")
Back to Journals » Journal of Blood Medicine » Volume 14
RNAi for the Treatment of People with Hemophilia: Current Evidence and Patient Selection
Authors Boyce S , Rangarajan S
Received 11 November 2022
Accepted for publication 31 March 2023
Published 22 April 2023 Volume 2023:14 Pages 317—327
DOI https://doi.org/10.2147/JBM.S390521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Sara Boyce,1 Savita Rangarajan2
1Haemophilia Comprehensive Care Centre, University Hospital Southampton, Southampton, UK; 2Faculty of Medicine, University of Southampton, Southampton, UK
Correspondence: Sara Boyce, Email [email protected]
Abstract: Severe hemophilia is associated with spontaneous, prolonged and recurrent bleeding. Inadequate prevention and treatment of bleeding can lead to serious morbidity and mortality. Due to the limitations of intravenous clotting factor replacement, including the risk of inhibitory antibodies, innovative novel therapies have been developed that have dramatically changed the landscape of hemophilia therapy. Ribonucleic acid interference (RNAi) has brought the opportunity for multiple strategies to manipulate the hemostatic system and ameliorate the bleeding phenotype in severe bleeding disorders. Fitusiran is a RNAi therapeutic that inhibits the expression of the natural anticoagulant serpin antithrombin. Reduction in antithrombin is known to cause thrombosis if coagulation parameters are otherwise normal and can rebalance hemostasis in severe hemophilia. Reports from late stage clinical trials of fitusiran in hemophilia A and B participants, with and without inhibitory antibodies to exogenous clotting factor, have demonstrated efficacy in preventing bleeding events showing promise for a future “universal” prophylactic treatment of individuals with moderate-severe hemophilia.
Keywords: hemophilia, RNAi, siRNA, fitusiran, inhibitor, ribonucleic acid interference, small integral RNA
Introduction
Hemophilia A and B are inherited X-linked recessive bleeding disorders caused by mutations in coagulation factor VIII (hemophilia A) and IX (hemophilia B). These are essential proteins of the blood coagulation system that involves a cascade of zymogen activation that leads to the production of the serine protease thrombin, and ultimately cross-linked fibrin to form a stable blood clot.1 Functional deficiency in coagulation factors VIII and IX results in abnormal hemostasis and excessive bleeding. In untreated severe hemophilia, where factor levels are <1% of normal, spontaneous bleeding occurs that frequently affects joints and can involve critical organs. A similar bleeding phenotype, with less frequent spontaneous bleeding, can occur in moderate hemophilia where factor levels are 2–5%. Onset of bleeding often begins in infancy, and from early childhood recurrent spontaneous joint bleeding can develop with consequential chronic painful and disabling joint arthropathy.2 The current standard of treatment is replacement of the deficient factor VIII or IX with recombinant or plasma-derived factors. Clotting factor replacement is effective at preventing and treating the complications of hemophilia,3 but usually requires regular intravenous infusions 2–4 times a week with standard half-life factors. Extended half-life factors reduce the frequency of infusions, but individuals with severe hemophilia A still require intravenous prophylaxis 2–3 times a week.4,5 Frequent intravenous treatment is inconvenient and even distressing, particularly for infants, young children and their parents/caregivers where central venous access is often required that carries risk of infection.6 Lack of compliance comes hand-in-hand with difficulties of administering treatment and the complications of bleeding ensue.7 Additionally there is a risk of alloantibodies against the factor treatment, termed inhibitors, that occurs in approximately 30% of people with severe hemophilia A and 10% in severe hemophilia B.8,9 Inhibitors render standard treatment ineffective making patients more prone to severe and disabling bleeding, and treatment to eliminate inhibitors is costly, invasive and has a high failure rate of approximately 20–40% in hemophilia A10 and 70% in hemophilia B.11 Apart from immune tolerance therapy to eliminate inhibitors conventional treatment has been to administer bypassing agents, recombinant factor VIIa or activated prothrombin complex concentrate (aPCC) either on demand (OD) for bleeding, or as prophylaxis12 that, again, is costly and invasive, requiring good venous access.
Due to the limitations of traditional treatments for hemophilia A and B these disorders have become a target for innovative strategies to overcome these challenges. The explosion of novel treatments that can transform the lives of people with hemophilia (PwH) has made this a compelling area of medicine over recent years. A major breakthrough in the management of haemophilia A and inhibitors arrived with the development of a bispecific antibody that partially mimics factor VIII activity, emicizumab (F.Hoffman-La Roche manufactured by Genentech).13 This treatment is the first licensed subcutaneous treatment for hemophilia and has become an established prophylactic treatment in inhibitor and non-inhibitor patients.14,15
Making PwH free of regular prophylactic treatment altogether is now an attainable goal with gene therapy. As hemophilia is a monogenic disorder it has always been an attractive target for gene therapy and immense progress has been made in its clinical development.16 Though gene therapy has demonstrated great promise as a potential cure for hemophilia, immunological responses to the viral vector can lead to treatment failure and emerging long-term data has shown responses may not be sustained in hemophilia A.17 Additionally there are concerns regarding long-term safety of gene therapy, particularly viral integration into the host genome and oncogenesis.18 Gene therapy may be an opportunity to overcome the barriers to successful treatment of inhibitors and the efficacy of gene therapy in this patient population is being studied (ClinicalTrials.gov Identifier: NCT03734588).19,20
Emicizumab has had great success due to the convenience of administration and stable pharmacological effect, which has fueled the demand for other treatments administered in a similar manner that can encompass a broader patient population. This can be achieved by targeting the natural inhibitors of coagulation. Normal hemostasis relies on an intricate balance of the generation of thrombin though the activation of coagulation proteins and their inhibition with regulatory proteins.21 Deficiency of the key regulatory anticoagulant proteins are known to increase the risk of venous thrombosis in a person with otherwise normal coagulation proteins.22 In hemophilia these deficiencies have been shown to ameliorate the hemophilia bleeding phenotype.23 Manipulating natural inhibitors of coagulation has been demonstrated to effectively restore functional hemostasis using antibody neutralization of tissue factor pathway inhibitor24 and protease nexin 1,25 activated protein C specific serpin inhibition26 and antithrombin inhibition by nanobodies.27 Another strategy to rebalance hemostasis in hemophilia is to prevent the expression of the coagulation-inhibitory serpins is by utilizing ribonucleic acid interference (RNAi).
RNA Interference
RNAi, otherwise known as post-transcriptional gene silencing, is a highly conserved pathway whereby expression of a particular gene is inhibited through the targeting and destruction of an mRNA molecule by an RNA molecule.28 This ancient process protects eukaryotic cells against invasion by exogenous genes, such as viral DNA or double-stranded RNA, and regulates expression of protein-coding genes.29 Gene expression is inhibited when short RNA molecules, either micro RNAs (miRNA) or small integral RNAs (siRNA) bind to and functionally inactivate endogenous mRNA.
The concept of RNAi arose when, after attempts to overexpress the chalcone synthase gene in Petunia plants, expression of pigment was inhibited resulting in white flowers.30 The term RNAi was coined by biotechnologist Craig Mello and Andrew Fire in 1998 with the discovery that double-stranded RNAs were the cause of post-translational gene silencing in the nematode worm Caenorhabditis elegans.31 This RNAi phenomenon led to the discovery of siRNA, typically double-stranded RNA molecules 20–25 nucleotides in length, that could induce RNAi silencing in mammalian cells.32,33 It was recognized siRNAs can potentially suppress any disease-related gene of interest and it has been a long and complex process to harness the widespread therapeutic possibilities. Unmodified siRNA is rapidly degraded and will not accumulate into the intended tissue.29 In early clinical studies off-target effects were demonstrated with unmodified or slightly modified siRNA-targeting vascular endothelial growth factor A and R1 (VEGFA and VEGFR1) for local administration in age related macular degeneration.34 Targeted siRNA against VEGFA or VEGFR1 to suppress choroidal neovascularization from age related macular degeneration via Toll-like receptor 3 and its adaptor molecule TRIF induced secretion of proinflammatory cytokines IL-12 and IFNy.34,35 Further barriers against efficacy and safety include enzymatic degradation by endogenous nucleases, rapid clearance, immune recognition, and egress from the bloodstream into the desired tissue.29,36 Some of these barriers have been overcome with chemical modification of siRNAs that can reduce off-target toxicity and enhance specificity and activity without compromising the siRNA activity. Phosphonate and ribose modification prevents siRNA degradation and are utilized in the Alnylam Pharmaceuticals Standard Template Chemistry universal modification pattern.37 These modifications have made the theory of siRNA a clinically applicable reality and revolutionized therapeutics by circumventing the challenges of small molecules or antibodies having to recognize complex protein conformations. This technology was successfully applied with the first FDA approved (August 2018) siRNA, patisiran (ONPATTRO) to deliver chemically modified anti-transthyretin siRNA to treat hereditary amyloidogenic transthyretin amyloidosis with polyneuropathy.38 Patisiran employs lipids to protect entrapped siRNA from nuclease attack and renal clearance and transport to target tissue.29 After intravenous administration of patisiran ionizable lipid nanoparticles associate with apolipoproteins facilitating endocytosis into liver.39 During this period of siRNA development therapeutic applications in hemostasis had been explored. Lipid-based reagents had similarly allowed successful delivery of siRNAs to mouse livers in the first synthetic siRNA study on modifying the expression of liver coagulation proteins via hepatocyte nuclear factor 4a and CCAAT/enhancer binding protein 2 transcription factors.40 Using the same mode of delivery siRNA silencing of the genes for the serpins protein C and antithrombin led to severe coagulopathy and macrothrombosis in multiple tissues in mice.41
Another method to surmount the evolutionary defenses to keep invading RNA outside of the cell was the design of N-acetylgalactosamine (Ga1NAc)-siRNA conjugates.42 Suppressing the expression of proteins produced by hepatocytes requires facilitating the entry of siRNA circulating in the blood stream after parenteral administration into the cytoplasm of hepatocytes; GA1NAc was utilized for this purpose as it avidly binds to the Asiaglycoprotein, which is highly expressed on hepatocytes and endocytosed. GA1NAc-siRNA conjugates can thus enter the hepatocyte and target cellular RNA and influence protein expression. This was successfully utilized in givosiran, the second FDA approved siRNA to treat acute hepatic porphyrias.43
Fitusiran
Fitusiran (ALN-AT3, Alnylam/Sanofi) is a RNAi therapeutic developed for prophylaxis of bleeding in hemophilia A and B. This synthetic siRNA is covalently linked to a triantennary Ga1NAc ligand. The Ga1NAc-siRNA conjugate specifically targets antithrombin messenger RNA (mRNA) to lower antithrombin production (Figure 1). Antithrombin, encoded by the SERPINC1 gene, is a serpin produced by the liver that has an essential role in maintaining the balance of hemostasis. It functions as a natural anticoagulant serpin by potent inhibition of thrombin, and to other serine proteases including factors Xa and IXa. Antithrombin has a half-life of 2–3 days, normal plasma concentrations are 112–140 µg/mL and the standard reference range is 80–120% of normal.44 Individuals with inherited deficiency of antithrombin usually have levels of 40–60%;this carries a lifetime risk of venous thrombosis and is well known to cause a more severe thrombotic tendency than other inherited thrombophilias.45,46 Homozygous antithrombin deficiency usually causes in utero death, but rare cases have been reported.44 Examination of the small number of reported cases of individuals with severe hemophilia and co-inherited thrombophilia has indicated reduction in the severity of their bleeding phenotype.23,47,48 Murine studies showed subcutaneous injection of fitusiran caused potent and durable reduction of antithrombin in both wild-type and hemophilia A mice, with an associated increased in thrombin generation and enhanced hemostasis.49 In the Phase I dose escalation study to evaluate the safety and tolerability of subcutaneous fitusiran in healthy volunteers and non-inhibitor moderate-severe adult hemophilia A and B participants antithrombin levels were reduced by approximately 50% at doses between 0.015mg/kg and 1.8mg/kg (Table 1).50 Peak fitusiran plasma levels occurred 2–6 hours after administration and rapidly decreased with a mean elimination half-life of 2.6–5.3 hours. Plasma levels of fitusiran increased and antithrombin decreased in a dose-dependent manner in the hemophilia patients (Table 1). Lowering of antithrombin was associated with increased thrombin generation at similar levels in hemophilia A and B patients. When antithrombin levels was reduced >75% from baseline peak thrombin generation values were at the lower end of the range observed in the healthy participants and thrombin generation values were similar to those described in mild hemophilia.51 There was a prolonged pharmacological effect with AT recovery at a median slope of 10–15% per month after fitusiran discontinuation indicating this treatment offers consistent hemostasis over time. Further exploratory analysis suggested this translates as fewer bleeding episodes in patients, and reported episodes were successfully treated with clotting factor replacement. Fitusiran was well tolerated with injection site reactions and arthralgia being the most reported adverse events. Transient increases in in liver aminotransferase was observed in 36% of participants and one participant had reactivation of hepatitis C infection that was thought unrelated to the study drug. No patients developed anti-fitusiran antibodies.
 |
Table 1 Comparison of Median Nadir Lowering of at with Varying Fitusiran Dosing Regimens |
 |
Figure 1 Diagram of the mechanism of action of fitusiran. After entering the hepatocyte the Ga1NAc- siRNA conjugate binds with a ribonucleoprotein forming a RNA-induced silencing complex (RISC). It targets AT3 mRNA and silences the gene, thus inhibiting antithrombin production. Consequently FIXa, Xa and thrombin lack inhibition and there is increased generation of thrombin to form blood clots. Created with BioRender.com. |
Due to the small sample size the phase I study was extended to evaluate the long-term safety and tolerability. In this extended study fatal cerebral venous sinus thrombosis occurred after factor VIII concentrate was repeatedly administered after an initial misdiagnosis of subarachnoid hemorrhage and all fitusiran trials were temporarily halted.52 The US Food and Drug Administration allowed the studies to continue after the trial protocol was amended to protect participants from the risk of thrombosis. For bleeding episodes lower doses of factor must be used; factor VIII 10–20 IU/kg or factor IX 20–30 IU/kg.53 In vitro data on thrombin generation of bypassing agents and antithrombin reduction means it is likely lower doses are required54 so aPCC 30–50 units/kg (standard dose 50–100 units/kg) or Factor VIIa < 45 mg/kg (standard dose 90mg/kg) should be given for bleeding in participants with inhibitors. Should a thrombotic episode occur in a PwH receiving fitusiran AT concentrate should be administered aiming for AT levels of 80–120%, in combination with anticoagulation and clotting factor or BPA replacement.
The trial program was extended to an open label Phase I, part D, study of efficacy in hemophilia participants with inhibitors.55 Participants were treated with once monthly fixed subcutaneous doses of fitusiran at 50mg or 80mg. Again, the most common adverse events were mild and transient injection site reactions and increases in liver aminotransferases that were all assessed as mild to moderate reactions that did not require fitusiran suspension. The hepatotoxicity of Ga1NAc- siRNA has been shown as mainly due to RNAi-mediated off target gene silencing.56 Due to the proportion of PwH with a history of hepatitis C infection, with 12/17 participants affected, this is thought to contribute to the risk of transaminitis related to this liver-specific therapeutic. Elevated d-dimer was measured as a potential predictor of thrombosis and was observed in 47%, more with the 80mg dose (55%) compared to the 50mg dose (33%). As d-dimer is also an acute phase reactant the clinical significance of this is unknown. Peak plasma levels were similar to the phase I study at 4 hours. The mean nadir AT level was 18% in the 50mg dose group and 12.5% in the 80mg dose group corresponding as 82% and 87.4% reduction from baseline (Table 1). As demonstrated previously reduced AT correlated with increased thrombin generation. Overall bleeding episodes were reduced with an improvement in quality of life; 65% participants reported no bleeding episodes (Table 2). The median aPCC dose used was 28.6 U/kg and no thrombotic events occurred. This study suggested a fixed monthly fitusiran dose is safe and efficacious for long-term treatment of inhibitor and non -inhibitor hemophilia patients.
 |
Table 2 Dosing Regimens and Bleeding Events in Published Fitusiran Studies |
The ATLAS trial program continued to expand (Table 3) and recently the findings of the Phase III ATLAS-A/B (ALN-AT3SC-004) study of the safety and efficiency of fitusiran in hemophilia A and B participants aged >12 years without inhibitors and previously receiving standard OD factor treatment were presented.57 Participants were randomized to receive their usual OD treatment or monthly fitusiran 80mg s/c. Of the 79 participants that received fitusiran 50.6% did not have any bleeding episodes requiring factor treatment compared with 5% in the OD arm. The median annualized bleeding rate (ABR) was 0.0, with a clinically meaningful and statistically significant 89.9% reduction in ABR compared to the OD arm. No thrombotic events occurred during the study.
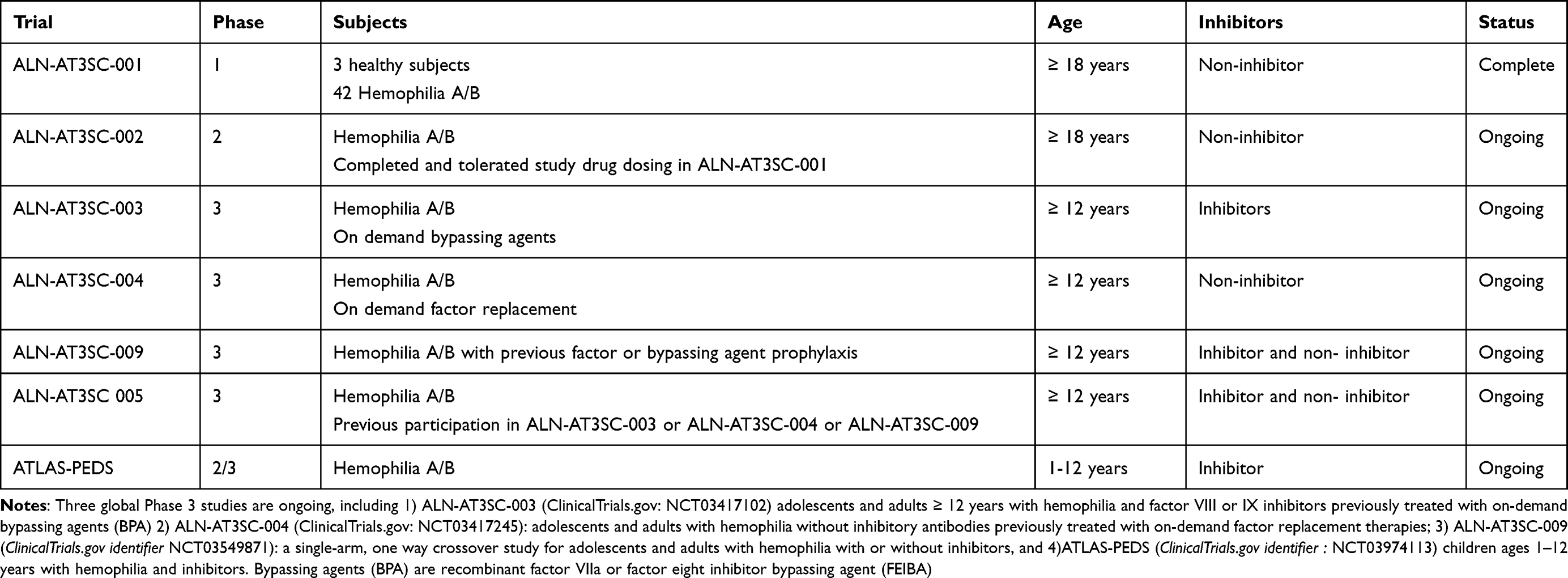 |
Table 3 A Summary of the ATLAS program Fitusiran Studies |
The ATLAS-INH (ALN-AT3SC-003) study evaluated the safety and efficacy of fitusiran in inhibitor patients >12 years and randomized participants to receive either 80mg fitusiran monthly or OD bypassing treatment.58 Again, there was a significantly statistical reduction in ABR of 90.8% and 65.8% and fitusiran-receiving participants had zero bleeds compared with 5.8% in the OD arm. Seven patients in the fitusiran arm had treatment emergent serious adverse effects, including subclavian vein thrombosis and thrombosis. One patient with a spinal vascular disorder and thrombosis had study treatment discontinuation.
Fitusiran in Other Rare Inherited Bleeding Disorders
As fitusiran does not target a specific coagulation factor it should theoretically prevent bleeding in other rare bleeding disorders. Addition of fitusiran to the plasma of patients severely deficient in factor V, VII or X has demonstrated improvement in thrombin generation.59 AT-targeting RNAi may therefore be a therapeutic option for patients with rare bleeding disorders requiring prophylaxis where there is limited or no availability of clotting factor concentrate.
Other Strategies for RNAi Therapeutics in Hemophilia
Protein S
Protein S (PS) is a natural anticoagulant serpin encoded by the PROS1 gene. It regulates the generation of thrombin by acting as a cofactor for activated protein C in the inactivation of coagulation factors Va and factor VIIIa.60 It is also a cofactor for tissue factor pathway inhibitor that inhibits factor Xa.61 Homozygous PS deficiency can cause purpura fulminans and fatal disseminated intravascular coagulation (DIC).62 Heterozygous PS deficiency is associated with venous thromboembolic events.22 Like AT this regulator of hemostasis is an attractive target for RNAi therapeutics in the prevention of hemophilia-associated bleeding. Murine PS siRNA administered to hemophilia A (F8−/−) and hemophilia B (F9−/−) mice reduced bleeding in tail-bleeding assays, prevented intraarticular bleeding and PS expression was reduced in both the plasma and synovium.63 High expression of PS and TFPI by synovial cells was found indicating this PS as a RNAi therapeutic target may further protect the joints in individuals prone to hemophilic arthropathy. Further studies to evaluate the safety of Ga1NAc conjugated to PS siRNA showed it was well tolerated mice with no evidence of DIC making further progress towards Phase 1 trials.64
Heparin Cofactor II
Heparin cofactor II (HCII) is a serpin secreted by the liver and encoded by the SERPIND1 gene.65 Like AT, HCII has an essential role in regulating hemostasis through inhibition of thrombin.66 HCII inactivates thrombin less efficiently than AT but glycosaminoglycans, such as heparin and dermatan sulfate enhance its effect. Dermatan sulfate is found mainly in the walls of blood vessels and has a particularly potent catalytic effect catalyzing HCII. This indicates HCII primarily exerts its effect in connective tissue. Like the better described inherited thrombophilias, deficiency of HCII has been associated with clinical thrombosis,67 but other studies have shown the frequency of HCII deficiency in people with thrombosis is similar to healthy individuals.68 The effect of RNAi on HCII has been studied in mice by conjugating siRNA-HCII with Ga1NAc.69 This study suggested that by inhibition of HCII thrombin generation could be upregulated and hemostatic ability improved thus making HCII a potential target for hemophilia treatment.
Selection of Hemophilia Patients for RNAi Therapy
The development of plasma fractionation in the 1970s made the quality of life and life expectancy of PwH begin to increase. Transmission of hepatitis B and C and HIV through contaminated blood products to treat hemophilia tragically caused the loss of many lives. Now with recombinant clotting factors and successful antiviral therapies a significant rise in life expectancy is being seen.70 As the hemophilia population ages, with associated co-morbidities, there are added complexities to their care. The ATLAS trial participants do not represent the entire hemophilia population as they were carefully selected by their clinicians and trial criteria, with key exclusion of inherited thrombophilia and thrombotic history. Fitusiran, therefore, cannot be presumed to be a “one size fits all” prophylactic treatment for severe hemophilia. As Phase IV and real-world data are accumulated clinicians are likely to select patients in line with the ATLAS program enrollment criteria. As inherited thrombophilia affects over 7% of certain populations some PwH will not be able to receive fitusiran based on thrombophilia testing alone.71,72 Data is needed to examine if the least prothrombotic and more frequent factor V Leiden and prothrombin G20210A mutations in conjunction with siRNA antithrombin suppression translates as a clinical thrombotic risk. The risk of thrombosis demonstrated in the ATLAS trials can be minimized through screening patients for thrombotic risk factors, modification of cardiovascular risk factors and cautious treatment of bleeding episodes. As well as the need to have a normal baseline AT prior to commencing fitusiran the AT response should be monitored in clinical practice to select which patients can safely continue fitusiran. Evaluation of the thrombotic events related to fitusiran suggest there is association with AT levels of <10%. Therefore, as with ongoing trials, patients with AT levels of <15% should have fitusiran withdrawn. It has been reported to achieve the desirable ABR while minimizing adverse thrombotic events there should be an upper AT threshold with target levels of 35.73 Future studies aim to further mitigate the risk of thrombosis by starting with 50mg dosing 2 monthly, and adjusting the dose and frequency based on the AT levels. The outcome of this study will guide clinicians on whether lower initial doses and tight control of AT target levels will minimize thrombosis risk enough to encompass a broader population of hemophilia patients, particularly those with cardiovascular risk factors, including rising age.
The lifestyle of a PwH may impact their selection for treatment. Though it has been established the risk of spontaneous bleeding with fitusiran is low, in clinical practice we are so far unable to precisely quantity the risk of bleeding from activities with high-risk physical impact. PwH who frequently play sports that carry a significant risk of head injury may be advised to use clotting factors prior to activity so there is confidence of a normal level of hemostasis, e.g., factor VIII or IX levels measurable at >50%. The same may apply to a PWH with an occupation that puts them at risk of major injury and bleeding.
It also needs to be considered increased bleeding risk can emerge in the ageing population, such as hemorrhagic stroke, bowel angiodysplasia or malignancy.74 These co-morbidities alongside hemophilia may require the need for prophylaxis for the first time in the life of a PwH, or frequency of prophylaxis may need to be increased. Venous access is often challenging in the elderly, and vein fragility along with restricted joint movements due to osteoarthritis or hemophilic arthropathy may render self-treatment with intravenous infusions unfeasible and makes subcutaneous fitusiran, every 1–2 months, as attractive and convenient option. In these cases the bleeding and thrombotic risks would need to be carefully weighed against each other before a treatment decision is made.
Difficulties with venous access is not restricted to the elderly and is a major challenge in the early years of life. Traditional treatment of inhibitors, that most commonly affects children, requires high frequency intravenous infusions of clotting factor and/or BPAs. Recent data has reported the frequency of inhibitors in hemophilia B to be higher than previous findings;9 emicizumab cannot be used in this population and it is therefore likely they will be prioritized for fitusiran due to the unmet need and the high potential to turnaround the quality of life of the PwH and their caregivers.
PwH in the ATLAS trial programme were under the care of hemophilia centres and able to receive high quality medical care. This does not reflect global hemophilia care and access is a worldwide problem. Only 30% have access to clotting factor replacement therapy75 and up to 10% of children with severe hemophilia in the developing world sustain intracranial hemorrhage.76 Fitusiran could benefit PwH in developing countries with limited availability of medical care as the infrequent dosing and ease of administration has the potential to mitigate some of the difficulties with refrigerated storage and access to hospitals for instance with mobile monthly dosing clinics.
Conclusion
The goal of hemophilia therapy is to produce steady state hemostasis through the restoration of thrombin. With unprecedented progress in hemophilia treatments there has been a paradigm shift with expectations rising to aim for comparable quality of life to people without hemophilia.12 RNAi therapeutics have been a major contributor to this change in landscape with numerous possibilities of rebalancing hemostasis through targeted suppression of serpin mRNA. This not only provides opportunities in the improving the care of PwH, but other people with rare severe bleeding disorders that have limited treatment options. Fitusiran is in late stages of clinical development and has been studied in over 200 participants in the international ATLAS clinical program. It has demonstrable efficacy in preventing bleeds through targeted reduction in antithrombin and restoration of thrombin generation in adolescents and adults with severe-moderate hemophilia A and B. Fitusiran differs from hemophilia treatments licensed to date as it is not only effective if inhibitors are present but also in both hemophilia A and B. If fitusiran is shown to be effective and safe in children, as is currently being assessed in Phase II and III studies, it has the potential to be an almost universal prophylactic treatment for hemophilia, with some caveats in patient selection. As the effective dosing regimens of 50mg and 80mg given subcutaneously every 1–2 months are much less frequent than standard prophylaxis with clotting factor infusions it could to transform the quality of life of PwH, particularly for those with inhibitors. Long-term morbidity from hemophilic arthropathy may be prevented with the greatest benefit offered to those with severe hemophilia managed with OD clotting factor treatment. Infrequent injections with sustained pharmacological effects has the additional benefit of granting ‘hemophilia-free days’, where the PwH is relieved of the psychological burden of their disease.77
Overall fitusiran may transform lives of PwH across the world but should be prescribed after careful consideration, with particular attention to thrombotic risk factors, and with ongoing caution until there is enough data to enable clinicians to confidently select patients appropriately.
Disclosure
Dr Sara Boyce reports grants from Sangamo Therapeutics Ltd, personal fees, advisory board and speaker fees from CSL Behring, outside the submitted work. Dr Savita Rangarajan reports being a principal investigator in ongoing clinical Trials from Sanofi, during the conduct of the study; consultant from Reliance Life Sciences, advisory board from Pfizer, Sigilon, and Takeda, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Hoffman M, Monroe DM. Coagulation 2006: a Modern View of Hemostasis. Hematol Oncol Clin North Am. 2007;21(1):1–11. doi:10.1016/j.hoc.2006.11.004
2. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus Episodic Treatment to Prevent Joint Disease in Boys with Severe Hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
3. Collins PW, Blanchette VS, Fischer K, et al. Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost. 2009;7(3):413–420. doi:10.1111/j.1538-7836.2008.03270.x
4. Young G, Mahlangu JN. Extended half-life clotting factor concentrates: results from published clinical trials. Haemophilia. 2016;22:25–30. doi:10.1111/hae.13028
5. Collins P, Chalmers E, Chowdary P, et al. The use of enhanced half-life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia. 2016;22(4):487–498. doi:10.1111/hae.13013
6. Khair K, Ranta S, Thomas A, Lindvall K; PedNet study group. The impact of clinical practice on the outcome of central venous access devices in children with haemophilia. Haemophilia. 2017;23(4):e276–e281. doi:10.1111/hae.13241
7. García-Dasí M, Aznar JA, Jiménez-Yuste V, et al. Adherence to prophylaxis and quality of life in children and adolescents with severe haemophilia A. Haemophilia. 2015;21(4):458–464. doi:10.1111/hae.12618
8. Osooli M, Berntorp E. Inhibitors in haemophilia: what have we learned from registries? A systematic review. J Intern Med. 2015;277(1):1–15. doi:10.1111/joim.12301
9. Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2020;106(1):123–129. doi:10.3324/haematol.2019.239160
10. Peyvandi F, Makris M. Inhibitor development in haemophilia. Haemophilia. 2017;23:3. doi:10.1111/hae.13145
11. Meeks SL, Batsuli G. Hemophilia and inhibitors: current treatment options and potential new therapeutic approaches. Hematology. 2016;2016(1):657–662. doi:10.1182/asheducation-2016.1.657
12. Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia. Haemophilia. 2020;26(S6):1–158. doi:10.1111/hae.14046
13. Young G, Liesner R, Chang T, et al. A multicenter, open-label Phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127–2138. doi:10.1182/blood.2019001869
14. Callaghan MU, Negrier C, Paz-Priel I, et al. Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1-4 studies. Blood. 2021;137(16):2231–2242. doi:10.1182/blood.2020009217
15. Holstein K, Albisetti M, Bidlingmaier C, et al. Practical Guidance of the GTH Haemophilia Board on the Use of Emicizumab in Patients with Haemophilia A. Hämostaseologie. 2020;40(05):561–571. doi:10.1055/a-1127-6476
16. Leebeek FWG, Miesbach W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood. 2021;138(11):923–931. doi:10.1182/blood.2019003777
17. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N Engl J Med. 2020;382(1):29–40. doi:10.1056/NEJMoa1908490
18. Donsante A, Miller DG, Li Y, et al. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science. 2007;317(5837):477. doi:10.1126/science.1142658
19. Arruda VR, Samelson-Jones BJ. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J Thromb Haemost JTH. 2016;14(6):1121–1134. doi:10.1111/jth.13331
20. Batty P, Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum Mol Genet. 2019;28(R1):R95–R101. doi:10.1093/hmg/ddz157
21. Monroe DM, Hoffman M. What Does It Take to Make the Perfect Clot? Arterioscler Thromb Vasc Biol. 2006;26(1):41–48. doi:10.1161/01.ATV.0000193624.28251.83
22. Khan S, Dickerman JD. Hereditary thrombophilia. Thromb J. 2006;4(1):15. doi:10.1186/1477-9560-4-15
23. Shetty S, Vora S, Kulkarni B, et al. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol. 2007;138(4):541–544. doi:10.1111/j.1365-2141.2007.06693.x
24. Chowdary P. Inhibition of Tissue Factor Pathway Inhibitor (TFPI) as a Treatment for Haemophilia: rationale with Focus on Concizumab. Drugs. 2018;78(9):881–890. doi:10.1007/s40265-018-0922-6
25. Aymonnier K, Kawecki C, Venisse L, et al. Targeting protease nexin-1, a natural anticoagulant serpin, to control bleeding and improve hemostasis in hemophilia. Blood. 2019;134(19):1632–1644. doi:10.1182/blood.2019000281
26. Polderdijk SGI, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood. 2017;129(1):105–113. doi:10.1182/blood-2016-05-718635
27. Barbon E, Ayme G, Mohamadi A, et al. Single‐domain antibodies targeting antithrombin reduce bleeding in hemophilic mice with or without inhibitors. EMBO Mol Med. 2020;12:4. doi:10.15252/emmm.201911298
28. Setten RL, Rossi JJ, Han S. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421–446. doi:10.1038/s41573-019-0017-4
29. Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020;5(1):101. doi:10.1038/s41392-020-0207-x
30. Napoli C, Lemieux C, Jorgensen R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell. 1990;2:279–289. doi:10.1105/tpc.2.4.279
31. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi:10.1038/35888
32. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi:10.1038/35078107
33. Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci. 2001;98(17):9742–9747. doi:10.1073/pnas.171251798
34. Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591–597. doi:10.1038/nature06765
35. Barakat MR, Kaiser P. VEGF inhibitors for the treatment of neovascular age-related macular degeneration. Expert Opin Investig Drugs. 2009;18(5):637–646. doi:10.1517/13543780902855316
36. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi:10.1038/nrd2742
37. Maraganore J. Reflections on Alnylam. Nat Biotechnol. 2022;40(5):641–650. doi:10.1038/s41587-022-01304-3
38. Weng Y, Xiao H, Zhang J, Liang XJ, Huang Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol Adv. 2019;37(5):801–825. doi:10.1016/j.biotechadv.2019.04.012
39. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543–552. doi:10.1038/nrg3978
40. Safdar H, Cheung KL, Vos HL, et al. Modulation of Mouse Coagulation Gene Transcription following Acute In Vivo Delivery of Synthetic Small Interfering RNAs Targeting HNF4α and C/EBPα. PLoS One. 2012;7(6):e38104. doi:10.1371/journal.pone.0038104
41. Safdar H, Cheung KL, Salvatori D, et al. Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production. Blood. 2013;121(21):4413–4416. doi:10.1182/blood-2012-11-465674
42. Springer AD, Dowdy SF. GalNAc-siRNA Conjugates: leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018;28(3):109–118. doi:10.1089/nat.2018.0736
43. Sardh E, Harper P, Balwani M, et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N Engl J Med. 2019;380(6):549–558. doi:10.1056/NEJMoa1807838
44. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review: AT DEFICIENCY. Haemophilia. 2008;14(6):1229–1239. doi:10.1111/j.1365-2516.2008.01830.x
45. Lijfering WM, Brouwer JLP, Veeger NJGM, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood. 2009;113(21):5314–5322. doi:10.1182/blood-2008-10-184879
46. Croles F, Borjas-Howard J, Nasserinejad K, Leebeek F, Meijer K. Risk of Venous Thrombosis in Antithrombin Deficiency: a Systematic Review and Bayesian Meta-analysis. Semin Thromb Hemost. 2018;44(04):315–326. doi:10.1055/s-0038-1625983
47. Escuriola Ettingshausen C, Halimeh S, Kurnik K, et al. Symptomatic onset of severe hemophilia A in childhood is dependent on the presence of prothrombotic risk factors. Thromb Haemost. 2001;85(2):218–220. doi:10.1055/s-0037-1615679
48. Franchini M, Montagnana M, Targher G, et al. Interpatient Phenotypic Inconsistency in Severe Congenital Hemophilia: a Systematic Review of the Role of Inherited Thrombophilia. Semin Thromb Hemost. 2009;35(03):307–312. doi:10.1055/s-0029-1222609
49. Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21(5):492–497. doi:10.1038/nm.3847
50. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med. 2017;377(9):819–828. doi:10.1056/NEJMoa1616569
51. Dargaud Y, Béguin S, Lienhart A, et al. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost. 2005;93(03):475–480. doi:10.1160/TH04-10-0706
52. Alnylam Reports Patient Death in Fitusiran Clinical Study; 2017. Available from: https://www.hemophilia.org/news/.
53. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135–140. doi:10.2147/JBM.S159297
54. Livnat T, Sehgal A, Qian K, et al. Thrombin generation in plasma of patients with haemophilia A and B with inhibitors: effects of bypassing agents and antithrombin reduction. Blood Cells Mol Dis. 2020;82:102416. doi:10.1016/j.bcmd.2020.102416
55. Pasi KJ, Lissitchkov T, Mamonov V, et al. Targeting of antithrombin in hemophilia A or B with investigational siRNA therapeutic fitusiran—Results of the phase 1 inhibitor cohort. J Thromb Haemost. 2021;19(6):1436–1446. doi:10.1111/jth.15270
56. Janas MM, Schlegel MK, Harbison CE, et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat Commun. 2018;9(1):723. doi:10.1038/s41467-018-02989-4
57. Srivastava A, Rangarajan S, Kavakli K, et al. Fitusiran, an Investigational siRNA Therapeutic Targeting Antithrombin for the Treatment of Hemophilia: first Results from a Phase 3 Study to Evaluate Efficacy and Safety in People with Hemophilia a or B without Inhibitors (ATLAS-A/B). Blood. 2021;138(Supplement2):LBA–3. doi:10.1182/blood-2021-155018
58. Young G, Srivastava A, Kavakli K, Ross C, Sathar J, Tran H. Efficacy and safety of fitusiran prophylaxis, an siRNA therapeutic, in a multicentre phase 3 study (ATLAS-INH) in people with haemophilia A or B, with inhibitors. ASH Plenary Sci Sess. 2021:543.
59. Sridharan G, Liu J, Qian K, Goel V, Huang S, Akinc A. In silico modelling of the impact of antithrombin lowering on thrombin generation in rare bleeding disorders. Blood. 2017;130(Supplement 1):3659.
60. Ten Kate MK, Van Der Meer J. Protein S deficiency: a clinical perspective. Haemophilia. 2008;080512004759292. doi:10.1111/j.1365-2516.2008.01775.x
61. Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci. 2006;103(9):3106–3111. doi:10.1073/pnas.0504240103
62. Mahasandana C, Veerakul G, Tanphaichitr VS, Suvatte V, Opartkiattikul N, Hathaway WE. Homozygous protein S deficiency: 7-year follow-up. Thromb Haemost. 1996;76(6):1122. doi:10.1055/s-0038-1650718
63. Prince R, Bologna L, Manetti M, et al. Targeting anticoagulant protein S to improve hemostasis in hemophilia. Blood. 2018;131(12):1360–1371. doi:10.1182/blood-2017-09-800326
64. Prince R, Schaeper U, Dames S, et al. Targeting Protein S Using Small Interfering RNA Is Well Tolerated and Protects Mice with Hemophilia a from Acute Hemarthrosis. ASH Oral Abstr. 2020:54.
65. Tollefsen DM. Heparin Cofactor II Modulates the Response to Vascular Injury. Arterioscler Thromb Vasc Biol. 2007;27(3):454–460. doi:10.1161/01.ATV.0000256471.22437.88
66. Rau J, Mitchell J, Fortenberry Y, Church F. Heparin Cofactor II: discovery, Properties, and Role in Controlling Vascular Homeostasis. Semin Thromb Hemost. 2011;37(04):339–348. doi:10.1055/s-0031-1276582
67. Lopaciuk S, Bykowska K, Kopeć M. Prevalence of heparin cofactor II deficiency in patients with a history of venous thrombosis. Pol J Pharmacol. 1996;48(1):109–111.
68. Tollefsen DM. Heparin Cofactor II Deficiency. Arch Pathol Lab Med. 2002;126(11):1394–1400. doi:10.5858/2002-126-1394-HCID
69. Lin W, Zhu R, Zhang Z, et al. RNAi targeting heparin cofactor II promotes hemostasis in hemophilia A. Mol Ther Nucleic Acids. 2021;24:658–668. doi:10.1016/j.omtn.2021.03.022
70. Kempton CL, Makris M, Holme PA. Management of comorbidities in haemophilia. Haemophilia. 2021;27(S3):37–45. doi:10.1111/hae.14013
71. Ridker PM. Ethnic Distribution of Factor V Leiden in 4047 Men and Women: implications for Venous Thromboembolism Screening. JAMA. 1997;277(16):1305. doi:10.1001/jama.1997.03540400055031
72. Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. The Lancet. 1995;346(8983):1133–1134. doi:10.1016/S0140-6736(95)
73. Pipe S, Srivastava A, Klamroth R, et al. Fitusiran, an Investigational siRNA Therapeutic Targeting Antithrombin: analysis of Antithrombin Levels and Thrombin Generation from a Phase 3 Study in People with Hemophilia A or B Without inhibitors [abstract]. ISTH Congr. 2022.
74. Philipp C. The Aging Patient with Hemophilia: complications, Comorbidities, and Management Issues. Hematology. 2010;2010(1):191–196. doi:10.1182/asheducation-2010.1.191
75. Pierce GF, Haffar A, Ampartzidis G, et al. First-year results of an expanded humanitarian aid programme for haemophilia in resource-constrained countries. Haemophilia. 2018;24(2):229–235. doi:10.1111/hae.13409
76. Andersson NG, Auerswald G, Barnes C, et al. Intracranial haemorrhage in children and adolescents with severe haemophilia A or B - The impact of prophylactic treatment. Br J Haematol. 2017;179(2):298–307. doi:10.1111/bjh.14844
77. Krumb E, Hermans C. Living with a “hemophilia‐free mind” – the new ambition of hemophilia care? Res Pract Thromb Haemost. 2021;5(5):e12567. doi:10.1002/rth2.12567
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.