")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Robotic voltammetry with carbon nanotube-based sensors: a superb blend for convenient high-quality antimicrobial trace analysis
Authors Theanponkrang S, Suginta W, Weingart H, Winterhalter M, Schulte A
Received 1 October 2014
Accepted for publication 1 November 2014
Published 27 January 2015 Volume 2015:10(1) Pages 859—868
DOI https://doi.org/10.2147/IJN.S75237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Somjai Theanponkrang,1,2 Wipa Suginta,2,3 Helge Weingart,4 Mathias Winterhalter,4 Albert Schulte1,2
1School of Chemistry, 2Biochemistry–Electrochemistry Research Unit, Institute of Science, 3School of Biochemistry, Suranaree University of Technology, Nakhon Ratchasima, Thailand; 4Life Sciences, School of Engineering and Science, Jacobs University Bremen, Bremen, Germany
Abstract: A new automated pharmacoanalytical technique for convenient quantification of redox-active antibiotics has been established by combining the benefits of a carbon nanotube (CNT) sensor modification with electrocatalytic activity for analyte detection with the merits of a robotic electrochemical device that is capable of sequential nonmanual sample measurements in 24-well microtiter plates. Norfloxacin (NFX) and ciprofloxacin (CFX), two standard fluoroquinolone antibiotics, were used in automated calibration measurements by differential pulse voltammetry (DPV) and accomplished were linear ranges of 1–10 µM and 2–100 µM for NFX and CFX, respectively. The lowest detectable levels were estimated to be 0.3±0.1 µM (n=7) for NFX and 1.6±0.1 µM (n=7) for CFX. In standard solutions or tablet samples of known content, both analytes could be quantified with the robotic DPV microtiter plate assay, with recoveries within ±4% of 100%. And recoveries were as good when NFX was evaluated in human serum samples with added NFX. The use of simple instrumentation, convenience in execution, and high effectiveness in analyte quantitation suggest the merger between automated microtiter plate voltammetry and CNT-supported electrochemical drug detection as a novel methodology for antibiotic testing in pharmaceutical and clinical research and quality control laboratories.
Keywords: antibiotics, electroanalysis, automation, microtiter plates, pharmaceutical screening, pharmacoanalytics
Introduction
Drug discovery and clinical analysis are areas in which efficient, accurate, cost-effective, and convenient analytical methods are needed. A balance between the different criteria is best reached by systematic parameter optimization for the specific detection scheme of choice and, if possible, automation.1 Accomplishments in automation of electroanalysis schemes include computer-controlled microfluidic devices with voltammetric detection in flow-through electrochemical chambers,2 non-manually operated electrode microarrays,3 microtiter plates with multiplexed three-electrode systems on vial bottoms,4 and electrochemical analyzers with robotic sampling and pipetting.5,6 Our own recent contribution to automatic electroanalysis is timed voltammetry, with a three-electrode assembly that moves sequentially between microtiter plate wells for nonmanual analyte determinations at user-specified points. With graphite pencil “leads” used as working electrodes (PL-WEs), this setup was capable of accurate robotic differential pulse voltammetry (DPV) of vitamin C,7 automated amperometric total antioxidant capacity measurements,8 and robotic trace lead and cadmium stripping voltammetry.9 Motivated by pharmaceutical and clinical interest in drug assays, our next aim was to develop easily handled antibiotic analysis by microtiter plate-based voltammetry. Two antibiotics, norfloxacin (NFX) and ciprofloxacin (CFX), served as model analytes in evaluating this approach. Voltammetric antibiotic quantification is, however, often limited by poor redox conversion of this group of analytes at bare carbon or noble metal electrodes, which badly affects the onset of faradaic current signals and thus compromises sensitivity and accuracy. This can be improved by coating the sensor with materials that facilitate electron transfer for drug electroxidation or reduction.10–16 Based on earlier reports of success with carbon nanotube (CNT)-assisted NFX17,18, CFX19,20 and other analyte22–24 voltammetry, electrophoretic CNT deposition21 was chosen to modify the PL-WEs in the three-electrode assembly of a robotic workstation for NFX and CFX voltammetry. Through parameter optimization for WE modification, CNT layers were obtained that enabled stable and accurate NFX/CFX detection during voltammetric microtiter plate runs.
We report the details of the sensor testing by voltammetry, results of robotic voltammetric drug calibrations in preloaded microtiter plates, and NFX/CFX quantification by robotic drug voltammetry in model, dissolved tablet, and, for NFX, human serum samples with added analyte. In contrast to previously described options for automated electroanalysis, the practice described in this study does not need complicated microfluidics or complex electrode structures. Its application in pharmacoanalytics for nonmanual antibiotic sample evaluation is thus practical, provided that the target molecules are electroactive and hence suitable for electrochemical detection.
Material and methods
Chemicals and materials
Analytical grade NFX and CFX powders were from Sigma-Aldrich GmbH (Munich, Germany) while their tablet formulations came from QUALIMED Co, Ltd (Bangkok, Thailand) (NFX, type Norfar®, 400 mg/tablet) and MILLIMED Co, Ltd (Bangkok Thailand) (CFX, type Cobay®, 500 mg/tablet). Stock solutions of 1 mM drug were prepared by dissolving their powders in 0.01 M HCl. NFX was measured in acetate buffer (0.1 M in 0.1 M KCl, pH 4.5), while phosphate buffers (0.1 M in 0.1 M KCl, pH 4.0) were used for CFX. The electrolyte for cyclic voltammetry (CV) and DPV electrode tests was 1 mM K3[Fe(CN)6] in 0.1 M KCl.NaNO3 (anhydrous, 99%) and N2 gas (99.99%, Linde Thailand, Thailand) were used to make and condition the electrolyte for the voltammetric evaluation of WE capacitances. If no specific source and quality is stated, chemicals were analytical grade and obtained via Italmar (Thailand) Co, Ltd (Bangkok, Thailand). Drug stock and test solutions and buffers used ultrapure deionized water. Human serum was made from the whole blood of a healthy volunteer following a standard isolation protocol.
Common heat-shrinking tubes, as used in electrical engineering for the isolation of wirings and joints, and 0.5-mm-diameter pencil leads (Pentel® HB, Pentel Co, Ltd, Bangkok, Thailand) from a local stationery shop were used for the fabrication of PL-WEs. The process of polymer tube sealing left ~2 mm of the pencil lead exposed at the bottom end, which brought ~3 mm2 into use as electroactive area. A 1-mm-diameter Ag wire (Goodfellow, Cambridge, UK) became the applied pseudo-Ag/AgCl reference electrode (RE) via 10-minute application of a DC voltage of 10 V vs a Pt wire in 1 M HCl/3 M KCl electrolyte, with consequent formation of a well-adhering layer of AgCl. A coiled 0.5-mm-diameter Pt wire (Goodfellow) served as counterelectrode (CE), with an immersed area of about 500 mm2. For thin-film surface modification of PL-WEs, aqueous suspensions of purified single-walled CNTs, with 1.0–3.0 at.% carboxylic acid entities (P3-SWNT; Carbon Solutions, Inc, Riverside, CA, USA), were used.
Instrumentation
The PL-WEs for NFX and CFX trials in the robotic voltammetry mode were obtained by inserting 3-cm-long pieces of the thin graphite sticks into heat-shrinking tubes of marginally larger diameter. It was ensured that 4 mm of the pencil leads projected at the top to act as contact point to the WE potentiostat cable and 2 mm came out at the bottom to serve as the sensor. A hot air stream from a laboratory heat gun was then used to trigger the desired temperature-induced tube shrinking, which, with no involvement of an inert composite sealant material as, for instance, epoxy- or silicon-based resins, reproducibly sealed the conductive microfilament into a tightly gripping polymeric cover. A home-made WE/CE/RE holder was used to form a movable three-electrode assembly, which was small enough to enter the microtiter plate vials via a micropositioning system control without the risk of damage (Figure 1).
 | Figure 1 The device (A) for robotic antibiotic electroanalysis in 24-well microtiter plates (B). |
The potentiostat was a PalmSens® device (PalmSens BV, Utrecht, the Netherlands), and a set of computer-controlled micropositioners (Owis GmbH, Staufen im Breisgau, Germany) enabled vertical (z) electrode and horizontal (x/y) microtiter plate movements. The hardware and software of the workstation moved the electrode assembly in a user-defined sequence from well to well and made voltammetric recordings during halts of preset lengths. Manual voltammetric sensor tests used conventional beaker-type electrochemical cells and the PalmSens® potentiostat. A function generator (Agilent Technologies, Inc, Santa Clara, CA, USA) served as power supply for CNT electrodeposition on PL-WEs, and a scanning electron microscope (eSEM Dual Beam™ Quanta 3D FEG, FEI, Hillsboro, OR, USA) was used for inspection of the electrodes.
CNT modification of PL-WEs
Before use, PL-WEs were cleaned by dipping in 4 M nitric acid for 1 minute, then rinsed with deionized water, and air-dried. The electrolyte for electrical field-induced CNT deposition onto PL-WE surfaces was a 20 mg/mL suspension of the carboxylated graphitic filaments in water, ultrasonicated for 30 minutes. As illustrated in Figure 2A, CNT film formation on PL-WE surfaces was accomplished in a small beaker-type two-electrode cell by 10-minute application of a constant potential of +1 V, against a platinum wire CE.
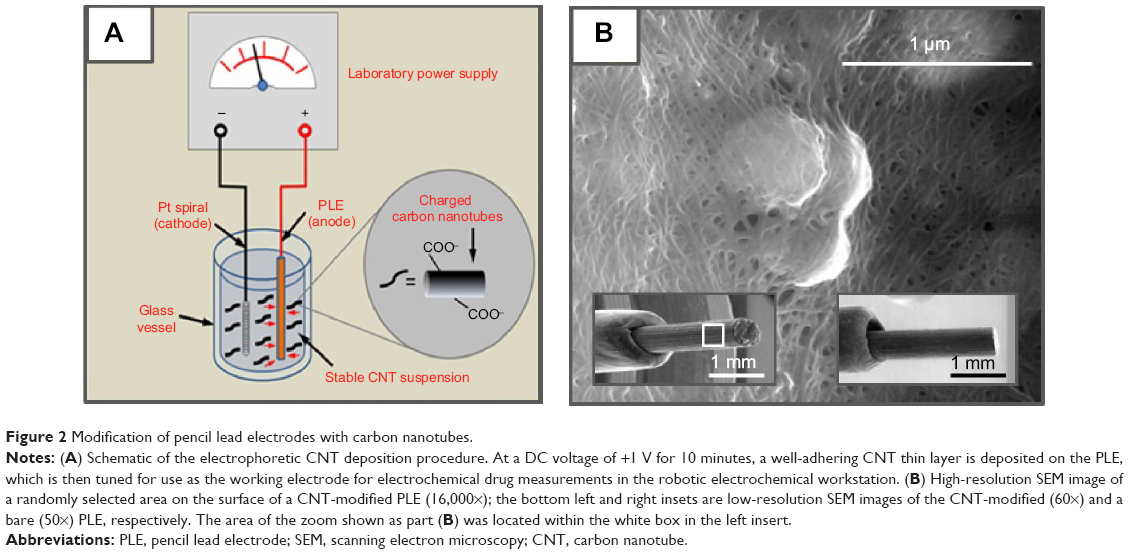 | Figure 2 Modification of pencil lead electrodes with carbon nanotubes. |
Results and discussion
CNT sedimentation and defect-free thin film formation on the PL-WE substrates were first examined by SEM. In low-magnification SEM micrographs, the ultrathin superficial nanomaterial covering was not resolved and CNT-modified and bare PL-WEs appeared virtually identical (insets of Figure 2B). At higher magnification, however, the dense nanoporous CNT network on the outside of treated PL-WEs became clearly evident (Figure 2B). Electrodeposited CNT layers were uniformly and firmly stacked on the surface of the PL-WE with no sign of cracking or delamination. The visible nanoporosity of the films suggested an increased electroactive surface area compared with the unmodified surfaces.
The results of subsequent manual voltammetric assessment of the intrinsic surface properties of bare and CNT-modified PL-WEs are shown in Figures 3 and 4. Cyclic potential scans with the electrodes in redox-mediator-free, 99.99% pure, N2-purged electrolyte produced quasi-rectangular voltammograms, typical of a purely capacitive electrode response (Figure 3A and C). As expected, the anodic and cathodic charging currents I were directly proportional to scan speed v (graphs not shown). Plots of ΔI (ie, the difference between the two currents extracted from the voltammograms at the same potential) against v were also linear (R2>0.999, Figure 3B and D).
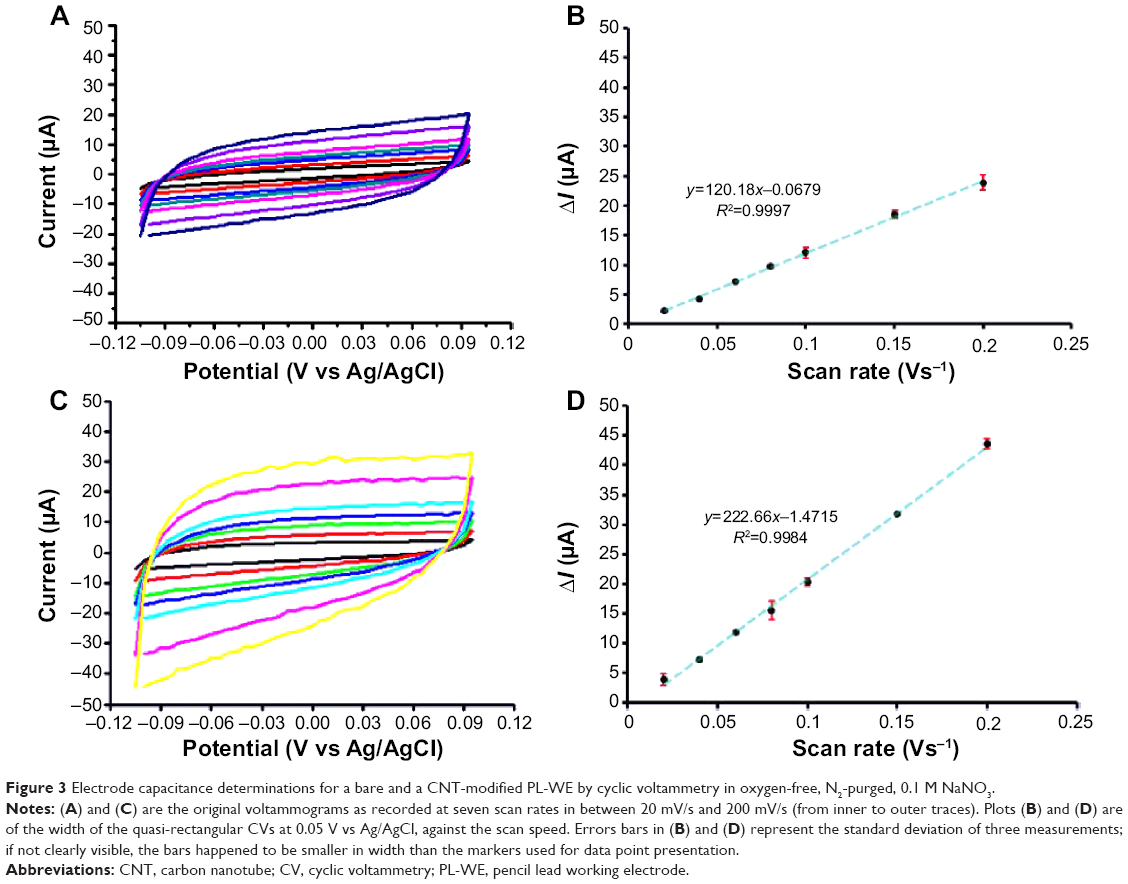 | Figure 3 Electrode capacitance determinations for a bare and a CNT-modified PL-WE by cyclic voltammetry in oxygen-free, N2-purged, 0.1 M NaNO3. |
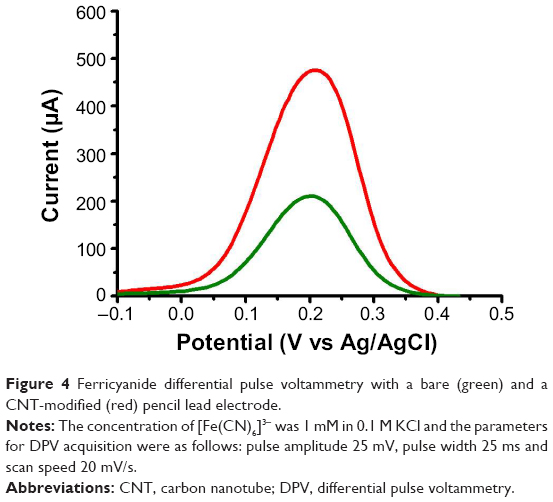 | Figure 4 Ferricyanide differential pulse voltammetry with a bare (green) and a CNT-modified (red) pencil lead electrode. |
Measurement of ΔI as a function of v allowed calculation of the individual electrode double-layer capacitance C from the formula C = ΔI/(2v). The average values for otherwise identical PL-WEs, with and without CNT, were 111±7 μF (n=3) and 67±5 μF (n=3), respectively. Since the capacitance of an electrode is directly proportional to its effective area, the ~1.7-fold larger C-value for the CNT-modified PL-WE confirmed the observation from SEM inspection that the porosity of the thin-layer films of the carbon filaments created an electrode structure with enlarged contact area for the electrolyte.
The increase in sensitivity produced by the CNT coating is shown in Figure 4. Here we compare the standard ferricyanide DPV at bare and CNT-modified PL-WEs. In each case, there was a well-defined symmetrical current peak for the reduction of ferricyanide at the graphitic surface; however, while the peak potentials were identical, their amplitudes differed significantly, with the CNT modification producing ~2.3-fold higher cathodic redox current. Thus, the increase in surface area shown by the SEM and CV inspections of the interfacial nanotube network in Figures 2 and 3 led to a clear voltammetric signal enhancement. Interestingly, a decrease in the charge transfer resistance Rct across the electrode/electrolyte interface has been reported in a number of published studies (eg, in Refs 22–24); this effect is another advantage of the CNT electrode covering and, though not specifically studied here, may have contributed to the observed increase in the ferricyanide reduction peak current.
In preliminary manual antibiotic voltammetry trials, DPVs were recorded for NFX and CFX with a bare and a CNT-modified PL-WE. For 100 μM NFX, a representative pair of DPV traces is shown in Figure 5. The bare PL-WE produced only a slight deflection for the anodic oxidation of NFX, while electroxidation at the CNT-modified PL-WE resulted in a pronounced peak, suitable for quantitative analysis. Apparently, the nanotube film enhanced the electrochemical oxidation of NFX and (data not shown) also CFX, the second antibiotic studied. Reliably, manual NFX and CFX DPV produced proportional current peaks for concentrations up to 10 μM and 100 μM, respectively. The minimum antibiotic levels producing analyzable current peaks with CNT PL-WEs as detectors were 0.3±0.1 μM (n=7) for NFX and 1.6±0.1 μM (n=7) for CFX. We concluded at this stage that operational analytical voltammetry of the selected antibiotics could be accomplished only with a current-enhancing CNT layer on the surface of the PL-WE. All subsequent robotic trials therefore used thin CNT-modified PL-WEs.
 | Figure 5 DPV of NFX with a bare (black) and a CNT-modified (red) pencil lead working electrode. |
The establishment of robotic antibiotic voltammetry began with tests of the stability of the system’s response to the analytes through complete microtiter plate voltammetry trials. As indicated in Figure 6, all sample wells in microtiter plate rows 2, 4, and 6 were filled with the antibiotic solution of a concentration within the manually determined linear response range. Use of the electrode assembly for an automated analytical plate run, with sensor cleaning between individual measuring steps, produced 12 antibiotic voltammograms (data not shown). Statistical analysis of all peak currents revealed virtually constant values for both 8 μM NFX (3.71±0.11 μA, n=12) and 50 μM CFX (2.81±0.12 μA, n=12) during the 3-hour (NFX) and 2-hour (CFX) plate runs. This superb signal stability meant that analyte degradation and sensor fouling could be excluded, at least for up to 3 hours of recording.
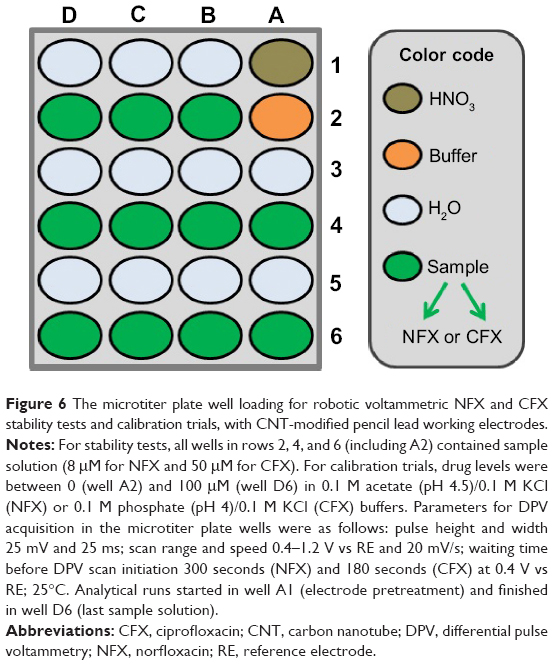 | Figure 6 The microtiter plate well loading for robotic voltammetric NFX and CFX stability tests and calibration trials, with CNT-modified pencil lead working electrodes. |
Calibration curves were acquired automatically with the microtiter plate loadings outlined in Figure 6. Typical plate calibration runs began with dipping the PL-WE in HNO3 (well A1), followed by dip-cleaning in deionized H2O (well A3), and, finally the acquisition of the first voltammogram in buffer alone (well A2). The steps of acid sensor conditioning, water cleaning, and use were repeated throughout the test until all the sample wells had been tested and the resultant DPVs stored. Completion of one full plate run thus navigated the three-electrode assembly in the following sequence through the 24 individual wells: A1, A3, A2, A1, A3, A4, A1, A5, A6, A1, B1, B2, A1, B3, B4, A1, B5, B6, A1, C1, C2, A1, C3, C4, A1, C5, C6, A1, D1, D2, A1, D3, D4, D1, D5, D6. Representative sets of automatically acquired NFX and CFX DPVs and the related standard plots of the extracted voltammetric peak currents against the corresponding analyte concentrations are shown in Figure 7. With the parameter set used, linearity of the DPV response, with R2 values above 0.99, was maintained up to 10 μM and 100 μM for NFX and CFX, respectively. Typical average values for the current sensitivity of CNT PL-WEs against NFX and CFX were 0.421±0.007 μA/μM (NFX, n=3) and 0.070±0.001 μA/μM (CFX, n=3). Also, the minimum detectable NFX and CFX concentrations in robotic voltammetry PL-WEs were equivalent to the measures of the manual analog. It is worth mentioning here that the current peak maxima of NFX and CFX DPVs are in the order of 100 mV distant to each other on the potential scale. This significant anodic peak separation technically would be suitable for simultaneous NFX and CFX determination; however, direct dual detection of the two drugs was not the focus of the current work but may be explored in future application of the methodology.
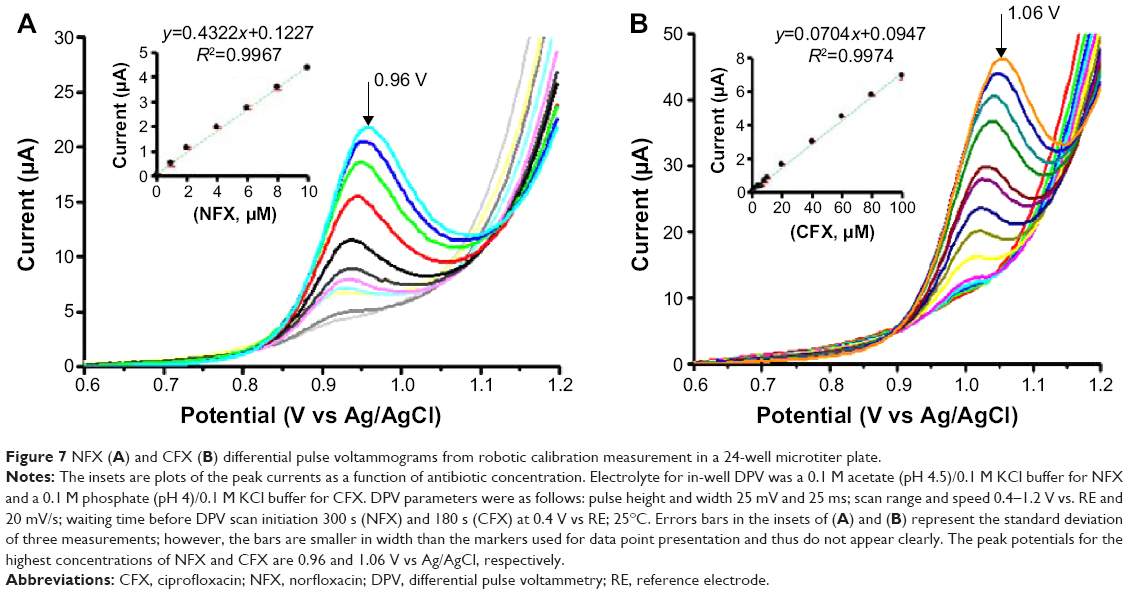 | Figure 7 NFX (A) and CFX (B) differential pulse voltammograms from robotic calibration measurement in a 24-well microtiter plate. |
Table 1 presents the analytical figures of merit of NFX and CFX DPV in the novel robotic mode and measured with CNT-modified PL-WEs, in comparison with equivalent data for manual voltammetric detection of the two analytes with various previously reported CNT-modified and CNT-free electrochemical sensors, which used polished disks of glassy carbon (GC), pyrolytic carbon (PC), carbon paste (CP), and gold (Au) or mercury (Hg) films on carbon or noble metal electrode disks as precursor structures. In automatic operation, the performance of CNT-modified PL-WEs reached in terms of linear range and sensitivity for both analytes about the level of the cited manual procedures. Slightly poorer detection limits and linear ranges probably reflect differences in the sensor’s base material, which was common pencil graphite in this study but GC, PC, CP, Au, or Hg in the previous reports. Graphite pencil “leads” contain up to 20% clay as added binder material, and the presence of this nonconducting inorganic ingredient may to a small extent reduce electron charge transfer on the graphitic planes across the PL-WE surfaces. Our experience showed, however, that notwithstanding this effect, the results of robotic NFX/DPV at CNT-modified PL-WEs were satisfactory, provided the target sample levels were within the detectable range. Confirmed was in the automated calibration trials an analytical performance that was as good as possible with the GC/PC/CP/Au/Hg equivalents. And reach of the indispensable signal stability of PL-WEs for the entire time of their use in the robotic electrochemical workstation was evident from robotic 2-3 hours voltammetric microtiter plate runs with, for instance, no significant current decay for the same NFX/CFX levels in all sample plate wells. This was an important new technical accomplishment because PL-WEs in terms of design and ease of fabrication turned out to be most practical for implantation into the movable three-electrode assembly of the robotic electrochemical microtiter plate assay. The potential of the methodology was thus further explored for antibiotic determinations in various test solutions. Buffer solutions of defined NFX and CFX concentrations were screened first, to test the recovery of analyte. Simulations of biological samples were made from pharmaceutical NFX and CFX formulations dissolved in the measuring buffers or from fresh human blood serum spiked with NFX.
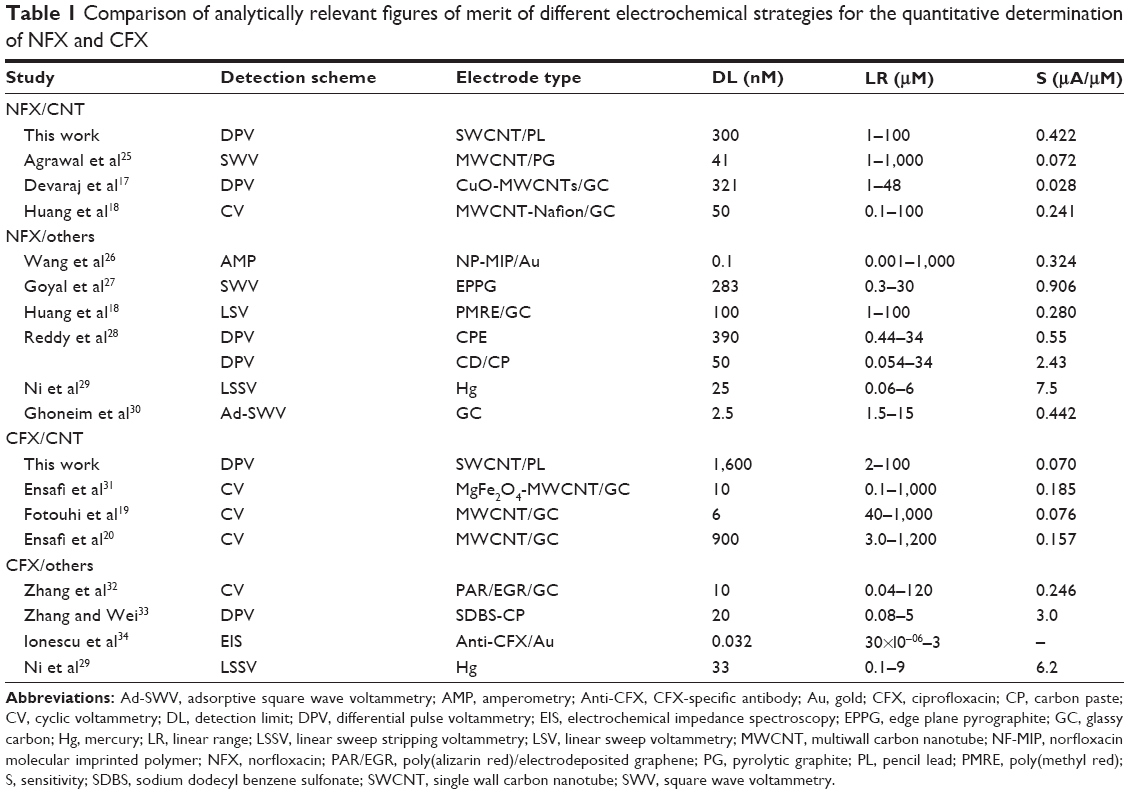 | Table 1 Comparison of analytically relevant figures of merit of different electrochemical strategies for the quantitative determination of NFX and CFX |
Microtiter plate loading for robotic voltammetric antibiotic quantification in the standard addition mode is shown in Figure 8. The wells in row 1 contained HNO3 (A1) and H2O (B1, C1, D1) and all vessels in row 2 contained buffer solution. The wells in the rows 3–6 contained the samples, either untreated or spiked with antibiotic. With this layout, the four samples could be processed in 3-hour (NFX) or 2-hour (CFX) automated runs, without any operator involvement. A representative example of the resulting raw data is shown in Figure 9A as a set of four DPVs that were successively acquired during robotic NFX determination in human blood serum. A plot of the peak currents of the presented DPVs against added NFX levels was linear (Figure 9B), and the regression line (R2=0.997) extrapolation to zero current and/or a calculation based on the corresponding line equation (y=1.61x+3.36) allowed determination of the endogenous NFX level in the original sample. Actually, the final concentration of NFX added to the serum was 2.00 μM, while that measured was 2.09 μM. The recovery, defined here as the ratio of the value obtained from robotic DPV analysis divided by the known value of serum sample supplementation, was thus 103.5%±5.1%. The outcome of all robotic voltammetry NFX and CFX sample measurements, in terms of apparent recoveries, is summarized in Table 2. Values in the range 97%–104% recovery were consistent with the standard method performance requirements recommended by the Association of Analytical Communities for the range of analyte concentration.35 Thus, the quality of the proposed automated electrochemical microtiter plate assay, with CNT-modified PL-WEs, was comparable with traditional manual voltammetric methods for measuring the two chosen antibiotics. The convenience of automation is an additional asset, and use of the proposed methodology for analysis of sample libraries is thus feasible.
 | Figure 8 Microtiter plate loading as used for robotic voltammetric NFX and CFX quantification trials with CNT-modified pencil lead working electrodes and the standard addition method. |
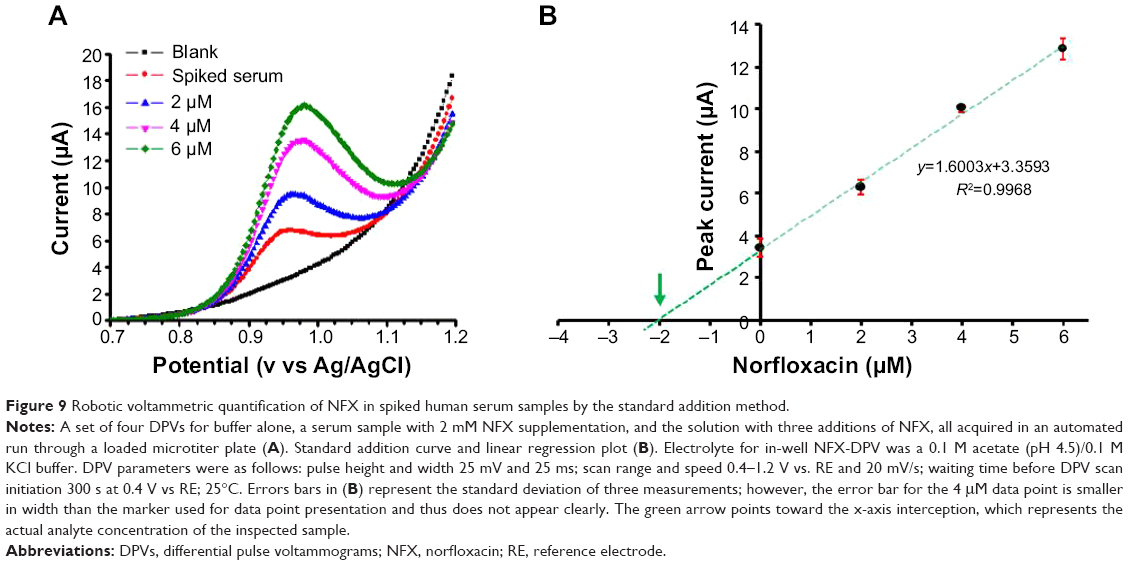 | Figure 9 Robotic voltammetric quantification of NFX in spiked human serum samples by the standard addition method. |
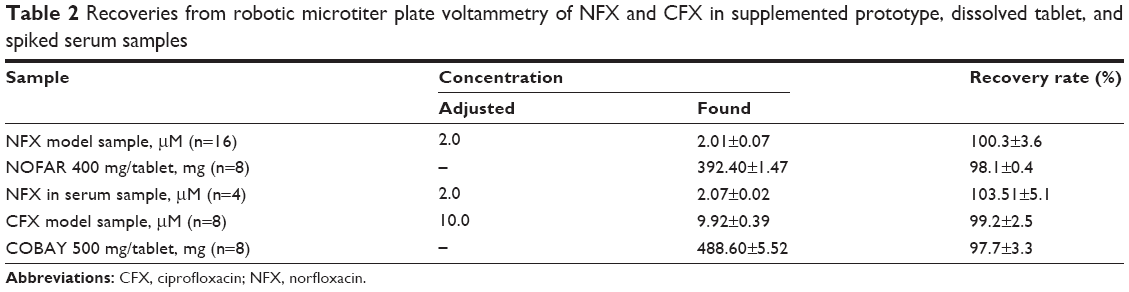 | Table 2 Recoveries from robotic microtiter plate voltammetry of NFX and CFX in supplemented prototype, dissolved tablet, and spiked serum samples |
Conclusion
A novel microtiter plate-based robotic voltammetric antibiotic assay that uses CNT-modified PL-WEs as tailored sensors for analyte quantification has been developed. For two model antibiotics, NFX and CFX, the automated electroanalysis offered good response stability and a practical linear response range. Near-perfect recoveries during measurements of standard solutions, dissolved pharmaceutical tablets, and spiked human serum samples confirmed the reliability of the methodology. An important factor in achieving this electroanalytical performance was the modification of the working electrode surface with a thin film of CNTs, as only then was reliable electrochemical antibiotic detection possible. Simple automation of the voltammetric analytical procedure microtiter plate stage conferred the additional benefits of manual error exclusion, convenience, and operator time-saving. In summary, this novel analytical approach may be a useful alternative to labor-intensive, manual, multiple-sample antibiotic screening in pharmaceutical work and, considering that antibiotics are ubiquitous pollutants of ground and surface water and soil,36,37 valuable also for application to large sets of environmental samples in governmental and private control and protection agencies. In relation to the current worldwide fears about the dramatic rise in the number of multidrug-resistant infectious microorganisms38 and the related global “apocalyptic” threat of unchecked antibiotic resistance development by bacterial pathogens,39 our planned future work will target applications in pharmaceutical and environmental antibiotic studies and also address the utilization of the technique for the pharmacokinetic study of bacterial antibiotic efflux pumps.
Acknowledgments
The authors wish to thank the Suranaree University of Technology (SUT) for funding of this project with research grant SUT1-102-54-12-14. They are grateful to the Biochemistry Laboratory of SUT’s Center for Scientific and Technological Equipment for general support. The National Research Council of Thailand (NRCT) is thanked for sponsoring the robotic workstation through the “High-throughput screening/analysis: Tools for drug discovery, disease diagnosis and health safety” scheme. WS additionally received financial support from The Thailand Research Fund (grant no RMU5380055), and AS gratefully acknowledges financial support from the Thailand Research Fund (TRF) through Grant TRF-BRG568000013. All authors also express their sincere thanks to Dr David Apps, Biochemistry Reader (retired), Centre for Integrative Physiology, Edinburgh University, Scotland, for a critical reading of the manuscript and valuable language improvements, and Prof. Wolfgang Schuhmann and Sandra Schmidt from the Ruhr University Bochum, Bochum, Germany, for their help with the SEM inspection of PL-WE surfaces.
Author contributions
Research conception and design was done by Wipa Suginta, Helge Weingart, Mathias Winterhalter, and Albert Schulte. All authors participated in the data acquisition, analysis and interpretation, and manuscript drafting and revising, and approved the version to be published. They all agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Disclosure
The authors report no conflicts of interest in this work.
References
Stanley R. Automation in analytical chemistry – from rule of thumb to fully automated methods. Some philosophies and social consequences. J Auto Chem. 1984;6:6–13. | ||
Chailapakul O, Ngamukot P, Yoosamran A, Siangproh W, Wangfuengkanagul N. Recent electrochemical and optical sensors in flow-based analysis. Sensors. 2006;6:1383–1410. | ||
Seidel M, Niessner R. Automated analytical microarrays: a critical review. Anal Bioanal Chem. 2008;391:1521–1544. | ||
Ley C, Zengin Cekic S, Kochius S, et al. An electrochemical microtiter plate for parallel spectroelectrochemical measurements. Electrochim Acta. 2013;89:98–105. | ||
Hynek D, Prasek J, Businova P, et al. Automated voltammetric determination of lead (II) ions using sensor array. Int J Electrochem Sci. 2013;8:4441–4446. | ||
Kremplova M, Krejcova L, Hynek D, et al. Automated electrochemical detection of iron ions in erythrocytes from MeLiM minipigs suffering from melanoma. Int J Electrochem Sci. 2012;7:5893–5909. | ||
Intarakamhang S, Leson C, Schuhmann W, Schulte A. A novel automated electrochemical ascorbic acid assay in the 24-well microtiter plate format. Anal Chim Acta. 2011;687:1–6. | ||
Intarakamhang S, Schulte A. Automated electrochemical free radical scavenger screening in dietary samples. Anal Chem. 2012;84:6767–6774. | ||
Intarakamhang S, Schuhmann W, Schulte A. A robotic heavy metal anodic stripping voltammetry: ease and efficacy for trace lead and cadmium electroanalysis. J Solid State Electrochem. 2013;17:1535–1542. | ||
Zhang F, Gu S, Ding Y, Zhang Z, Li L. A novel sensor based on electropolymerization of β-cyclodextrin and L-arginine on carbon paste electrode for determination of fluoroquinolones. Anal Chim Acta. 2013;770:53–61. | ||
Ojani R, Raoof JB, Zamani S. A novel voltammetric sensor for amoxicillin based on nickel-curcumin complex modified carbon paste electrode. Bioelectrochemistry. 2012;85:44–49. | ||
Peng JY, Hou CT, Liu XX, Li HB, Hu XY. Electrochemical behavior of azithromycin at graphene and ionic liquid composite film modified electrode. Talanta. 2011;86:227–232. | ||
Jain R, Rather JA. Voltammetric determination of antibacterial drug gemifloxacin in solubilized systems at multi-walled carbon nanotubes modified glassy carbon electrode. Colloids Surf B Biointerfaces. 2011;83:340–346. | ||
Vega D, Agüí L, González-Cortés A, Yáñez-Sedeño P, Pingarrón JM. Voltammetry and amperometric detection of tetracyclines at multi-wall carbon nanotube modified electrodes. Anal Bioanal Chem. 2007;389:951–958. | ||
Loetanantawong B, Suracheep C, Somasundrum M, Surareungchai W. Electrocatalytic tetracycline oxidation at a mixed-valent ruthenium oxide-ruthenium cyanide-modified glassy carbon electrode and determination of tetracyclines by liquid chromatography with electrochemical detection. Anal Chem. 2004;76:2266–2272. | ||
Ensafi AA, Allafchian AR, Rezaei B. A sensitive and selective voltammetric sensor based on multiwall carbon nanotubes decorated with MgCr2O4 for the determination of azithromycin. Colloids Surf B Biointerfaces. 2013;102:468–474. | ||
Devaraj M, Deivasigamani RK, Jeyadevan S. Enhancement of the electrochemical behavior of CuO nanoleaves on MWCNTs/GC composite film modified electrode for determination of norfloxacin. Colloids Surf B Biointerfaces. 2013;102:554–561. | ||
Huang KJ, Liu X, Xie WZ, Yuan HX. Electrochemical behavior and voltammetric determination of norfloxacin at glassy carbon electrode modified with multi walled carbon nanotubes/nafion. Colloids Surf B Biointerfaces. 2008;64:269–274. | ||
Fotouhi L, Alahyari M. Electrochemical behavior and analytical application of ciprofloxacin using a multi-walled nanotube composite film-glassy carbon electrode. Colloids Surf B Biointerfaces. 2010;81:110–114. | ||
Ensafi AA, Taei M, Khayamian T, Hasanpour F. Simultaneous voltammetric determination of enrofloxacin and ciprofloxacin in urine and plasma using multiwall carbon nanotubes modified glassy carbon electrodes by least-squares support vector machines. Anal Sci. 2010;26:803–808. | ||
Boccaccini AR, Cho J, Roether JA, Thomas BJC, Minay EJ, Shaffer MSP. Electrophoretic deposition of carbon nanotubes. Carbon NY. 2006;44:3149–3160. | ||
Gong ZQ, Sujari ANA, Ab Ghani S. Electrochemical fabrication, characterization and application of carboxylic multi-walled carbon nanotube modified composite pencil graphite electrodes. Electrochim Acta. 2012;65:257–265. | ||
Vural T, Kuralay F, Bayram C, Abaci S, Denkbas EB. Preparation and physical/electrochemical characterization of carbon nanotube-chitosan modified pencil graphite electrode. Appl Surf Sci. 2010;257:622–627. | ||
Kalachar HCB, Nayaka YA, Vinayaka KS, Viswanatha R, Vasanth Kumar MS. Electrochemical studies on usnic acid from Usnea pseudosinensis using multi walled carbon nanotube modified pencil graphite electrode. Int J Anal Bioanal Chem. 2012;2:179–184. | ||
Agrawal B, Chandra P, Goyal RN, Shim YB. Detection of norfloxacin and monitoring its effect on caffeine catabolism in urine samples. Biosens Bioelectron. 2013;47:307–312. | ||
Wang Z, Li J, Liu X, Yang J, Lu X. Preparation of an amperometric sensor for norfloxacin based on molecularly imprinted grafting photopolymerization. Anal Bioanal Chem. 2013;405:2525–2533. | ||
Goyal RN, Singh Rana AR, Chasta H. Electrochemical sensor for the sensitive determination of norfloxacin in human urine and pharmaceuticals. Bioelectrochemistry. 2012;83:46–51. | ||
Reddy TM, Balaji K, Reddy SJ. Voltammetric behavior of some fluorinated quinolone agents and their differential pulse voltammetric determination in drug formulations and urine samples using a β-cyclodextrin-modified carbon-paste electrode. J Anal Chem. 2007;62:168–175. | ||
Ni Y, Wang Y, Kokot S. Simultaneous determination of three fluoroquinolones by linear sweep stripping voltammetry with the aid of chemometrics. Talanta. 2006;69:216–225. | ||
Ghoneim MM, Radi A, Beltagi AM. Determination of norfloxacin by square-wave adsorptive voltammetry on a glassy carbon electrode. J Pharm Biomed Anal. 2001;25:205–210. | ||
Ensafi AA, Allafchian AR, Mohammadzadeh R. Characterization of MgFe2O4 nanoparticles as a novel electrochemical sensor: application for the voltammetric determination of ciprofloxacin. Anal Sci. 2012;28:705–710. | ||
Zhang X, Wei Y, Ding W. Electrocatalytic oxidation and voltammetric determination of ciprofloxacin employing poly(alizarin red)/graphene composite film in the presence of ascorbic acid, uric acid and dopamine. Anal Chim Acta. 2014;835:29–36. | ||
Zhang S, Wei S. Electrochemical determination of ciprofloxacin based on the enhancement effect of sodium dodecyl benzene sulfonate. Bull Korean Chem Soc. 2007;28:543–546. | ||
Ionescu RE, Jaffrezic-Renault N, Bouffier L, et al. Impedimetric immunosensor for the specific label free detection of ciprofloxacin antibiotic. Biosens Bioelectron. 2007;23:549–555. | ||
The Association of Analytical Communities (AOAC). Guidelines for single laboratory validation of chemical methods for dietary supplements and botanicals. 2013. Available from: http://www.eoma.aoac.org/app_k.pdf. Download from November, 2013. | ||
Kümmerer K. Antibiotics in the aquatic environment – A review – Part I. Chemosphere. 2009;75:417–434. | ||
Li WC. Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environ Pollut. 2014;187:193–204. | ||
Woolhouse M, Farrar J. An intergovernmental panel on antimicrobial resistance. Nature. 2014;509:555–557. | ||
Torjesen I. Antimicrobial resistance presents an “apocalyptic” threat similar to that of climate change, CMO warns. BMJ. 2013;346:f1597. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.