heart J. 2019;40(48):3898–3900. doi:10.1093/eurheartj/ehz781
3. Pucci C, Martinelli C, Ciofani G. Innovative approaches for cancer treatment: current perspectives and new challenges. Ecancermedicalscience. 2019;13:961. doi:10.3332/ecancer.2019.961
4. El-Agamy SE, Abdel-Aziz AK, Esmat A, et al. Chemotherapy and cognition: comprehensive review on doxorubicin-induced chemobrain. Cancer Chemother Pharmacol. 2019;84(1):1–14. doi:10.1007/s00280-019-03827-0
5. Rivankar S. An overview of doxorubicin formulations in cancer therapy. J Cancer Res Ther. 2014;10(4):853–858. doi:10.4103/0973-1482.139267
6. Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. 2017;8(28):46663–46680. doi:10.18632/oncotarget.16944
7. Shabalala S, Muller CJF, Louw J, et al. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017;180(160):160–170. doi:10.1016/j.lfs.2017.05.003
8. Wu Z, Zhao X, Miyamoto A, et al. Effects of steroidal saponins extract from ophiopogon japonicus root ameliorates doxorubicin-induced chronic heart failure by inhibiting oxidative stress and inflammatory response. Pharm Biol. 2019;57(1):176–183. doi:10.1080/13880209.2019.1577467
9. Altieri P, Barisione C, Lazzarini E, et al. Testosterone antagonizes doxorubicin-induced senescence of cardiomyocytes. J Am Heart Assoc. 2016;5(1):e002383. doi:10.1161/JAHA.115.002383
10. Hsu CH, Altschuler SJ, Wu LF. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell. 2019;178(2):361–373 e312. doi:10.1016/j.cell.2019.05.041
11. Kilic Eren M, Kilincli A, Eren O. Resveratrol induced premature senescence is associated with DNA damage mediated SIRT1 and SIRT2 down-regulation. PLoS One. 2015;10(4):e0124837. doi:10.1371/journal.pone.0124837
12. Ghosh AK, Rai R, Park KE, et al. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget. 2016;7(45):72443–72457. doi:10.18632/oncotarget.12494
13. Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol. 2012;165(5):1288–1305. doi:10.1111/j.1476-5381.2011.01729.x
14. Hatzelmann A, Morcillo EJ, Lungarella G, et al. The preclinical pharmacology of roflumilast–a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2010;23(4):235–256. doi:10.1016/j.pupt.2010.03.011
15. Antoniu SA. New therapeutic options in the management of COPD - focus on roflumilast. Int J Chron Obstruct Pulmon Dis. 2011;6:147–155. doi:10.2147/COPD.S7336
16. Chapman RW, House A, Jones H, et al. Effect of inhaled roflumilast on the prevention and resolution of allergen-induced late phase airflow obstruction in Brown Norway rats. Eur J Pharmacol. 2007;571(2–3):215–221. doi:10.1016/j.ejphar.2007.05.074
17. Mata M, Sarria B, Buenestado A, et al. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax. 2005;60(2):144–152. doi:10.1136/thx.2004.025692
18. Wohlsen A, Hirrle A, Tenor H, et al. Effect of cyclic AMP-elevating agents on airway ciliary beat frequency in central and lateral airways in rat precision-cut lung slices. Eur J Pharmacol. 2010;635(1–3):177–183. doi:10.1016/j.ejphar.2010.03.005
19. van Empel VP, Bertrand AT, Hofstra L, et al. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67(1):21–29. doi:10.1016/j.cardiores.2005.04.012
20. Takahashi T, Tang T, Lai NC, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114(5):388–396. doi:10.1161/CIRCULATIONAHA.106.632513
21. Kwak HJ, Park KM, Choi HE, et al. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008;20(5):803–814. doi:10.1016/j.cellsig.2007.12.011
22. Massie BM, Shah NB. The heart failure epidemic: magnitude of the problem and potential mitigating approaches. Curr Opin Cardiol. 1996;11(3):221–226. doi:10.1097/00001573-199605000-00001
23. McGregor E, Dunn MJ. Proteomics of the heart: unraveling disease. Circ Res. 2006;98(3):309–321. doi:10.1161/01.RES.0000201280.20709.26
24. Xin W, Lu X, Li X, et al. Attenuation of endoplasmic reticulum stress-related myocardial apoptosis by SERCA2a gene delivery in ischemic heart disease. Mol Med. 2011;17(3–4):201–210. doi:10.2119/molmed.2010.00197
25. Rachmat J, Sastroasmoro S, Suyatna FD, et al. Ischemic preconditioning reduces apoptosis in open heart surgery. Asian Cardiovasc Thorac Ann. 2014;22(3):276–283. doi:10.1177/0218492313481223
26. Maejima Y, Adachi S, Ito H, et al. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008;7(2):125–136. doi:10.1111/j.1474-9726.2007.00358.x
27. Niemann B, Chen Y, Teschner M, et al. Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J Am Coll Cardiol. 2011;57(5):577–585. doi:10.1016/j.jacc.2010.09.040
28. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585–621. doi:10.1016/0014-4827(61)90192-6
29. Spyridopoulos I, Isner JM, Losordo DW. Oncogenic ras induces premature senescence in endothelial cells: role of p21(Cip1/Waf1). Basic Res Cardiol. 2002;97(2):117–124. doi:10.1007/s003950200001
30. Martinez DE, Borniego ML, Battchikova N, et al. SASP, a Senescence-Associated Subtilisin Protease, is involved in reproductive development and determination of silique number in Arabidopsis. J Exp Bot. 2015;66(1):161–174. doi:10.1093/jxb/eru409
31. Passos JF, Nelson G, Wang C, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6(1):347. doi:10.1038/msb.2010.5
32. Samarakoon R, Higgins SP, Higgins CE, et al. The TGF-beta1/p53/PAI-1 signaling axis in vascular senescence: role of caveolin-1. Biomolecules. 2019;9(8):341. doi:10.3390/biom9080341
33. Lee SH, Lee JH, Lee HY, et al. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52(1):24–34. doi:10.5483/BMBRep.2019.52.1.290
34. Hwang JW, Yao H, Caito S, et al. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med. 2013;61:95–110. doi:10.1016/j.freeradbiomed.2013.03.015
35. Liu MH, Shan J, Li J, et al. Resveratrol inhibits doxorubicin-induced cardiotoxicity via sirtuin 1 activation in H9c2 cardiomyocytes. Exp Ther Med. 2016;12(2):1113–1118. doi:10.3892/etm.2016.3437
36. Dolinsky VW. The role of sirtuins in mitochondrial function and doxorubicin-induced cardiac dysfunction. Biol Chem. 2017;398(9):955–974.
37. Ruan Y, Dong C, Patel J, et al. SIRT1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell Physiol Biochem. 2015;35(3):1116–1124. doi:10.1159/000373937
38. Dallons M, Schepkens C, Dupuis A, et al. New insights about doxorubicin-induced toxicity to cardiomyoblast-derived H9C2 cells and dexrazoxane cytoprotective effect: contribution of in vitro1H-NMR metabonomics. Front Pharmacol. 2020;11:79. doi:10.3389/fphar.2020.00079
39. Enayetallah AE, Puppala D, Ziemek D, et al. Assessing the translatability of in vivo cardiotoxicity mechanisms to in vitro models using causal reasoning. BMC Pharmacol Toxicol. 2013;14(1):46. doi:10.1186/2050-6511-14-46
40. Molina CE, Leroy J, Richter W, et al. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol. 2012;59(24):2182–2190. doi:10.1016/j.jacc.2012.01.060
41. Eschenhagen T. PDE4 in the human heart - major player or little helper? Br J Pharmacol. 2013;169(3):524–527. doi:10.1111/bph.12168
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.