")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Rituximab use in adult primary glomerulopathy: where is the evidence?
Authors Mallat S, Itani H, Abou-Mrad R , Abou Arkoub R, Tanios B
Received 4 June 2016
Accepted for publication 14 July 2016
Published 29 August 2016 Volume 2016:12 Pages 1317—1327
DOI https://doi.org/10.2147/TCRM.S114316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Samir G Mallat,1 Houssam S Itani,2 Rana M Abou-Mrad,3 Rima Abou Arkoub,4 Bassem Y Tanios1
1Division of Nephrology, Department of Internal Medicine, American University of Beirut Medical Center, 2Division of Nephrology, Department of Internal Medicine, Makassed General Hospital, Beirut, Lebanon; 3Specialist Nephrology, FMC, Abu Dhabi, UAE; 4Division of Nephrology, The Ottawa Hospital, Ottawa, ON, Canada
Abstract: Rituximab is a chimeric anti-CD20 antibody that results in depletion of B-cell lymphocytes. It is currently used in the treatment of a variety of autoimmune diseases, in addition to CD20-positive lymphomas. The use of rituximab in the treatment of the adult primary glomerular diseases has emerged recently, although not yet established as first-line therapy in international guidelines. In patients with steroid-dependent minimal change disease or frequently relapsing disease, and in patients with idiopathic membranous nephropathy (IMN), several retrospective and prospective studies support the use of rituximab to induce remission, whereas in idiopathic focal and segmental glomerulosclerosis (FSGS), the use of rituximab has resulted in variable results. Evidence is still lacking for the use of rituximab in patients with immunoglobulin A nephropathy (IgAN) and idiopathic membranoproliferative glomerulonephritis (MPGN), as only few reports used rituximab in these two entities. Randomized controlled trials (RCTs) are warranted and clearly needed to establish the definitive role of rituximab in the management of steroid-dependent and frequently relapsing minimal change disease, IMN, both as first-line and second-line treatment, and in MPGN. We await the results of an ongoing RCT of rituximab use in IgAN. Although current evidence for the use of rituximab in patients with idiopathic FSGS is poor, more RCTs are needed to clarify its role, if any, in the management of steroid-resistant or steroid-dependent FSGS.
Keywords: rituximab therapy, primary glomerulopathy, adult glomerunephritis, membranous nephropathy, minimal change disease, focal and segmental glomerulosclerosis, immunoglobulin A nephropathy, idiopathic membranoproliferative glomerulonephritis
Introduction
Rituximab is a chimeric monoclonal antibody (murine/human), designed to bind specifically to the CD20 receptor – lymphocyte differentiation antigen B – present in the cell membrane of pre-B-cells and mature B but not in the plasma cells. The binding of rituximab to CD20 causes a depletion of B lymphocytes through three mechanisms: antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, and induction of apoptosis.1
Rituximab is currently approved for the treatment of CD20-positive lymphoma and rheumatoid arthritis; however, it is increasingly being used off-label in a wide variety of autoimmune disorders.2
In this review, we will focus on the role of rituximab in the management of adult patients with primary glomerular disease. We will emphasize on the pathophysiological aspects and recent clinical trials supporting the use of rituximab in the management of minimal change disease (MCD), idiopathic membranous nephropathy (IMN), and primary focal and segmental glomerulosclerosis (FSGS), immunoglobulin A nephropathy (IgAN), and idiopathic membranoproliferative glomerulonephritis (MPGN).
For this review we searched MEDLINE and PubMed for reviews, case reports, case series, retrospective, and prospective studies, using keywords such as “Rituximab”, “Therapy”, “glomerulonephritis”, “minimal change disease”, “focal and segmental glomerulosclerosis”, “idiopathic membranous nephropathy”, “IgA nephropathy”, and “membranoproliferative glomerulonephritis”. We included some of the yielded case reports; however, we tried to include all relevant retrospective and prospective studies published to date, testing the use of rituximab in these pathologies. In addition, we searched the clinical trial registry ClinicalTrials.gov for ongoing trials related to our review.
Rituximab in MCD
MCD accounts for up to 20% of cases of nephrotic syndrome in the adult population.3 Although most of the patients with nephrotic syndrome related to MCD will respond to corticosteroid therapy, up to a third will become corticosteroid-dependent or have a frequently relapsing disease.
For this subset of patients, current guidelines suggest using other regimens such as oral cyclophosphamide, calcineurin inhibitors (CNIs) (tacrolimus or cyclosporine), or mycophenolate mofetil (MMF) in patients who are intolerant to the abovementioned agents. These guidelines noted the need for randomized clinical trials to establish the role of rituximab in this setting.4
Historically, MCD has been considered a lymphocyte T-cell pathology. However, recent advances in our understanding of the pathways involved in the pathogenesis of the disease identified a more complex pathogenesis with participation of innate immunity, B-cells, and regulatory T-cells.5
Moreover, it has been shown that rituximab binds to the sphingomyelin phosphodiesterase acid-like 3b protein in glomerular podocytes and regulates acid sphingomyelinase activity, stabilizing the actin cytoskeleton, and preventing apoptosis of podocytes, thus providing another rationale for the use of rituximab in the management of MCD.6
The first case that reported the use of rituximab in the management of MCD in adults was in 2007. In this report, a patient with a multirelapsing nephrotic syndrome secondary to MCD (>30 relapses), who failed treatment with all other potentially steroid-sparing drugs including cyclophosphamide, cyclosporine, and MMF, was treated with rituximab at 375 mg/m2 every week for 4 weeks. Long-term remission was achieved starting 3 weeks after therapy, and was still maintained after 28 months at the time of writing of the report.7 Since the publications of this report, several other case reports of the successful use of rituximab in the setting of frequent relapses/steroids dependence were published.8
Following these case reports, several retrospective and prospective trials have been published.
In one retrospective case series, 17 patients with steroid-dependent or frequently relapsing MCD despite several immunosuppressive therapy were treated with rituximab and analyzed. The infusion protocol for rituximab differed between patients, and some of the patients received a second course of rituximab during follow-up because of CD19 cell recovery. Rituximab achieved a sustained response with no relapse in 65% of patients after 2 years. No infectious or hematologic complications were observed during follow-up.9
In another retrospective analysis involving 41 patients with steroid-dependent or multiple relapsing MCD, rituximab achieved complete clinical response (defined as urine protein over creatinine ratio [UPCR] of <0.3 g/g and withdrawal of all immunosuppressive treatment) in 61% of patients, and a partial clinical response in 17% (defined as UPCR of <0.3 g/g and withdrawal of at least one immunosuppressive drug). Twenty-two percent of patients did not respond to rituximab therapy. No serious adverse events were noted secondary to rituximab treatment.10
In a prospective trial involving 25 patients with steroid-dependent MCD, rituximab was administered twice at 6 months interval, at a dose of 375 mg/m2. All patients were on prednisolone, 20 patients were on cyclosporine A, three patients were on MMF, and five patients were on mizoribine. Twelve months after the first rituximab infusion, only four patients out of 25 relapsed, only four patients remained on prednisolone, and only six patients remained on cyclosporine. Furthermore, the mean doses of both prednisolone and cyclosporine A were reduced significantly from 26.4±11.5 mg/day at baseline to 1.1±2.8 mg/day at 12 months for prednisolone and from 110±43 mg/day at baseline to 30±48 mg/day at 12 months for cyclosporine A. Most of the relapses developed simultaneously with the recovery of the B-cell count (CD19 or CD20), supporting that suppression of B-cell is involved in the pathophysiology of MCD.11
In an extended prospective follow-up of the previous trial to evaluate the long-term effects of rituximab, infusions of rituximab at 375 mg/m2 were continued at 6 months interval for 24 months. The cohort was then divided into a treatment continuation group (20 patients), in whom rituximab infusions were maintained at 6 months intervals beyond the initial four doses, and treatment discontinuation group (five patients), in whom rituximab therapy was stopped after the initial four doses. Complete remission (urine protein <0.3 g/day) was maintained in all 20 patients of the continuation therapy group, from 36 to 54 months after the first rituximab infusion. Only one patient out of the five of the discontinuation therapy group developed relapse requiring resumption of rituximab infusions. Interestingly, and despite B-cell repletion, ten patients with B-cell repletion (including four from the treatment discontinuation group and six from the treatment continuation group) maintained complete remission, suggesting that single infusion protocols of rituximab may be an effective and safe treatment regimen for patients with steroid-dependent MCD, and that B-cell repletion does not always correlate with disease relapse.12
In summary, current evidence supports the use of rituximab in steroid-dependent and frequently relapsing MCD; however, properly designed randomized controlled trials (RCTs) are needed to establish the superiority and safety of rituximab as compared to other currently used agents in this setting, such as cyclophosphamide, cyclosporine, and MMF. Table 1 summarizes the characteristics of the discussed studies of rituximab in MCD.
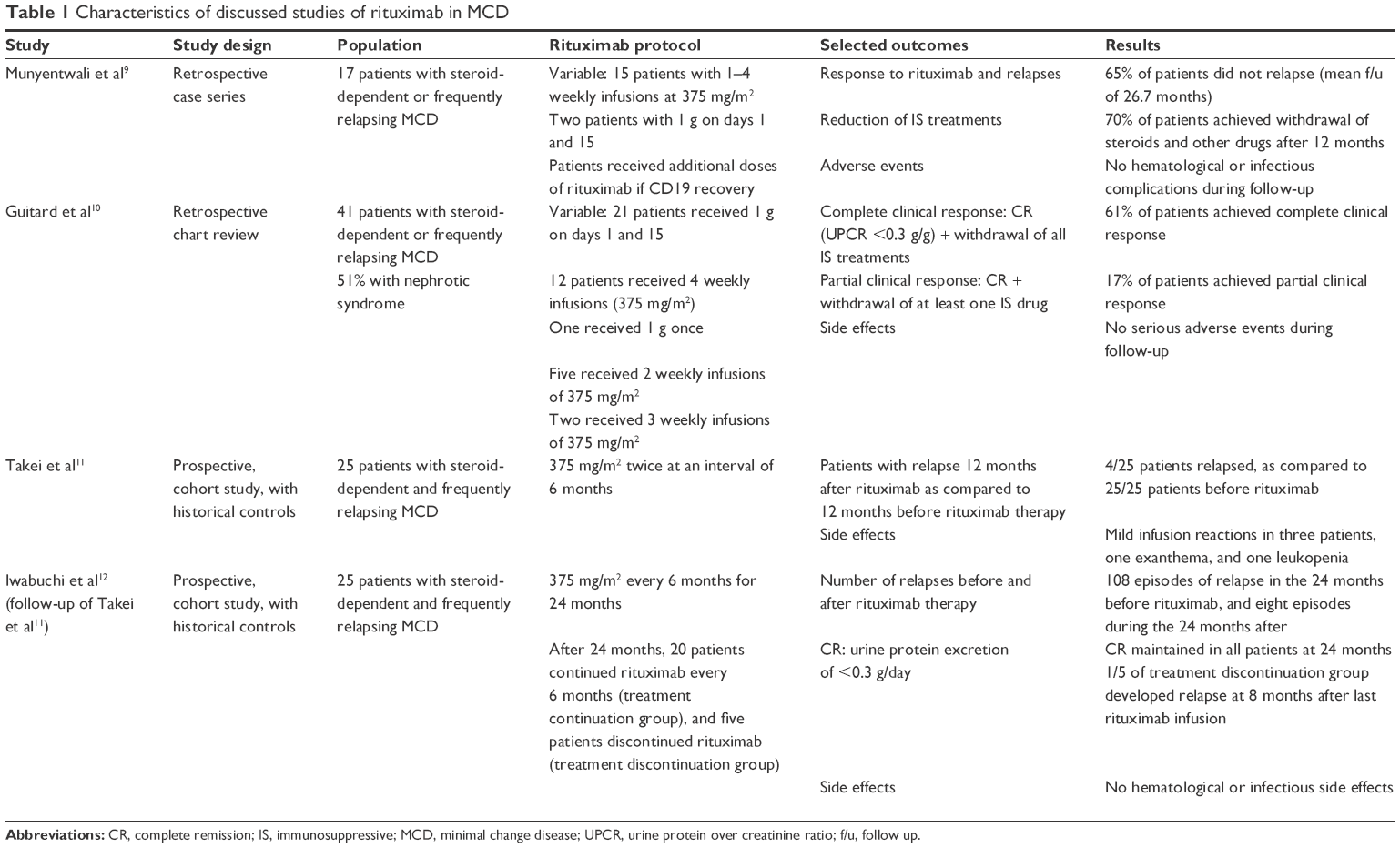 | Table 1 Characteristics of discussed studies of rituximab in MCD |
Rituximab in IMN
Membranous nephropathy is considered to be the most common cause of nephrotic syndrome in adults.13 Seventy percent of cases are labeled as primary or idiopathic.14 Spontaneous remission develops in 30%–50% of patients with IMN and nephrotic syndrome.15 Thus, immunosuppressive therapy is reserved only for cases with high risk of progression.16 Currently recommended first-line immunosuppressive agents, such as steroids and cyclophosphamide, are not free of harm. Serious complications like bone marrow suppression, infection, iatrogenic diabetes, infertility, and malignancy, can occur.17
In the light of the recent discovery of autoantibodies against the podocyte enzyme, M-type phospholipase A2 receptor (PLA2R), our understanding of the pathogenesis of IMN has improved dramatically.18 These autoantibodies are present in most patients with IMN. Antibodies to other podocyte antigens, thrombospondin type 1 domain containing protein 7 may be present as well.19 Accordingly, this major breakthrough heightened the value of the B-cell depleting agent rituximab as an attractive therapeutic option.
In fact, in a cohort of 35 patients with IMN, 71% (25/35) had positive anti-PLA2R antibodies (PLA2R-Ab) and they declined or disappeared in 68% (17/25) within 12 months after rituximab therapy. Decline in the autoantibody levels was translated into a higher rate of remission of proteinuria.20
Most of the current literature on the use of rituximab in IMN comes from observational studies and one recently published RCT.
The earliest report was published in 2002 by Remuzzi et al. In this study, four weekly doses of rituximab (375 mg/m2) were given to eight patients with IMN who had nephrotic range proteinuria (>3.5 g/24 h) for at least 6 months without remission, despite full-dose angiotensin-converting-enzyme inhibitors (ACEI). Mean proteinuria decreased from 8.6 g to 3.8 g/24 h during the treatment period. Two patients achieved complete remission (proteinuria <1 g/24 h), and three patients achieved partial remission.21
A different rituximab regimen was employed in a prospective observational study in 15 severely nephrotic patients (urine protein excretion range between 6.1 and 23.5 g/24 h) refractory to maximally tolerated doses of ACEIs and/or angiotensin receptor blockers (ARBs) for at least 6 months. Patients received two infusions of rituximab at a dose of 1 g on days 1 and 15. Those who remained with significant proteinuria (>3 g/24 h) at 6 months, and had recovered B-cell counts were given a second course of the same treatment (ten patients). Mean proteinuria decreased from 13±5.7 to 6±7.3 g/24 h at 12 months. Furthermore, there was a 60% complete and partial remission rate at 1 year.22
In a nonrandomized study, Ruggenenti et al prospectively monitored the outcomes of 100 consecutive patients with IMN treated with 4 weekly rituximab infusion. Duration of follow-up was at least 6 months. Thirty-two patients had already been treated at other institutions with steroids alone or in combination with alkylating agents, CNIs, or other immunosuppressant. Twenty patients (60%) had transient partial remissions. Nevertheless, no patient was in complete or partial remission after completion of the last course of immunosuppression and no one had any complete or partial remission on subsequent follow-up. At baseline evaluation, median serum creatinine was 1.2 mg/dL, median serum albumin was 2.2±0.6 g/dL, and median proteinuria was 9.1 g/24 h. Median duration of proteinuria before rituximab administration was 25.5 and 65.4 months for the 32 patients with second-line therapy. Blood pressure was well controlled and all patients were on ACEIs. Over a median follow-up of 29 months after rituximab administration, 65 patients achieved complete or partial remission. The median time to remission was 7.1 months. Similar proportion of patients achieved complete or partial remission among those given rituximab as first-line (47 of 68) or second-line (18 of 32) therapy. Rituximab was well tolerated and there was no treatment-related adverse events apart from some reactions to the first infusion.23
The efficacy of rituximab in the setting of CNI-dependent IMN was assessed in a small pilot prospective study. Thirteen patients with ≥4 CNI-responsive relapses while being weaned off and with glomerular filtration rate >60 mL/min were given 4 weekly doses of rituximab (375 mg/m2). As a result, proteinuria decreased significantly from 2.5±0.76 g at baseline to 0.85±0.17 g at 6 months. CNIs and other immunosuppressant drugs could be stopped in all patients. Three patients relapsed but responded to a repeated course of rituximab. At 30 months, all patients were in remission.24
And finally, the results of the only randomized trial to date of rituximab in the management of IMN have been recently published.
GEMRITUX was a prospective, multicenter, RCT at 31 French hospitals. Seventy-seven patients with IMN and nephrotic syndrome despite 6 months of nonimmunosuppressive antiproteinuric treatment (NIAT) were randomized to either continue NIAT alone or the addition of rituximab 375 mg/m2 on days 1 and 8. At 6 months there was no difference between the two groups with regards to the primary outcome of complete or partial remission, as 13 patients in the NIAT-rituximab group and eight patients in the NIAT group, achieved either complete or partial remission. However, rituximab achieved PLA2R-Ab depletion in 56% of patients after 3 months, and more patients in the rituximab reached a composite endpoint of decreased proteinuria of >50% and increased serum albumin of >30%, as compared to placebo (41% vs 13%, P<0.01). Another important finding during this trial was that patients with positive PLA2R-Ab at baseline, who achieved PLA2R-Ab depletion at month 3 after rituximab, had a higher chance of complete or partial remission, suggesting that PLA2R-Ab depletion may be a strong predictor of response to rituximab therapy in this setting. In contrast, B-cell depletion (which was achieved in all patients treated with rituximab) did not predict response to rituximab therapy. In the extended observational period of the trial (up to 24 months), significantly more patients in the NIAT-rituximab group achieved partial or complete remission as compared to NIAT group (64.9% and 34.2%, respectively, odds ratio, 3.5; 95% confidence interval, 1.7–9.2; P<0.01). There was no difference in side effects between the two groups.25
In summary, rituximab may be an effective alternative in the management of IMN. The results of GEMRITUX is a major step toward establishing the role of rituximab in the management of IMN, and importantly this trial found no increased side effects of rituximab as compared to placebo. However, more RCTs with longer follow-up (beyond 6 months as in GEMRITUX) are still needed to confirm the benefit and safety of rituximab both as first-line or second-line therapy as compared to the commonly used regimens of corticosteroids/cyclophosphamide, cyclosporine, and tacrolimus. Table 2 summarizes the characteristics of the discussed studies of rituximab in IMN.
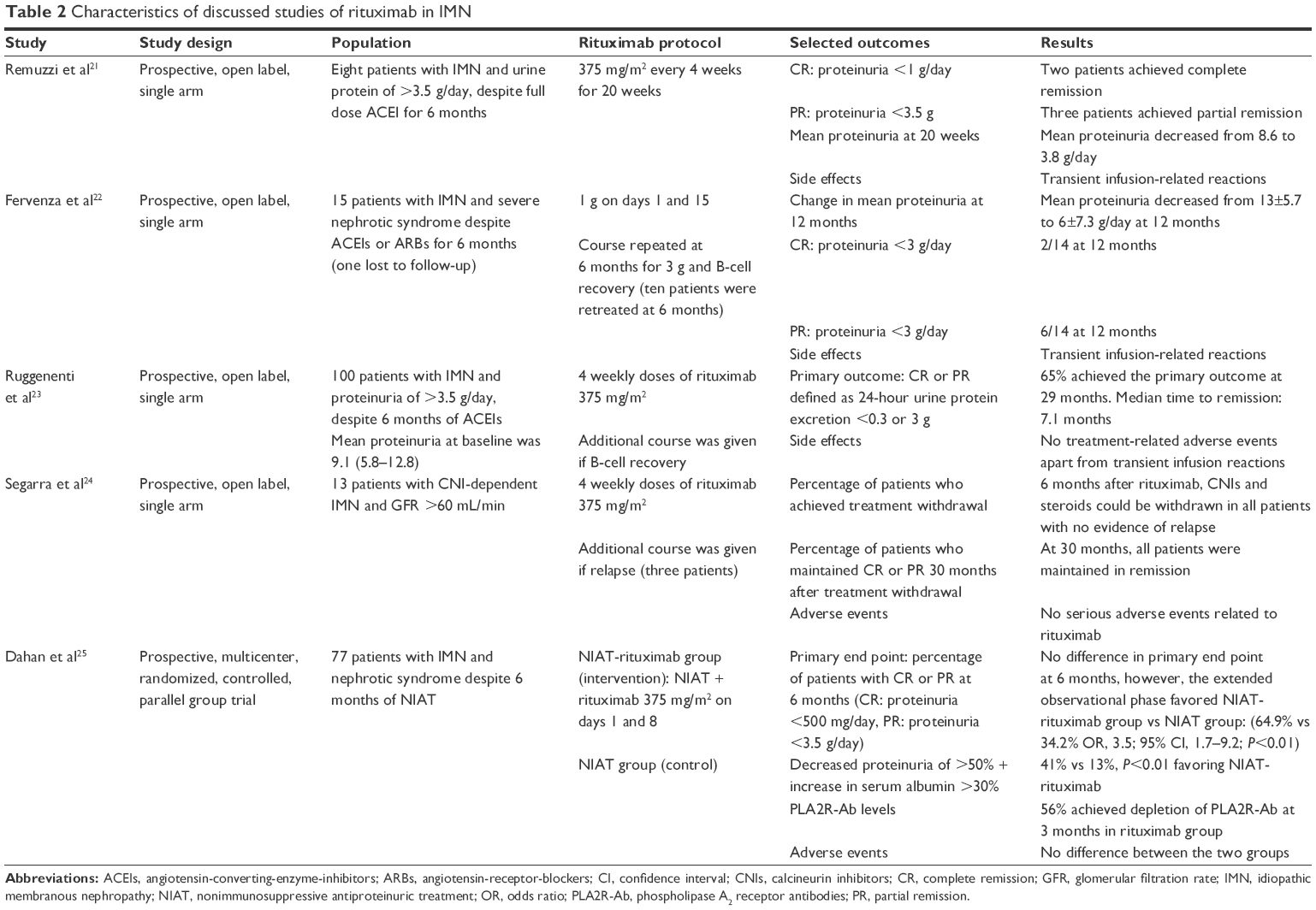 | Table 2 Characteristics of discussed studies of rituximab in IMN |
Rituximab in FSGS
The 2012 Kidney Disease: Improving Global Outcomes (KDIGO) guidelines recommend the use of corticosteroids as first-line therapy for idiopathic FSGS with features of the nephrotic syndrome. Patients who remain resistant to steroids after at least 4 months, are treated with cyclosporine or high-dose dexamethasone with MMF. According to KDIGO, available evidence was insufficient to support the use of rituximab in FSGS.4
Only few reports have evaluated the use of rituximab in adult patients with FSGS, with variable results.
Fernandez-Fresnedo et al reported the use of rituximab in eight patients with FSGS resistant to steroids and other therapies. Only two patients had a sustained and significant reduction in proteinuria at 1 year and one patient had a significant but transient effect. All other five patients failed to respond to rituximab therapy.26
In another report from Japan, two patients with steroid-resistant FSGS did not respond to a single dose of rituximab. However, two patients with steroid-dependent FSGS achieved complete remission after a single dose of rituximab, which allowed discontinuation of steroids and CNIs. The two patients eventually relapsed, which coincided with CD19/20 positive B-cell count recovery, but they both responded again with sustained remission upon readministration of rituximab.27
In a recent study, rituximab was administered to 30 patients with steroid-dependent or frequently relapsing idiopathic nephrotic syndrome, and included five adult patients with FSGS. Over 1 year observation period, relapses decreased by approximately fivefold compared with the year preceding rituximab treatment. In addition, there was a subsequent reduced need for immunosuppressant medications in cases of recurrent disease.28
In summary, currently available evidence do not support a role for rituximab in the management of FSGS. RCTs are warranted to assess the possible benefit of rituximab in the management of steroid-dependent and frequently relapsing FSGS. Table 3 summarizes the characteristics of the discussed studies of rituximab in FSGS.
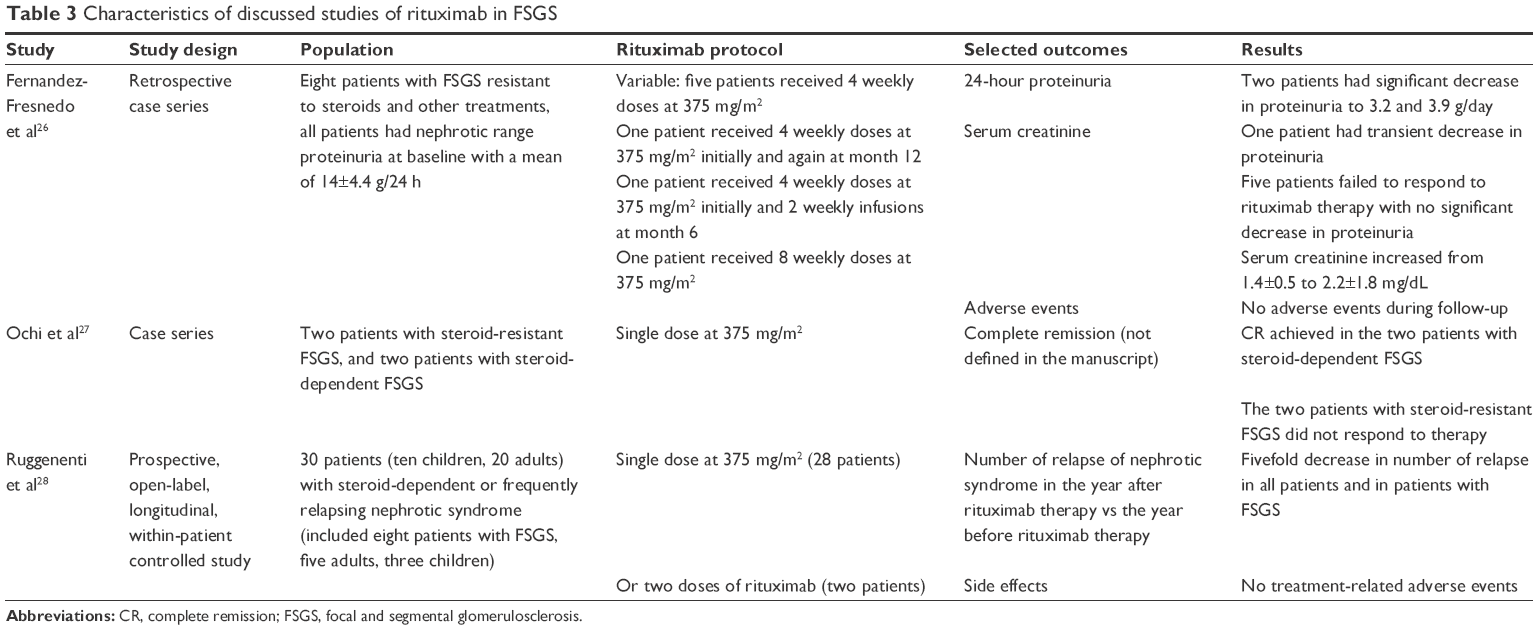 | Table 3 Characteristics of discussed studies of rituximab in FSGS |
Rituximab in IgAN
IgAN is the most common form of idiopathic glomerulonephritis worldwide. Recent studies have demonstrated that IgAN is an immune-mediated disease, with deposition of under-galactosylated, dimeric, or polymeric IgA in the glomerular mesangium. In addition, IgG or IgA autoantibodies directed against these abnormal IgA complexes might also contribute to the development and progression of IgAN, providing a plausible rationale for the use of immunosuppressive therapy in the treatment of IgAN.29 Current guidelines, however, recommend antiproteinuric and antihypertensive therapy with ACEIs or ARBs as an initial therapeutic approach in IgAN patients with persistent proteinuria of >1 g/day, and a 6-month course of corticosteroids if the former approach was not successful after 3–6 months. Combination immunosuppressive therapy and the addition of cyclophosphamide and azathioprine was not advocated except in cases of rapidly progressive crescentic glomerulonephritis.4
The recently published Stop-IgA trial randomized 162 patients with IgAN, who had persistent proteinuria of at least 750 mg/day, despite a 6-month course of antiproteinuric supportive therapy, to continued supportive therapy alone, vs addition of immunosuppression. Corticosteroids monotherapy was used for patient with estimated glomerular filtration rate (eGFR) ≥60 mL/min, and a combination immunosuppressive therapy with corticosteroids, cyclophosphamide, followed by azathioprine maintenance for 3 years was used for patients with an eGFR between 30 and 59 mL/min. The results of this trial were disappointing, and although more patients in the immunosuppressive therapy group achieved a decrease in protein-to-creatinine ratio to <0.2 g/g, immunosuppression led to more side effects, and there was no difference between the two groups with regards to change in eGFR at 3 years. One of the limitations of this trial is the exclusion of patients with proteinuria of >3.5 g/day, who might have a better response to immunosuppressive therapy.30
Rituximab use in IgAN was reported in only a few patients. In a recent case series, rituximab was used to treat three kidney transplant recipients with biopsy-proven recurrence of IgAN. Recurrence of IgAN occurred at a median of 20 months posttransplantation, and the patients had a mean proteinuria of 4.8 g/day. Four monthly doses of rituximab at 375 mg/m2 were given, and achieved a mean decrease of proteinuria of 3.1 g/day at month 6. Only one patient was treated initially with an ACEI, which was stopped after only 1 week due to increased serum creatinine level.31
In a prospective, single-arm trial, a single dose of rituximab at 375 mg/m2 was given to treat 24 patients with primary glomerulonephritis, and included five patients with IgAN. After 6 months of the rituximab dose, there was no significant change in proteinuria in patients with IgAN (1.0±0.8 g/day at baseline vs 0.9±0.8 g/day at 6 months). Interpretation of the results of this trial was limited by the short follow-up of 6 months, and the possible need for several doses of rituximab to achieve response in a slowly progressive disease like IgAN.32
We await the results of a prospective, multicenter, randomized, controlled trial: rituximab in the treatment of progressive IgA nephropathy. In this trial, 54 patients with biopsy-proven IgAN, and proteinuria of >1 g/day, while on an ACEI, ARB, or a direct renin inhibitor, will be randomized to rituximab 1 g, on days 1, 15, 168, and 182, or to placebo. Change in proteinuria and kidney function will be assessed at 12 months.33
In summary, although the pathogenesis of IgAN may provide a plausible rationale for the use of rituximab, evidence is still lacking to date. The awaited results of the RCT presented in this section will help in clarifying the role of rituximab in the management of IgAN. Table 4 summarizes the characteristics of the discussed studies of rituximab in FSGS.
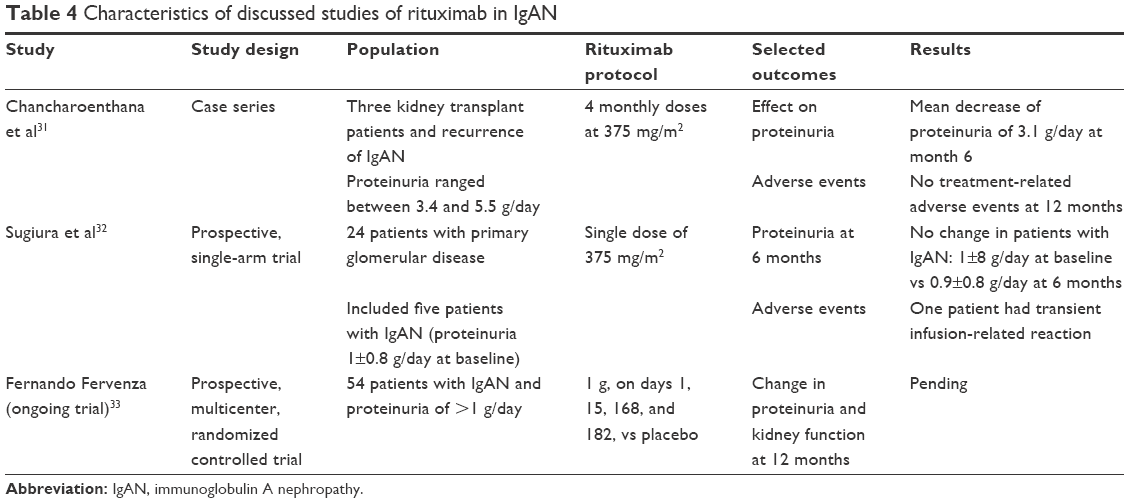 | Table 4 Characteristics of discussed studies of rituximab in IgAN |
Rituximab in idiopathic MPGN
MPGN is a pattern of glomerular injury resulting from predominantly subendothelial and mesangial deposition of immune complexes and/or complement factors and their products.29
Current classification based on immunofluorescence findings divides etiology of MPGN into immune complex-mediated, characterized by capillary wall and mesangial deposition of C3 and immunoglobulin, and complement-mediated, characterized by capillary wall and mesangial deposition of C3, with negative immunoglobulins. Complement-mediated MPGN is usually secondary to abnormalities in alternate complement pathway.
Most cases of immune complex-mediated MPGN are secondary to infections (such as hepatitis C with or without cryoglobulinemia), autoimmune diseases such as systemic lupus erythematosus and monoclonal gammopathies. The diagnosis of idiopathic MPGN is established after all of these secondary causes are excluded. Therefore, truly idiopathic MPGN is decreasing in frequency, and currently considered a rare entity.34
The optimal therapy for patients with idiopathic MPGN is not clearly defined, and the latest KDIGO guidelines found very weak evidence to suggest the use of cyclophosphamide or MMF in combination with corticosteroids, in the treatment of patients with idiopathic MPGN with the nephrotic syndrome and progressive decline of kidney function.4
Few reports described the use of rituximab in idiopathic MPGN. In a retrospective case review study of 24 adult patients with primary glomerulonephritis who received rituximab, two patients with idiopathic MPGN were included. One patient presented with rapidly progressive glomerulonephritis and received two infusions of rituximab at 375 mg/m2 each in addition to corticosteroids. Dialysis dependency was achieved after 5 months, and complete remission 19 months later (defined as UPCR <30 mg/mmol and serum albumin >35 g/L). The other patient presented with nephrotic syndrome, and achieved partial remission 29 months after a single dose of rituximab.35
In a prospective trial involving 24 patients with primary glomerulonephritis, a single dose of rituximab at 1 g was administered. The trial included one patient with idiopathic MPGN, who presented with nephrotic syndrome. The urine protein decreased from 9.8 g/day at baseline to 1.8 g/day at 6 months after the rituximab injection.32 In another prospective trial, six patients with MPGN (four idiopathic and two with cryoglobulinemia), two doses of rituximab were administered at 1 g on days 1 and 15. The two patients with MPGN with cryoglobulinemia achieved complete remission after 12 months, while the four patients with idiopathic MPGN achieved partial remission (defined as reduction in the urine protein excretion of >50% to between 0.3 and 3.5 g/day without a doubling in the serum creatinine concentration).36
In summary, and despite the paucity of data, preliminary results for rituximab in the management of idiopathic MPGN are encouraging, which warrants a well-designed RCT comparing rituximab to other immunosuppressive agents in the management of idiopathic MPGN. Table 5 summarizes the characteristics of the discussed studies of rituximab in idiopathic MPGN.
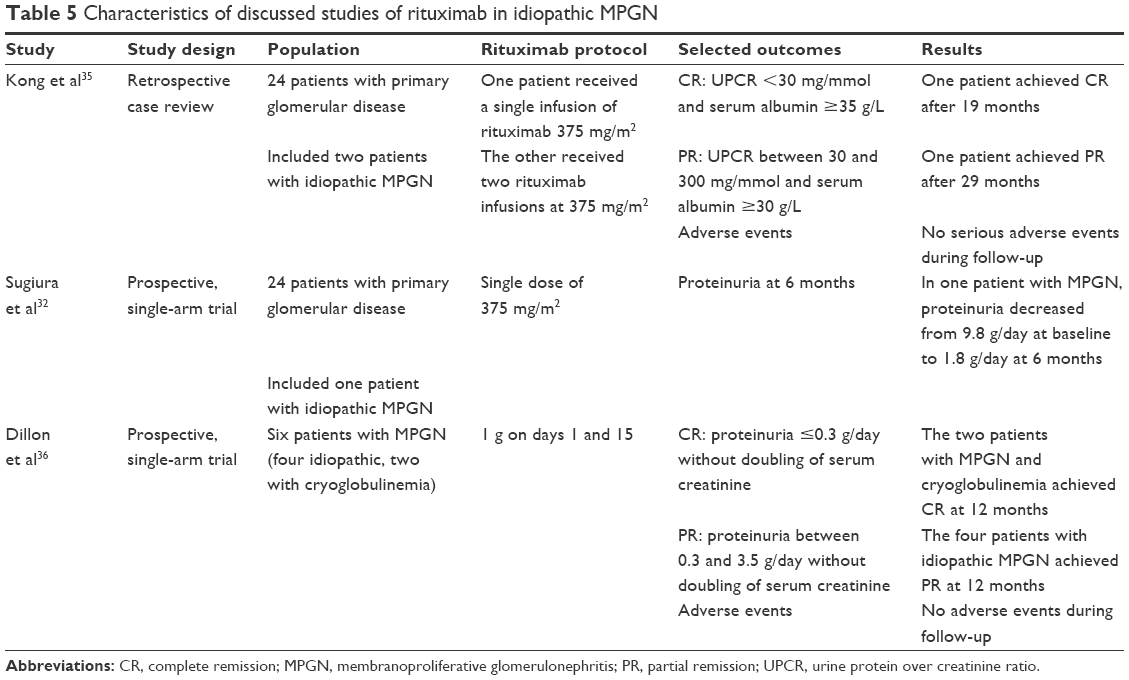 | Table 5 Characteristics of discussed studies of rituximab in idiopathic MPGN |
Discussion
The available evidence supports the use of rituximab in the management of patients with MCD who are steroid-dependent or with frequently relapsing disease, and in the management of IMN.
In patients with steroid-dependent or frequently relapsing MCD, there is a need for well-designed RCTs with head-to-head comparison of the efficacy and safety of rituximab, vs other currently used agents, such as CNI, cyclophosphamide, and MMF. This trial should have an extended follow-up to properly assess sustained remission, episodes of relapse and safety of rituximab as compared to currently used second-line agents.
There is also a need for RCTs to establish the role and the risk–benefit ratio of rituximab as compared to steroids/cyclophosphamide or CNI, both as first-line or second-line agent in the management of IMN. Potential advantages of rituximab in this setting would include ease of administration, a more favorable safety profile as compared to cyclophosphamide therapy, and a less nephrotoxic potential as compared to CNI therapy. In fact, and apart from transient infusion reactions, most trials using rituximab in the treatment of primary glomerulopathies found no increased risk of serious hematologic or infectious adverse events. The results of GEMRITUX are encouraging, as it was the first RCT to test the efficacy and safety of rituximab therapy in the management of severe IMN, but this trial compared rituximab to placebo. The ongoing randomized controlled MENTOR trial will test another hypothesis, as it will assess the efficacy and safety of rituximab as first-line agent in the treatment of IMN, as compared to cyclosporine.37 More RCTs like GEMRITUX are needed, although with a longer follow-up (beyond 1 year), to better assess remission rates and eventual episodes of relapse after rituximab therapy. Properly designed trials, and the results of the MENTOR trial will help in clarifying the optimal role of rituximab in the management of IMN.
In addition, the optimal infusion regimen and duration of rituximab therapy needs to be further clarified, as different infusion protocols were used in different trials, and B-cell repletion may not be the optimal predictor of disease relapse. In the setting of IMN, GEMRITUX, and previous data demonstrated that PLA2R-Ab depletion and not B-cell depletion predicted response to rituximab. The possibility of tailoring the rituximab infusion protocol according to PLA2R-Ab levels deserves further study.20,25
In contrast, results in FSGS are not as encouraging, likely suggesting a different underlying mechanism in FSGS whereby B lymphocytes are not the main role players. More trials are needed to establish the role of rituximab, if any, in the management of steroid-resistant or steroid-dependent FSGS.
On the other hand, the few reports that used rituximab in the setting of idiopathic MPGN are encouraging and warrant a formal RCT to assess the efficacy of rituximab compared to currently used regimens of cyclophosphamide or MMF with corticosteroids.
Evidence for the use of rituximab in the setting of IgAN is still lacking, and although recent trials have failed to demonstrate a significant benefit from immunosuppressive therapy for most patients with IgAN, the results of an ongoing RCT will help in clarifying the role of rituximab in patients with IgAN.33
Disclosure
The authors report no conflicts of interest in this work.
References
Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–445. | ||
McDonald V, Leandro M. Rituximab in non-haematological disorders of adults and its mode of action. Br J Haematol. 2009;146(3):233–246. | ||
Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic syndrome: a comparison of renal biopsy findings from 1976–1979 and 1995–1997. Am J Kidney Dis. 1997;30(5):621–631. | ||
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl. 2012;2(Suppl 2):139–274. | ||
Bertelli R, Bonanni A, Di Donato A, Cioni M, Ravani P, Ghiggeri GM. Regulatory T cells and minimal change nephropathy: in the midst of a complex network. Clin Exp Immunol. 2016;183(2):166–174. | ||
Glassock RJ. Therapy of relapsing minimal-change disease in adults: a new approach? Kidney Int. 2013;83(3):343–345. | ||
François H, Daugas E, Bensman A, Ronco P. Unexpected efficacy of rituximab in multirelapsing minimal change nephrotic syndrome in the adult: first case report and pathophysiological considerations. Am J Kidney Dis. 2007;49(1):158–161. | ||
Takei T, Nitta K. Rituximab and minimal change nephrotic syndrome: a therapeutic option. Clin Exp Nephrol. 2011;15(5):641–647. | ||
Munyentwali H, Bouachi K, Audard V, et al. Rituximab is an efficient and safe treatment in adults with steroid-dependent minimal change disease. Kidney Int. 2013;83(3):511–516. | ||
Guitard J, Hebral AL, Fakhouri F, et al. Rituximab for minimal-change nephrotic syndrome in adulthood: predictive factors for response, long-term outcomes and tolerance. Nephrol Dial Transpl. 2014;29(11):2084–2091. | ||
Takei T, Itabashi M, Moriyama T, et al. Effect of single-dose rituximab on steroid-dependent minimal-change nephrotic syndrome in adults. Nephrol Dial Transpl. 2013;28(5):1225–1232. | ||
Iwabuchi Y, Takei T, Moriyama T, Itabashi M, Nitta K. Long-term prognosis of adult patients with steroid-dependent minimal change nephrotic syndrome following rituximab treatment. Medicine. 2014;93(29):e300. | ||
Austin HA 3rd, Antonovych TT, MacKay K, Boumpas DT, Balow JE. NIH conference. Membranous nephropathy. Ann Intern Med. 1992;116(8):672–682. | ||
Glassock RJ. Secondary membranous glomerulonephritis. Nephrol Dial Transplant. 1992;7(Suppl 1):64–71. | ||
Polanco N, Gutiérrez E, Covarsí A, et al; Grupo de Estudio de las Enfermedades Glomerulares de la Sociedad Española de Nefrología. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol. 2010;21(4):697–704. | ||
Hofstra JM, Fervenza FC, Wetzels JF. Treatment of idiopathic membranous nephropathy. Nat Revi Nephrol. 2013;9(8):443–458. | ||
Faurschou M, Sorensen IJ, Mellemkjaer L, et al. Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol. 2008;35(1):100–105. | ||
Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. New Engl J Med. 2009;361(1):11–21. | ||
Tomas NM, Beck LH Jr, Meyer-Schwesinger C, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. New Engl J Med. 2014;371(24):2277–2287. | ||
Beck LH Jr, Fervenza FC, Beck DM, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol. 2011;22(8):1543–1550. | ||
Remuzzi G, Chiurchiu C, Abbate M, Brusegan V, Bontempelli M, Ruggenenti P. Rituximab for idiopathic membranous nephropathy. Lancet. 2002;360(9337):923–924. | ||
Fervenza FC, Cosio FG, Erickson SB, et al. Rituximab treatment of idiopathic membranous nephropathy. Kidney Int. 2008;73(1):117–125. | ||
Ruggenenti P, Cravedi P, Chianca A, et al. Rituximab in idiopathic membranous nephropathy. J Am Soc Nephrol. 2012;23(8):1416–1425. | ||
Segarra A, Praga M, Ramos N, et al. Successful treatment of membranous glomerulonephritis with rituximab in calcineurin inhibitor-dependent patients. Clin J Am Soc Nephrol. 2009;4(6):1083–1088. | ||
Dahan K, Debiec H, Plaisier E, et al; GEMRITUX Study Group. Rituximab for severe membranous nephropathy: a 6-month trial with extended follow-up. J Am Soc Nephrol. Epub 2016 Jun 27. | ||
Fernandez-Fresnedo G, Segarra A, González E, et al. Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2009;4(8):1317–1323. | ||
Ochi A, Takei T, Nakayama K, et al. Rituximab treatment for adult patients with focal segmental glomerulosclerosis. Intern Med. 2012;51(7):759–762. | ||
Ruggenenti P, Ruggiero B, Cravedi P, et al; Rituximab in Nephrotic Syndrome of Steroid-Dependent or Frequently Relapsing Minimal Change Disease or Focal Segmental Glomerulosclerosis (NEMO) Study Group. Rituximab in steroid-dependent or frequently relapsing idiopathic nephrotic syndrome. J Am Soc Nephrol. 2014;25(4):850–863. | ||
Floege J, Amann K. Primary glomerulonephritides. Lancet. 2016;387(10032):2036–2048. | ||
Rauen T, Eitner F, Fitzner C, et al; STOP-IgAN Investigators. Intensive supportive care plus immunosuppression in IgA nephropathy. New Engl J Med. 2015;373(23):2225–2236. | ||
Chancharoenthana W, Townamchai N, Leelahavanichkul A, et al. Rituximab for recurrent IgA nephropathy in kidney transplantation: a report of 3 cases and proposed mechanisms. Nephrology. Epub 2016 Jan 12. | ||
Sugiura H, Takei T, Itabashi M, et al. Effect of single-dose rituximab on primary glomerular diseases. Nephron Clin Pract. 2011;117(2):c98–c105. | ||
Fernando Fervenza, Mayo Clinic. Rituximab in progressive IgA nephropathy. ClinicalTrials.gov [database on the Internet]. Bethesda, MD: U.S. National Library of Medicine. Available from: https://clinicaltrials.gov/ct2/show/NCT00498368. NLM Identifier: NCT00498368. Accessed July 2, 2016. | ||
Fervenza FC, Sethi S, Glassock RJ. Idiopathic membranoproliferative glomerulonephritis: does it exist? Nephrol Dial Transplant. 2012;27(12):4288–4294. | ||
Kong WY, Swaminathan R, Irish A. Our experience with rituximab therapy for adult-onset primary glomerulonephritis and review of literature. Int Urol Nephrol. 2013;45(3):795–802. | ||
Dillon JJ, Hladunewich M, Haley WE, Reich HN, Cattran DC, Fervenza FC. Rituximab therapy for Type I membranoproliferative glomerulonephritis. Clin Nephrol. 2012;77(4):290–295. | ||
Fervenza FC, Canetta PA, Barbour SJ, et al; Mentor Consortium Group. A multicenter randomized controlled trial of rituximab versus cyclosporine in the treatment of idiopathic membranous nephropathy (MENTOR). Nephron. 2015;130(3):159–168. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.