Back to Journals » Journal of Multidisciplinary Healthcare » Volume 10
Robin sequence: what the multidisciplinary approach can do
Authors Cohen SM, Greathouse ST, Rabbani CC, O'Neil J, Kardatzke MA, Hall TE, Bennett Jr WE, Daftary AS, Matt BH, Tholpady SS
Received 3 August 2016
Accepted for publication 11 November 2016
Published 27 March 2017 Volume 2017:10 Pages 121—132
DOI https://doi.org/10.2147/JMDH.S98967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Stephanie M Cohen,1 S Travis Greathouse,2 Cyrus C Rabbani,3 Joseph O’Neil,4 Matthew A Kardatzke,5 Tasha E Hall,6 William E Bennett Jr,7 Ameet S Daftary,8 Bruce H Matt,3 Sunil S Tholpady1
1Division of Plastic and Reconstructive Surgery, Indiana University School of Medicine, Indianapolis, 2Reid Health, Richmond, 3Department of Otolaryngology – Head and Neck Surgery, 4Section of Developmental Pediatrics, 5Section of Neonatal-Perinatal Medicine, Department of Pediatrics, 6Department of Orthodontics, 7Section of Children’s Health Services Research, Section of Pediatric and Adolescent Comparative Effectiveness Research, 8Section of Pediatric Pulmonology, Allergy and Sleep Medicine, Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, USA
Abstract: Robin sequence (RS) is a commonly encountered triad of micrognathia, glossoptosis, and airway obstruction, with or without a cleft palate. The management of airway obstruction is of paramount importance, and multiple reviews and retrospective series outline the diagnosis and treatment of RS. This article focuses on the multidisciplinary nature of RS and the specialists’ contributions and thought processes regarding the management of the RS child from birth to skeletal maturity. This review demonstrates that the care of these children extends far beyond the acute airway obstruction and that thorough monitoring and appropriate intervention are required to help them achieve optimal outcomes.
Keywords: cleft palate, micrognathia, mandibular distraction, retrognathia, laryngomalacia
Introduction
In 1822, the French naturalist and comparative anatomist Étienne Geoffroy Saint-Hilaire reported a case of micrognathia in a sheep associated with other congenital anomalies which he termed “sphenocephalus.”1 In 1844, cleft palate associated with tongue-based upper airway obstruction was reported by Fairbairn. The first of his two reported patients had significant problems with speech and the second died within 48 h of birth.2 The French stomatologist Pierre Robin wrote extensively on glossoptosis and mandibular hypotrophy and is credited with the eponymous triad. Pierre Robin sequence (PRS) or simply Robin sequence (RS) is traditionally described as retrognathia, glossoptosis, and airway obstruction.3
The incidence of RS is reported widely from 1 in 5,000 to 1 in 50,000 births based on the definition used.4,5 Cleft palate is associated with RS in 66%–90% of cases and is often included in the definition of RS.6 This was not part of Robin’s original description, which may lead to the sequence being underreported. The mortality associated with upper airway obstruction varies widely from 1.7% to 65%.7,8 The diagnosis of RS initiates an evaluation to determine whether this is an isolated occurrence or a known association or syndrome.9 The most common syndromes by which RS is identified are Stickler (34%) and velocardiofacial syndrome (11%), and in the literature, the occurrence of isolated RS accounts for 20%–40% of all cases.10–12
Developmental abnormalities primarily produce a hypoplastic or retro-positioned mandible. This leads to the base of the tongue being shifted into an abnormal posterior position that causes airway obstruction and a host of secondary issues. The exact etiology of the initial hypoplasia is not known in all cases, and the current genetic investigations demonstrate that it may significantly vary from case to case. The definition does not specifically require the etiologies to be similar, but the subsequent sequence of findings need to be consistent. Because the initial mandibular hypoplasia results in a sequence of later developmental and physiologic events, RS is defined as a sequence and not a syndrome. The secondary effects of airway obstruction at the tongue base include failure to thrive, developmental delay, tooth and jaw growth anomalies, reflux, feeding difficulties, CO2 retention, heart failure, brain damage, and sudden death.13–15
The presentation of RS is as variable as the specific etiologies that contribute to the final common pathway of RS. The multiple systems that are involved provide an avenue by which practitioners of multiple specialties can provide the initial diagnosis. Although not a syndrome itself, it can often be associated with syndromes resulting in multiple anatomical system involvement. Multiple specialties often need to be involved, including perinatology (maternal–fetal medicine), neonatology, pulmonary/sleep medicine, developmental pediatrics, plastic surgery, oral and maxillofacial surgery, orthodontics, otolaryngology, ophthalmology, genetics, general surgery, cardiology, neurology, dentistry, speech pathology, nutritionists, feeding specialists, audiology, and social work.16
Our specific multidisciplinary group grew from single specialty treatment to the inclusion of the above specialties because of the revolution over years that the small jaw had further reaching consequences than just airway obstruction. As a significant proportion of children have a cleft palate, they are seen under the auspices of the cleft and craniofacial clinic. Given the relatively low incidence of RS, it would also be difficult for any center to have sufficient patients to justify a robust RS clinic with specialists that are commonly encountered in a cleft care clinic.
Multiple reviews regarding the management of the RS patient as well as on the decision making regarding the choice of airway obstruction management have been written.17–22 The purpose of this review is to provide insight from all specialties regarding the thought process from conception to skeletal maturity of these intricate patients.
Prenatal and initial assessments – nonsurgical management (perinatology, neonatology, developmental pediatrics, pulmonary/sleep medicine, gastroenterology/feeding/nutrition)
Perinatology (maternal–fetal medicine)
In the multidisciplinary management of patients with RS, a perinatologist is responsible for the initial detection of RS in the prenatal period and coordination of delivery at a multidisciplinary center. Although not the most common path, the initial diagnosis of RS can sometimes be made prenatally, either by ultrasonography or by magnetic resonance imaging.23,24 Intuitively, identification of such children would be optimal as a difficult airway in a neonate can lead to hypoxia, cerebral damage, and possibly death.
The first described case was in 1986, where a fetus with RS was identified because of an affected sibling.25 As similar prior knowledge is difficult to obtain in most of the cases, specific measurements have been studied in order to provide a high specificity and sensitivity in prenatally diagnosing RS. In general, the severity of RS will dictate the sensitivity of the diagnostic test.23 Very severe cases that are apparent on imaging will subsequently result in very severe RS. As the severity decreases, the imprecision of the imaging and overlap of jaw sizes in normal and RS patients dictate that the test will become less sensitive and specific.24 Multiple case reports also show the association of polyhydramnios and a small jaw, possibly indicating that a tongue that is too large for the oral cavity could also cause obstruction in swallowing amniotic fluid.26,27
Finally, in most severe cases, ultrasound is used to identify children with RS, since problems that are large enough to be seen in utero are likely to translate into more severe forms of the disease. Although it is uncommonly diagnosed in this fashion, the authors usually recommend delivery at a center with neonatologists and anesthesiologists who are familiar with the management of complex airway obstruction.
Neonatology
In the multidisciplinary management of patients with RS, a neonatologist is responsible for initial recognition of RS, initial stabilization, as well as coordination of care in the early management of airway obstruction, feeding dysfunction, and identification of associated conditions.
Although airway obstruction is included in most definitions of RS, the timing of presentation and severity of compromise vary. For patients with mild obstruction, the initial management may be limited to prone positioning with or without supplemental oxygen. Prone positioning allows for the mandible and tongue base to fall forward, thus relieving the obstruction with reported success rates between 44% and 69%.6,28,29 For those in whom conservative measures are used, home cardiac-apnea monitoring could be used as an adjunct as the patients should be kept in prone positioning during sleep. In addition, provisions should be made for safe car transport in the prone positioning which can be performed with a specialized car bed.30
For infants with persistent obstruction in the prone position, additional short-term measures include nasal continuous positive airway pressure (CPAP) or nasopharyngeal airway (NPA) while awaiting a definitive airway intervention. Although these measures may be viewed as short-term interventions, a recent study reported a small group of infants supported with CPAP as outpatients.31 Although this treatment strategy is not likely to gain widespread use, interest has been increasing in the use of NPA for long-term airway management, NPA may allow for growth of the mandible with reports of as many as 36%–100% of RS patients being treated successfully by using this method.28,32–34 A recent Brazilian study reported that a group of infants treated with NPA for a median of 60 (±28) days showed comparable development at school age to those managed conservatively, despite the NPA group having a higher degree of obstruction.35
Although noninvasive measures are preferred, some infants with RS will not be adequately supported with these measures and for whom an invasive airway is needed. Endotracheal intubation provides a stable airway, but intubation in patients with RS may be challenging for even the most experienced clinicians. For infants in whom intubation is particularly challenging, a laryngeal mask airway (LMA) is a useful adjunctive measure and has been reported as a successful temporizing measure for patients with RS, with one study reporting LMA use for 4 days.36 Although an LMA is not intended for long-term ventilation, it may allow time for a more definitive airway solution.37
Once airway obstruction is adequately managed, a careful feeding assessment should be completed on all patients with RS since as many as 38%–62% of patients with RS require supplemental tube feedings. Supplemental feedings may initially be given by nasogastric tube, but for those with chronic feeding difficulties, a gastrostomy tube is often used. Those patients with a syndromic diagnosis or associated malformation are at highest risk for chronic feeding dysfunction, while significant feeding difficulty beyond 1 year of age in isolated RS is uncommon.19
Early management of patients with RS should include an evaluation for associated anomalies and syndromes. Initially, a thorough family history and physical examination help to identify any associated anomalies. Based on the physical examination, finding further imaging studies and genetic analysis should be performed. The neonatologist caring for patients with RS should have a heightened index of suspicion for associated conditions, since as many as 65% of patients have been shown to have other anomalies.38 Echocardiography is often performed since ~30% of RS patients have associated cardiac anomalies.39 Since the diagnoses associated with RS are diverse (currently 37 conditions listed by The Online Mendelian Inheritance in Man), a chromosomal microarray is often used to identify various genetic conditions.40 However, since many associated conditions exist and genetic testing modalities are changing rapidly, consultation with a geneticist is recommended for all patients with RS. Special consideration should be paid for evaluating Stickler syndrome as they account for 4%–36% of patients with RS. Association with 22q11.2 deletion syndrome has been reported in ~13% of patients with RS; however, a report using modern molecular testing showed a frequency of only 1.5%.38 Regardless of whether or not a syndrome is identified during the initial hospitalization, longitudinal follow-up with a medical geneticist is important to allow for the identification of associated syndromes as these may become more evident as the child ages. To this point, a recent study showed that after re-evaluation in childhood by clinical geneticists, the underlying diagnosis for patients with RS was changed in 25% of patients.40
In all, the neonatologist has the opportunity to provide initial stabilization as well as help to coordinate the early management of airway, feeding, and further evaluation of associated anomalies and syndromes.
Developmental pediatrics
In the multidisciplinary management of patients with RS, a pediatrician’s role is similar in part to the neonatologist, in that they are often responsible for the initial recognition of RS as well as coordination of care in the early management of airway obstruction, feeding dysfunction, and identification of associated conditions of those patients who are not initially evaluated in a neonatal setting. Depending on the primary care doctor’s expertise, the evaluation can be initiated in the birthing hospital, office, or at a tertiary pediatric facility, if available. During the first weeks of life, the presentation of an infant with a small appearing chin, cleft palate, and loud or noisy breathing, poor feeding, or excessive weight loss should help to identify infants at risk for RS. If not identified prenatally, the primary care provider should address these observations directly with the family and recommend further evaluation.
Airway management and breathing can pose significant challenges to the physician caring for the infant. An infant with RS may look unobstructed because the infant is unable to generate sufficient inspiratory pull to develop noisy breathing. These infants may become distressed while sleeping, feeding, or even with typical mother–baby activity. Such significant airway obstruction should be evaluated at the nearest facility with expertise to care for infants in respiratory distress. The infant needs continuous monitoring as significant airway obstruction can occur during the first 8 weeks of life.41
The primary care provider or developmental pediatrician in the craniofacial clinic system needs to be comfortable in obtaining consultations from various medical and pediatric professional services. While this process often begins in a neonatology setting, not all RS patients will be treated in this setting. Also, once the infant has been discharged from the neonatology setting, the developmental pediatrician takes over the coordination of the multidisciplinary care. At the first family meeting with the infant, a discussion of the complexity of RS should occur including the subspecialties that may need to have input in the infant’s medical plan. As mentioned in the “Neonatology” section, these include but are not limited to pulmonology, otolaryngology, plastic surgery/craniofacial surgery, genetics and genetic counseling, ophthalmology, cardiology, gastroenterology and nutrition, speech therapy, occupational therapy, and social work. The medical expertise of caregivers from these areas ensure the optimal growth and development of an infant born with RS. Early intervention cannot be emphasized enough. Cognitive, motor, and psychosocial development of children with RS has been compared with naturally developing children.42,43 Some authors have followed children with RS and identified early developmental challenges, especially speech, that are helped with early and consistent speech therapy intervention.44,45 Since development is considered to be similar to typically developing children, any identified deviations from typical development warrant a full evaluation.
Pulmonary/sleep medicine
In the multidisciplinary management of patients with RS, a pediatric pulmonologist is involved in the evaluation of ventilatory compromise in the presence of airway obstruction. Although upper airway obstruction is expected as a result of micrognathia, this can also be due to co-morbid airway lesions, treatment of which will be addressed more completely in the “Otolaryngology” section. This is particularly true in syndromic RS. Complete evaluation consists of a physical examination, anatomic assessment of airway compromise, and a functional assessment of the adequacy of ventilation.
Clinical practice varies widely between institutions depending on available specialist expertise, but typically includes bedside clinical assessment of airway compromise – assessing severity of stridor, chest and neck retractions in the supine and prone positions, feeding difficulty, as well as positional oxygen desaturations. Objective assessment consists of flexible nasopharyngoscopy, bronchoscopy, oximetry, polysomnography (PSG), and video swallow study in the presence of speech pathologist in order to assess oromotor coordination.
The reliability of airway endoscopy in ruling out airway lesions, other than base of tongue obstruction, depends on the clinical stability of the patient at the time of evaluation, technical skill, and experience of the bronchoscopist. The procedure is technically more difficult in neonates than in older children. Co-morbid airway lesions in RS include choanal atresia, pharyngomalacia, glossoptosis, laryngomalacia, subglottic stenosis, tracheomalacia, bronchomalacia, and tracheal stenosis – including complete tracheal rings. Aspiration bronchitis can be suggested by the airway evaluation and a bronchoalveolar lavage. Oximetry has been reported in RS, but no standardization exists for technique, or the proper threshold for clinically significant abnormalities. Also, artifacts in testing can significantly risk compromising data analysis.46
Because of the problems associated with other techniques, PSG has supplanted oximetry when institutional capability enables the test. Although accurate quantification of airway obstruction and gas exchange disturbances are possible by PSG, the literature does not offer clear guidance on which parameters and their respective severity thresholds are clinically significant. Studies have tried to correlate physical examination, anatomic severity, and PSG results with the finding that severity of clinical presentation is not necessarily correlated with PSG results.
A factor complicating PSG interpretation which is a less addressed issue, is that normative data for PSG parameters in the neonatal age group using current standardized data collection are lacking. In fact, PSG performed on all non-RS cleft palate patients averages around an apnea hypopnea index (AHI) of 20 per hour (while normal, although debatable is <2 events per hour). Outcome studies on RS patients who have undergone PSG have failed to provide guidance on an AHI threshold for predicting adverse outcomes.46
The most severe respiratory compromise is usually identified in the neonates, most likely due to a combination of immaturity in respiratory control and low functional residual capacity of the lung superimposed on the airway compromise. These factors are often exacerbated in the syndromic RS patient. Nonsyndromic RS patients often improve after the correction of airway obstruction post-intervention or with natural jaw growth but may have continued obstructive sleep apnea (OSA) and dysphagia throughout infancy and into the preschool years due to cleft palate and pharyngeal distortion. Syndromic RS patients may continue to have respiratory compromise for years depending on the nature and severity of their co-morbidities. Longitudinal follow-up typically involves serial PSG, airway endoscopy, and clinical assessments tailored to individual patients.
Gastroenterology/feeding/nutrition
In the multidisciplinary management of patients with RS, a gastroenterologist is involved in the evaluation and management of feeding dysfunction. Feeding dysfunction is primarily related to the micrognathia, cleft palate, and the need to alternate breathing and swallowing. The infant needs a free airway providing enough time to swallow between breaths. Feeding of infants with RS is challenging.42,43 The more significant the airway obstruction, the more difficult to initiate oral feeds to maintain growth and development. A nurse, physician, or feeding specialist should observe early feeds to determine safety of oral nutrition. Since the palate is often abnormal or nonfunctional, breast feeding may be difficult, if not impossible. Bottle feeding may also be challenging. Any system that removes the oral control of the milk flow and swallowing must be avoided as the failure to allow the infant to practice and improve oromotor skills while eating, will result in long term feeding dysfunction, even after correction of the anatomic defects.
Signs of dysphagia are prolonged bottle feeding, pulling back from an oral feeding system, feeding refusal, coughing or choking with feeds, or any other signs of distress while swallowing. If these signs are observed, oral feeds should be discontinued and nasogastric or orogastric tube feeds should be initiated. Families can be taught how to place nasogastric or orogastric tubes and how to feed an infant by using these means. Evaluation by feeding specialists and feeding therapy are essential for the infant to eventually become a successful feeder. Clinical evaluation of feeding, even by the most experienced feeding therapists, has shown to be erroneous and miss aspiration of milk into the airway 30% of the time.47 Fluoroscopic video assessment or flexible endoscopic evaluation of swallowing may be needed to guide feeding decisions and alternate methods of feeding if indicated. When aspiration is a concern, evaluation also needs to be performed to determine whether a component of reflux is involved. Traditional pH monitoring can be used; however, this only detects acid reflux. Newer methodologies, such as multichannel intraluminal impedance (MII-pH) provides more comprehensive evaluation of reflux, including ability for bolus clearance and reflux of milk or formula-based products.48 These methodologies may be helpful in determining the specific problem in these children.
Controversy exists on the use of surgically placed gastrostomy tubes. The use of these devices may provide a feeding method that can be safer than nasogastric feeding tubes. A surgically placed gastrostomy tube may allow feeding therapists full access to the child’s face unencumbered by a tube in the nose or mouth while providing adequate nutrition. Working with the child’s oromotor function is essential to the eventual resumption of total oral feeding capabilities. The role of the gastroenterologist/feeding specialist is to monitor and assist the family to help the infant maintain adequate growth, good pulmonary protection, and appropriate development during the first years of life.
Treatment – surgical/invasive management (cleft/craniofacial surgery, otolaryngology, and orthodontics)
Cleft/craniofacial surgery
In the multidisciplinary management of patients with RS, a cleft/craniofacial surgeon is usually a member of the multidisciplinary craniofacial team. From the evaluation to the diagnosis and finally to treatment, their involvement is focused on a few systems that can be surgically modified for patient benefit. The first system is the airway (and its evaluation begins from presentation through to skeletal maturity), the second system is the palate, the third is an ongoing speech evaluation, and the fourth is a facial esthetic evaluation.
Airway evaluation is the most critical, and two presentations are commonly seen in practice. The first is a neonate who struggles to breathe since birth and has been hospitalized. The second is the child seen in the cleft clinic with the failure to thrive, stridorous/heavy breathing, and difficulty in food intake. In both the cases, the first step for evaluation is a physical examination that usually demonstrates a cleft palate, as well as micrognathia. Cleft palate is different from the non-RS patient in that it is U shaped, as if the tongue left an imprint in the palate. The jaw is variably retrusive, ranging from obvious to very subtle. Although the maxillary to mandibular step off in occlusion is measured, a favorable occlusal relationship does not rule out RS.49
A computed tomography (CT) scan is also obtained routinely. One of the reasons is to delineate the three-dimensional (3D) anatomy of the mandible. From these images, decisions are made regarding mandibular osteotomies, distractor placement, and vector. Also, the scout films from CT images can demonstrate the tongue base position. A sleep study is obtained, with the above caveats of a lack of normative data as well as the knowledge that any child with a cleft palate will have a higher likelihood of having an abnormal result.46,50
Once these tests are obtained, a plan to secure the airway is formulated. In severe cases, when a child cannot be intubated, tracheostomy is performed. Beyond tracheostomy, there are operative and nonoperative options. Utilization of these methods depends largely on the expertise of the available multidisciplinary team.16 In mild cases, prone positioning or NP tube placement can sometimes be used while waiting for “catch up” mandibular growth.33
For more severe cases, either tongue–lip adhesions (TLAs) or mandibular distraction osteogenesis (MDO) is utilized. Studies show a definite preference exists, based on surgeon training and specialty, as well as the team experience in the management of RS.17 Some teams almost exclusively attempt nonsurgical management with tracheostomy as the airway salvage procedure, some teams perform a mix of procedures depending on severity with TLA reserved for less serious nonoperative failures, whereas other teams, use MDO as the first-line treatment for all patients with sleep study results above a certain threshold.51,52
In the mandible with glossoptosis, TLA serves to reduce the obstruction present in the airway because of the tongue. The anterior tongue is sutured to the lip (mucosa with or without muscle) and the posterior tongue to the midline chin with a circummandibular suture.53 Tongue and lip mechanics are altered with this nonanatomic procedure, with some studies demonstrating the problems of feeding and swallowing while others showing minimal problems and weight gain. Either way, the risks of the procedure are fairly minimal with dehiscence and scarring being the most common. The improvement seen with the procedure is likewise modest, with a significant portion of children failing the procedure and requiring MDO or tracheostomy.29,54 It must also be reversed and usually is during palate surgery (if indicated). Very few studies have been performed with pre- and post-operative sleep studies, but those that have been performed show that success is highly variable and is likely dependent on patient characteristics.55,56
A more recent technique, MDO, is a significantly more invasive option with much less time in clinical use. Originally described by Illizarov in the 1960s for limb deformities, neonatal mandibular distraction has only been used for the past 20 years in the treatment of RS.18,57 Distraction osteogenesis relies on the biologic principle that an osteotomized bone slowly pulled apart will fill the residual gap with bone. There are three components in this process: latency, activation, and consolidation. Latency is the period of time (usually between 1 and 5 days) after osteotomy and before activation. During activation, the bone is actively pulled apart at a rate of 1–2 mm per day. After the desired lengthening, the bone is formed during consolidation and the regenerate is mineralized (usually twice the time of activation). Thus for a 2 cm lengthening, the entire procedure takes ~60 days (overnight latency, 20 days for activation, and 40 days for consolidation).58 Distraction can be performed using external or internal devices. External devices can cause more scarring, but internal distractors require removal which carries similar risks as the index procedure. These risks include infection, tooth damage, facial nerve damage, and temporal mandibular joint (TMJ) ankyloses.17,52,59
MDO is highly efficacious in the treatment of tongue base airway obstruction as shown in multiple studies at multiple centers, using multiple devices and techniques.52,59,60 The original anatomic problem is corrected, and the tongue base is anteriorly repositioned. Although studies have tried to elucidate the necessary amount of lengthening, radiologic measurements do not easily correlate with clinical status. Thus, it is usually performed so that the lower jaw is slightly longer than the upper jaw. A relapse in length due to poor growth has been seen in some children and is the reason for overcorrection.17,18
After successful distraction in neonates, palatal surgery is contemplated in the RS patients with cleft palate. The operative method is still debated, and the choice is likely to depend on surgeon experience. Regardless of operation, the smaller airway in nonsurgical RS patient leads to a delay in operation beyond 1 year until the clinical judgment of the surgeon, the physical examination, and the sleep study all indicate that the palate can be closed (creating a smaller airway) in a safe fashion.61 Interestingly, when a neonate undergoes MDO, the palate can be safely closed at 10–12 months of age, which is the standard time of closure in non-RS patient.62
Palatal closure begins a time of observation, both for mandibular growth and for speech development. As the sites for osteotomy in MDO do not affect the presumptive growth centers of the mandible, growth continues normally. On rare occasion does the procedure need to be repeated (in our series of over 150 patients, two required re-distraction within 5 years of age). Children are monitored for signs of recurrent OSA including snoring, daytime somnolence, headaches, and changes in behavior.15 These symptoms would prompt a new evaluation for sleep apnea starting with a polysomnogram.
Similarly, with appropriate palatal surgery, healing then involves a time of observation of speech. Within the confines of a cleft and craniofacial center, these children are evaluated on a yearly basis for the development of speech and specifically the presence of velopharyngeal insufficiency. This is performed by the collaboration of a speech and language pathologist and a cleft-trained surgeon. Nasal endoscopy, usually tolerated around the age of 4 or 5 years, is capable of visualizing incomplete closure of the velum. The rate of fistula and impaired speech are within the same range as that for a non-RS child with cleft palate.63 This is not true in all centers, with others reporting a higher rate of velopharyngeal insufficiency, but these are likely due to the heterogeneous populations of RS patients as well as RS experience of surgeon .61,63,64
A yearly evaluation for airway and speech problems is performed. Upon skeletal maturity, a significant number of patients will require orthognathic surgery for their Angle class II dentition (upper jaw is more anterior than lower jaw). Special considerations of a smaller tooth bearing mandibular segment with a normal number of teeth pose challenges to orthodontist, but after orthodontic treatment, the performance of a bilateral sagittal split osteotomy of the mandible should provide a standard solution to retrognathia. In certain cases, where the teeth are aligned but the chin appears small, simple autogenic or alloplastic chin augmentation can be performed.65,66
Otolaryngology
In the multidisciplinary management of patients with RS, an otolaryngologist is involved in the management of other airway lesions as well as consideration of hearing in conjunction with a possible cleft palate. The goal of treating patients with RS is to achieve an adequate airway to allow the children to grow, by breathing at ease and eating safely. Other than airway, the connection between cleft palate and otitis media has been well established. A significant majority of cleft palate children will require myringotomy and placement of pressure equalizing (ventilation) tubes in an effort to minimize liquid (serous or mucoid) in the middle ear.67,68 This approach allows hearing to be maximized. Even after cleft repair, many children will require further ear tubes to maintain good hearing.
Patency of the complete airway is required for adequate ventilation. Evaluating the airway for multiple levels of obstruction makes logical sense. Andrews et al reported a 28% incidence of other (non-tongue base) airway lesions.69 In the general pediatric population with OSA, the incidence of laryngomalacia is 3.9%,70 and overall second airway lesions are present in 12%–51% of children.71–74 Examples include common problems like laryngomalacia, subglottic stenosis, true vocal fold motion impairment (previously referred to as true vocal fold paralysis or paresis), tracheomalacia, and bronchomalacia. Rare conditions like complete tracheal rings and vascular compression of the trachea or bronchi are also easily identified.75 Except for laryngomalacia, each of these problems, if severe enough, requires tracheostomy to stabilize and control the airway. The presence of any of these lesions has the potential to obviate the utility of surgery for the tongue base airway obstruction (TBAO).
Other nonoperative methods for managing the airway include a nasopharyngeal tube to bypass the tongue base. Abel et al33 reported an 86.5% success in nonoperative management of upper airway obstruction in RS. Although the NP airway is a temporary treatment, not cure, for the pharyngeal obstruction, it can provide an airway to allow the child to breathe, without intubation or tracheostomy, in the short term. Use of an NPA tube requires vigilance of the caregivers, to monitor and prevent or treat obstruction. In this method, airway obstruction can be caused because of the occlusion of the tube by mucus or dislodgement. This approach would generally also requires continuous monitoring, such as a cardiac apnea monitor or pulse oximeter. Additional equipment, such as a suction machine and associated disposables (canisters, tubing, suction catheters, etc), add to the complexity and cost.
Certainly, endoscopy can show the level and degree of airway obstruction.76 The actual severity of the entire breathing disorder is probably best quantified by use of PSG. As recommended by the American Academy of Pediatrics (AAP), PSG is the only test that quantifies the degree of severity of airway obstruction.77 However, it does not determine the site of obstruction; hence, endoscopy remains vital to determine the place of blockage.
Several methods exist to evaluate the airway. These range from radiographic methods (airway fluoroscopy) to flexible and rigid endoscopy. X-rays have disadvantages: ionizing radiation can lead to later malignancies, images are only two dimensional, and in black and white, instead of color.78 Endoscopy can provide dynamic, full color, 3D evaluation of the airway and remains as the preferred modality.
Rigid endoscopy, which has been available for more than a century, gives an excellent view of the airway with better optics (with the use of Hopkins rod telescopes) than flexible endoscopy.79 A disadvantage of rigid endoscopy is the concern for unnaturally distorting the airway anatomy, by pushing or propping structures into a more open position. This problem exists for flexible endoscopy as well: perioral endoscopy may keep the tongue from collapsing; transnasal endoscopy could prop up the soft palate anteriorly. Additionally, in children with severe micrognathia, passing a rigid endoscopy to the larynx could be limited by laryngeal size, pathway, and exposure. Flexible endoscopy can easily navigate the bends in the airway. Based on these factors, most practitioners will opt for flexible endoscopy to evaluate the entire airway, with one endoscope and one pass starting at the nostrils and continuing distally.
Based on comfort and judgment of the surgeon, either approach can be used to evaluate both secondary airway lesions and tongue-based obstruction. Endoscopic findings that are pathognomonic for RS include a tongue that is able to rotate dorsally around a pivot point of the tongue base, flipping through a cleft palate to obstruct the airway. Another finding that indicates the possibility for successful tongue-based surgery is a jaw thrust under direct visualization that appears to move the tongue base anteriorly out of the airway proper.
Endoscopy for airway evaluation has the advantage of identifying these secondary airway lesions, of which laryngomalacia is the most common congenital reason for stridor. Classic symptoms include an inspiratory noise (which can be high or low pitched). The noise worsens with anything that increases airflow through the larynx, by creating greater pressure drops. According to the Bernoulli principle, as the velocity of flow increases, the pressure drops exponentially. This principle can be applied to the laryngeal lumen where airflow increases as it enters the trachea from the pharynx, drawing in abnormally flaccid supraglottic tissue, thus causing obstruction of the airway.80 Thus during feeds, crying or anything that makes the child distressed, the noise, and obstruction can worsen. Positioning the child prone (opposite of the “back to sleep” campaign from the AAP) allows the larynx and tongue base to fall away from the posterior pharyngeal wall, thus opening up the airway.
Laryngomalacia can be classified as mild, moderate, or severe in three domains: endoscopic appearance, symptoms, and PSG data. Several classifications of the appearance of laryngomalacia on endoscopy exist, but these do not correlate well with clinical features of severity.81,82
Treatment of laryngomalacia depends on the overall effects on the child. In mild cases, expectant observation may be sufficient. Most children would be expected to grow out of symptomatic laryngomalacia (though they may still have laryngomalacia on endoscopy) by 18 months of age.83 As the severity of symptoms and PSG data increases, additional measure will be taken. Nasal cannula oxygen can act as mini-CPAP in small children.84 Inhibiting gastroesophageal, or more accurately, gastrolaryngeal (commonly called laryngopharyngeal or LPR) reflux may reduce inflammation in the larynx enough to provide an adequate airway.83 The airway does not have to be perfect, but merely good enough to allow the child to safely thrive. Thompson provides a useful algorithm for management.83
If a child cannot achieve a stable airway with appropriate growth and development, most pediatric otolaryngologists would offer a supraglottoplasty, where floppy tissue is removed to allow the airway to remain open. A number of variants in technique (cold steel vs laser; unilateral vs bilateral; excision of tissue vs tacking the anterior surface of the epiglottis to the posterior tongue base) exist, and like many operations, proponents vary in their preferences.82,85 Prior to the advent of endoscopic treatments, the only definitive treatment was tracheostomy, which completely bypasses the larynx for air flow.
Original descriptions of mandibular distraction contraindications listed laryngomalacia as a reason for tracheostomy. Some thought that the two airway lesions would combine to damage the beneficial effects of distraction. Work in our institution has shown that laryngomalacia is not a reason for tracheostomy, and that mandibular distraction may benefit children with laryngomalacia by tethering and stiffening up the anterior supraglottic structures bound to the mandible. Thus, pulling the larynx forward can prevent the larynx from suctioning against the posterior pharyngeal wall, but it would not necessarily prevent the supra-arytenoid tissue from still flopping into the laryngeal introitus.52 Thus, the standard protocol is an airway evaluation in the operating room by otolaryngology to confirm TBAO and to demonstrate any secondary lesions. The presence of laryngomalacia is treated with laser supraglottoplasty at the discretion of the surgeon. This approach optimizes the airway, ameliorates the airway obstruction, and prevents the need for tracheostomy in this patient population.
Orthodontics
An orthodontist’s role in providing care for the RS patient is primarily focused on both skeletal and dental development. Primary developmental concerns include narrow maxilla transversely secondary to clefting, bimaxillary retrognathia with increased severity in the mandible, and tendency toward vertical facial growth.86 Primary dental concerns include class II malocclusion, severe crowding, hypodontia, and tooth bud injury secondary to mandibular distraction. Although the orthodontist is responsible for correcting the malocclusion and establishing a functional dental relationship, one should note it is important to note that the standard treatment timeline may always be altered if airway becomes compromised. Collaboration with an orthodontist at these critical junctures can optimize functional outcomes since possible jaw surgery can lead to significant malocclusion.
RS patients are commonly treated with multi-phased orthodontic therapy for optimal results. These patients should begin orthodontic observation by the time the primary dentition has erupted and should be monitored continuously throughout the critical stages of development. As clinician preferences and expertise vary, many RS patients will begin being monitored by an orthodontist as early as infancy. As an RS child develops into a stage of mixed (primary and permanent) dentition, the orthodontist will evaluate their unique needs.
An individualized phase I orthodontic treatment plan will typically be utilized in order to reduce the severity of malocclusion. Commonly used approaches may include an orthodontic expansion appliance as well as orthodontic appliances (braces) in order to alleviate crowding and create symmetry of the dental arches. Dependent on the severity of crowding, the patient may need to be evaluated for extraction therapy; however, this is typically avoided until phase II if possible.
Upon completion of phase I treatment, an RS patient is monitored through adolescence for skeletal growth and dental development. Catch up growth of the mandible may be possible, which is common for a large majority of patients to require orthognathic surgery at skeletal maturity. The anteroposterior discrepancy of the maxilla and mandible as well as facial esthetics will commonly dictate the necessity and extent of orthognathic surgery. Special consideration for orthodontic treatment can be given to those patients who present with a mild anteroposterior discrepancy.
Phase II orthodontics is utilized to finalize the placement of the permanent dentition. Final decisions regarding missing and malformed dentition, crowding, and skeletal discrepancies should be made prior to proceeding with tooth movement. Dental movements for the orthognathic patient will be opposite to those of the orthodontics alone patient. Extractions may be utilized in order to pre-surgically increase the anteroposterior discrepancy resulting in a larger mandibular advancement. For an orthodontics patient, class II orthodontic correctors such as intraoral elastics or forsus springs may be utilized to establish a class I occlusion. An elective surgical procedure may be utilized to enhance the prominence of the chin if esthetically beneficial.
Conclusion
Although the optimal management of the acute RS patient has been exhaustively described, there are still staunch proponents of both operative and nonoperative management of the airway. Within the group of operative management proponents, there is a disagreement regarding the optimal procedure. Because of heterogeneity in the severity of RS, long-term prospective studies that are preferentially randomized would provide the best-level evidence on the airway management of these patients.
New investigations into the long-term consequences of RS are being undertaken at multiple institutions that are independent of the airway management question. One such area is the question of the optimal management on the difficulties of feeding. These feeding difficulties have anecdotally (at the authors’ center) been significantly ameliorated via mandibular distraction, but the literature varies in both the management and the effect of airway management on the feeding difficulty.
Another area of interest is the orthodontic issues regarding having a jaw that is too small and a normal number of teeth. As stated earlier, extraction is a method that can reduce this overcrowding, but does not affect the overall esthetics. A management plan that integrates the functional need to space the teeth appropriately and the esthetic concerns of a very convex face would provide benefit to many RS patients.
Long-term neurophysiologic outcomes are also of importance, as the ability to breathe without increased energy expenditure may translate to a better performance at skeletal maturity from a number of parameters. The timing as well as the type of intervention is likely to affect this parameter. Ongoing studies are measuring the outcomes of these population within differing age groups.
As can be seen, the management of RS patients is much more complex than simple airway management. This study has included an algorithm to outline the multidisciplinary management of RS (Figure 1). The management of these patients in the acute setting has coalesced a group of specialists interested in the management of these patients over the longer period of growth to skeletal maturity. Future studies involving the aforementioned areas, as well as others, will likely provide a standardized pathway for the care and management of these complex patients.
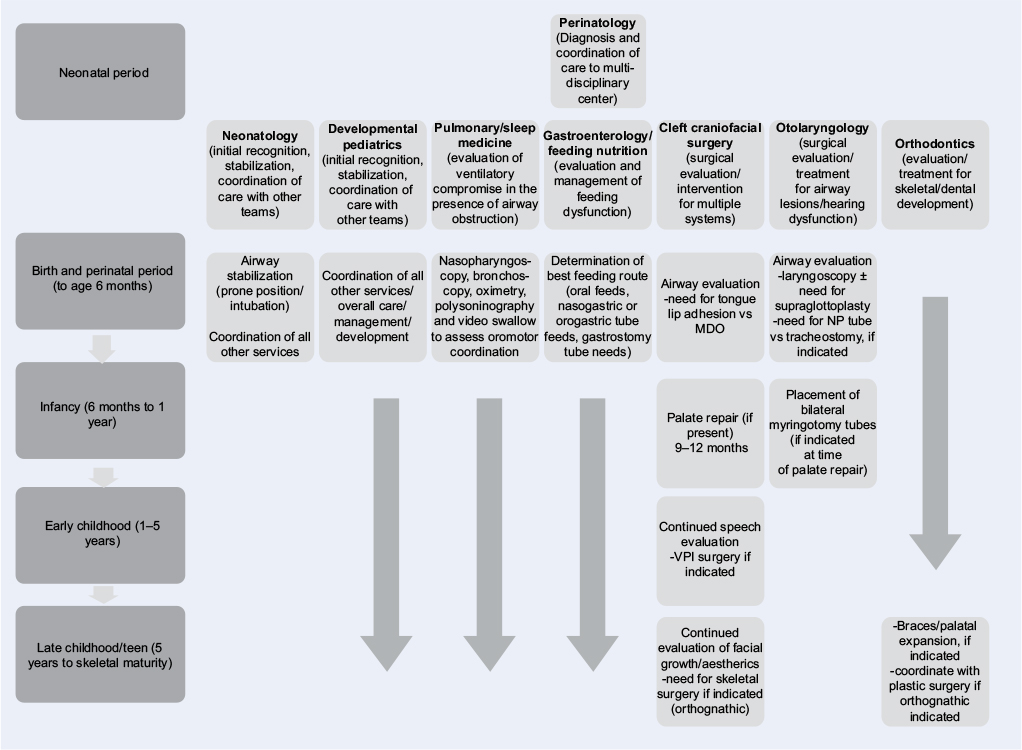 | Figure 1 A flowchart algorithm of assessment and treatment management of Robin sequence. Abbreviations: MDO, mandibular distraction osteogenesis; NP, nasopharyngeal; VPI, velopharyngeal incompetence. |
Disclosure
The authors report no conflicts of interest in this work.
References
Hilaire S. Sphenocephalus. Philos Anat. 1822;2:97–98. | ||
Fairbairn P. Suffocation in an infant from retraction of the base of the tongue, connected with defect of the frenum. Month J Med Sci. 1846;6:280–281. | ||
Robin P. La chute de la base de la langue considérée comme une nouvelle cause de gêne dans la respiration nasopharyngienne. Bull Acad Med (Paris). 1923;89:37–40. | ||
Bush PG, Williams AJ. Incidence of the Robin Anomalad (Pierre Robin syndrome). Br J Plast Surg. 1983;36(4):434–437. | ||
Isolated Pierre Robin Sequence. Available from: https://ghr.nlm.nih.gov/condition/isolated-pierre-robin-sequence - statistics. Accessed July 5, 2016. | ||
Caouette-Laberge L, Bayet B, Larocque Y. The Pierre Robin sequence: review of 125 cases and evolution of treatment modalities. Plast Reconstr Surg. 1994;93(5):934–942. | ||
Blechner M, Williamson AA. Consequences of obstructive sleep apnea in children. Curr Probl Pediatr Adolesc Health Care. 2016;46(1):19–26. | ||
Marcus CL, Carroll JL, Koerner CB, Hamer A, Lutz J, Loughlin GM. Determinants of growth in children with the obstructive sleep apnea syndrome. J Pediatr. 1994;125(4):556–562. | ||
Saal HM. Robin sequence. In: Cassidy SB, Allanson JE, editors. Management of Genetic Syndromes. 2nd ed. Hoboken (NJ): John Wiley and Sons; 2005. | ||
Pierre Robin Associated Conditions. Available from: http://www.pierrerobin.org/associated-conditions.htm. Accessed July 5, 2016. | ||
Pierre Robin Stickler Syndrome. Available from: http://www.pierrerobin.org/stickler-syndrome.htm. Accessed July 5, 2016. | ||
Pierre Robin Rare Diseases. Available from: https://rarediseases.info.nih.gov/gard/4347/pierre-robin-sequence/resources/1. Accessed July 5, 2016. | ||
Chan KC, Shi L, So HK, et al. Neurocognitive dysfunction and grey matter density deficit in children with obstructive sleep apnoea. Sleep Med. 2014;15(9):1055–1061. | ||
Grigg-Damberger M, Ralls F. Cognitive dysfunction and obstructive sleep apnea: from cradle to tomb. Curr Opin Pulm Med. 2012;18(6):580–587. | ||
Marcus CL, Brooks LJ, Draper KA, et al. Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics. 2012;130(3):576–584. | ||
Filip C, Feragen KB, Lemvik JS, et al. Multidisciplinary aspects of 104 patients with Pierre Robin sequence. Cleft Palate Craniofac J. 2015;52(6):732–742. | ||
Almajed A, Viezel-Mathieu A, Gilardino MS, Flores RL, Tholpady SS, Cote A. Outcome following surgical interventions for micrognathia in infants with Pierre Robin sequence: a systematic review of the literature. Cleft Palate Craniofac J. Epub 2016 Jul 14. | ||
Breik O, Tivey D, Umapathysivam K, Anderson P. Mandibular distraction osteogenesis for the management of upper airway obstruction in children with micrognathia: a systematic review. Int J Oral Maxillofac Surg. 2016;45(6):769–782. | ||
Evans KN, Sie KC, Hopper RA, Glass RP, Hing AV, Cunningham ML. Robin sequence: from diagnosis to development of an effective management plan. Pediatrics. 2011;127(5):936–948. | ||
Bookman LB, Melton KR, Pan BS, et al. Neonates with tongue-based airway obstruction: a systematic review. Otolaryngol Head Neck Surg. 2012;146(1):8–18. | ||
Gangopadhyay N, Mendonca DA, Woo AS. Pierre Robin sequence. Semin Plast Surg. 2012;26(2):76–82. | ||
Scott AR, Tibesar RJ, Sidman JD. Pierre Robin Sequence: evaluation, management, indications for surgery, and pitfalls. Otolaryngol Clin North Am. 2012;45(3):695–710, ix. | ||
Rogers-Vizena CR, Mulliken JB, Daniels KM, Estroff JA. Prenatal features predictive of Robin sequence identified by fetal magnetic resonance imaging. Plast Reconstr Surg. 2016;137(6):999e–1006e. | ||
Lind K, Aubry MC, Belarbi N, et al. Prenatal diagnosis of Pierre Robin Sequence: accuracy and ability to predict phenotype and functional severity. Prenat Diagn. 2015;35(9):853–858. | ||
Pilu G, Romero R, Reece EA, Jeanty P, Hobbins JC. The prenatal diagnosis of Robin anomalad. Am J Obstet Gynecol. 1986;154(3):630–632. | ||
Matsumoto M, Yanagihara T, Hanaoka U, et al. Antenatal three-dimensional sonographic features of Pierre-Robin syndrome. Case report. Gynecol Obstet Invest. 2001;51(2):141–142. | ||
Soulier M, Sigaudy S, Chau C, Philip N. Prenatal diagnosis of Pierre-Robin sequence as part of Stickler syndrome. Prenat Diagn. 2002;22(7):567–568. | ||
Evans AK, Rahbar R, Rogers GF, Mulliken JB, Volk MS. Robin sequence: a retrospective review of 115 patients. Int J Pediatr Otorhinolaryngol. 2006;70(6):973–980. | ||
Kirschner RE, Low DW, Randall P, et al. Surgical airway management in Pierre Robin sequence: is there a role for tongue-lip adhesion? Cleft Palate Craniofac J. 2003;40(1):13–18. | ||
Bull MJ, Stroup KB, Everly JS, Weber K, Doll JP. Child safety seat use for infants with Pierre Robin sequence. Arch Pediatr Adolesc Med. 1994;148(3):301–305. | ||
Amaddeo A, Abadie V, Chalouhi C, et al. Continuous positive airway pressure for upper airway obstruction in infants with Pierre Robin sequence. Plast Reconstr Surg. 2016;137(2):609–612. | ||
Meyer AC, Lidsky ME, Sampson DE, Lander TA, Liu M, Sidman JD. Airway interventions in children with Pierre Robin Sequence. Otolaryngol Head Neck Surg. 2008;138(6):782–787. | ||
Abel F, Bajaj Y, Wyatt M, Wallis C. The successful use of the nasopharyngeal airway in Pierre Robin sequence: an 11-year experience. Arch Dis Child. 2012;97(4):331–334. | ||
Anderson KD, Cole A, Chuo CB, Slator R. Home management of upper airway obstruction in Pierre Robin sequence using a nasopharyngeal airway. Cleft Palate Craniofac J. 2007;44(3):269–273. | ||
Alencar TR, Marques IL, Bertucci A, Prado-Oliveira R. Neurological development of children with isolated Robin sequence treated with nasopharyngeal intubation in early infancy. Cleft Palate Craniofac J. Epub 2016 Apr 4. | ||
Gandini D, Brimacombe J. Laryngeal mask airway for ventilatory support over a 4-day period in a neonate with Pierre Robin sequence. Paediatr Anaesth. 2003;13(2):181–182. | ||
Marston AP, Lander TA, Tibesar RJ, Sidman JD. Airway management for intubation in newborns with Pierre Robin sequence. Laryngoscope. 2012;122(6):1401–1404. | ||
Gomez-Ospina N, Bernstein JA. Clinical, cytogenetic, and molecular outcomes in a series of 66 patients with Pierre Robin sequence and literature review: 22q11.2 deletion is less common than other chromosomal anomalies. Am J Med Genet A. 2016;170(4):870–880. | ||
Costa MA, Tu MM, Murage KP, Tholpady SS, Engle WA, Flores RL. Robin sequence: mortality, causes of death, and clinical outcomes. Plast Reconstr Surg. 2014;134(4):738–745. | ||
The Inheritance of Man. Available from: https://www.omim.org/entry/261800. Accessed July 5, 2016. | ||
Tolarova MM. Pierre Robin Sequence. Medscape 2014; Available from: http://emedicine.medscape.com/article/995706-overview. Accessed July 5, 2016. | ||
Daniel M, Bailey S, Walker K, et al. Airway, feeding and growth in infants with Robin sequence and sleep apnoea. Int J Pediatr Otorhinolaryngol. 2013;77(4):499–503. | ||
Baujat G, Faure C, Zaouche A, Viarme F, Couly G, Abadie V. Oroesophageal motor disorders in Pierre Robin syndrome. J Pediatr Gastroenterol Nutr. 2001;32(3):297–302. | ||
Drescher FD, Jotzo M, Goelz R, Meyer TD, Bacher M, Poets CF. Cognitive and psychosocial development of children with Pierre Robin sequence. Acta Paediatr. 2008;97(5):653–656. | ||
Thouvenin B, Djadi-Prat J, Chalouhi C, et al. Developmental outcome in Pierre Robin sequence: a longitudinal and prospective study of a consecutive series of severe phenotypes. Am J Med Genet A. 2013;161A(2):312–319. | ||
Reddy VS. Evaluation of upper airway obstruction in infants with Pierre Robin sequence and the role of polysomnography – review of current evidence. Paediatr Respir Rev. 2016;17:80–87. | ||
Miller CK. Feeding issues and interventions in infants and children with clefts and craniofacial syndromes. Semin Speech Lang. 2011;32(2):115–126. | ||
Shin MS. Esophageal pH and combined impedance-pH monitoring in children. Pediatr Gastroenterol Hepatol Nutr. 2014;17(1):13–22. | ||
Cohen M Jr. Dysmorphology, syndromology, and genetics in plastic surgery. In: McCarthy JG, editor. Plastic Surgery. Philadelphia (PA): WB Saunders; 1990:69–112. | ||
Lee VS, Evans KN, Perez FA, Oron AP, Perkins JA. Upper airway computed tomography measures and receipt of tracheotomy in infants with Robin sequence. JAMA Otolaryngol Head Neck Surg. 2016;142(8):750–757. | ||
Rogers GF, Murthy AS, LaBrie RA, Mulliken JB. The GILLS score: part I. Patient selection for tongue-lip adhesion in Robin sequence. Plast Reconstr Surg. 2011;128(1):243–251. | ||
Tahiri Y, Viezel-Mathieu A, Aldekhayel S, Lee J, Gilardino M. The effectiveness of mandibular distraction in improving airway obstruction in the pediatric population. Plast Reconstr Surg. 2014;133(3):352e–359e. | ||
Bartlett SP, Losee JE, Baker SB. Reconstruction: craniofacial syndromes. In: Mathes SJ, editor. Plastic Surgery. Vol IV, 2nd editor. Philadelphia (PA): Elsevier; 2006:495–520. | ||
Bijnen CL, Don Griot PJ, Mulder WJ, Haumann TJ, Van Hagen AJ. Tongue-lip adhesion in the treatment of Pierre Robin sequence. J Craniofac Surg. 2009;20(2):315–320. | ||
Sedaghat AR, Anderson IC, McGinley BM, Rossberg MI, Redett RJ, Ishman SL. Characterization of obstructive sleep apnea before and after tongue-lip adhesion in children with micrognathia. Cleft Palate Craniofac J. 2012;49(1):21–26. | ||
Denny AD, Amm CA, Schaefer RB. Outcomes of tongue-lip adhesion for neonatal respiratory distress caused by Pierre Robin sequence. J Craniofac Surg. 2004;15(5):819–823. | ||
Boston M, Rutter MJ. Current airway management in craniofacial anomalies. Curr Opin Otolaryngol Head Neck Surg. 2003;11(6):428–432. | ||
Fritz MA, Sidman JD. Distraction osteogenesis of the mandible. Curr Opin Otolaryngol Head Neck Surg. 2004;12(6):513–518. | ||
Ow AT, Cheung LK. Meta-analysis of mandibular distraction osteogenesis: clinical applications and functional outcomes. Plast Reconstr Surg. 2008;121(3):54e–69e. | ||
Miloro M. Mandibular distraction osteogenesis for pediatric airway management. J Oral Maxillofac Surg. 2010;68(7):1512–1523. | ||
Basta MN, Silvestre J, Stransky C, et al. A 35-year experience with syndromic cleft palate repair: operative outcomes and long-term speech function. Ann Plast Surg. 2014;73(Suppl 2):S130–S135. | ||
Collares MV, Duarte DW, Sobral DS, Portinho CP, Faller GJ, Fraga MM. Neonatal mandibular distraction osteogenesis reduces cleft palate width and lengthens soft palate, influencing palatoplasty in patients with Pierre Robin sequence. J Craniofac Surg. 2016;27(5):1267–1272. | ||
Hardwicke JT, Richards H, Cafferky L, Underwood I, ter Horst B, Slator R. Outcomes of cleft palate repair in patients with Pierre Robin sequence: a matched case-control study. Plast Reconstr Surg. 2016;137(3):927–935. | ||
Wan T, Chen Y, Wang G. Do patients with isolated Pierre Robin sequence have worse outcomes after cleft palate repair: a systematic review. J Plast Reconstr Aesthet Surg. 2015;68(8):1095–1099. | ||
Suri S, Ross RB, Tompson BD. Mandibular morphology and growth with and without hypodontia in subjects with Pierre Robin sequence. Am J Orthod Dentofacial Orthop. 2006;130(1):37–46. | ||
Hermann NV, Kreiborg S, Darvann TA, Jensen BL, Dahl E, Bolund S. Early craniofacial morphology and growth in children with nonsyndromic Robin Sequence. Cleft Palate Craniofac J. 2003;40(2):131–143. | ||
Paradise JL, Bluestone CD. Diagnosis and management of ear disease in cleft palate infants. Trans Am Acad Ophthalmol Otolaryngol. 1969;73(4):709–714. | ||
Dhillon RS. The middle ear in cleft palate children pre and post palatal closure. J R Soc Med. 1988;81(12):710–713. | ||
Andrews BT, Fan KL, Roostaeian J, Federico C, Bradley JP. Incidence of concomitant airway anomalies when using the University of California, Los Angeles, protocol for neonatal mandibular distraction. Plast Reconstr Surg. 2013;131(5):1116–1123. | ||
Thevasagayam M, Rodger K, Cave D, Witmans M, El-Hakim H. Prevalence of laryngomalacia in children presenting with sleep-disordered breathing. Laryngoscope. 2010;120(8):1662–1666. | ||
Friedman EM, Vastola AP, McGill TJ, Healy GB. Chronic pediatric stridor: etiology and outcome. Laryngoscope. 1990;100(3):277–280. | ||
Mancuso RF, Choi SS, Zalzal GH, Grundfast KM. Laryngomalacia. The search for the second lesion. Arch Otolaryngol Head Neck Surg. 1996;122(3):302–306. | ||
Dickson JM, Richter GT, Meinzen-Derr J, Rutter MJ, Thompson DM. Secondary airway lesions in infants with laryngomalacia. Ann Otol Rhinol Laryngol. 2009;118(1):37–43. | ||
Yuen HW, Tan HK, Balakrishnan A. Synchronous airway lesions and associated anomalies in children with laryngomalacia evaluated with rigid endoscopy. Int J Pediatr Otorhinolaryngol. 2006;70(10):1779–1784. | ||
Filston HC, Ferguson TB Jr, Oldham HN. Airway obstruction by vascular anomalies. Importance of telescopic bronchoscopy. Ann Surg. 1987;205(5):541–549. | ||
Sher AE, Shprintzen RJ, Thorpy MJ. Endoscopic observations of obstructive sleep apnea in children with anomalous upper airways: predictive and therapeutic value. Int J Pediatr Otorhinolaryngol. 1986;11(2):135–146. | ||
Marcus CLB, Draper LJ, Gozal D, et al. Pediatric Clinical Practice Guidelines & Policies: A Compendium of Evidence-Based Research for Pediatric Practice. 7th ed. Elk Grove Village (IL): American Academy of Pediatrics; 2007. | ||
Pearce MS, Salotti JA, Little MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet. 2012;380(9840):499–505. | ||
Ellis H. The Hopkins rod-lens system. J Perioper Pract. 2007;17(6):272–274. | ||
Alipour F, Scherer RC. Pressure and velocity profiles in a static mechanical hemilarynx model. J Acoust Soc Am. 2002;112(6):2996–3003. | ||
Olney DR, Greinwald JH Jr, Smith RJ, Bauman NM. Laryngomalacia and its treatment. Laryngoscope. 1999;109(11):1770–1775. | ||
Richter GT, Thompson DM. The surgical management of laryngomalacia. Otolaryngol Clin North Am. 2008;41(5):837–864, vii. | ||
Thompson DM. Laryngomalacia: factors that influence disease severity and outcomes of management. Curr Opin Otolaryngol Head Neck Surg. 2010;18(6):564–570. | ||
Shoemaker MT, Pierce MR, Yoder BA, DiGeronimo RJ. High flow nasal cannula versus nasal CPAP for neonatal respiratory disease: a retrospective study. J Perinatol. 2007;27(2):85–91. | ||
Reddy DK, Matt BH. Unilateral vs. bilateral supraglottoplasty for severe laryngomalacia in children. Arch Otolaryngol Head Neck Surg. 2001;127(6):694–699. | ||
Hermann NV, Kreiborg S, Darvann TA, Jensen BL, Dahl E, Bolund S. Craniofacial morphology and growth comparisons in children with Robin Sequence, isolated cleft palate, and unilateral complete cleft lip and palate. Cleft Palate Craniofac J. 2003;40(4):373–396. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.