Back to Journals » Journal of Inflammation Research » Volume 19
Revisiting the Subtle Relationship Between Metabolic Syndrome and Osteoarthritis
Authors Ren HC ![]() , Wang YC, Cheng JB
, Wang YC, Cheng JB ![]() , Tao HC, Feng H, Feng ML
, Tao HC, Feng H, Feng ML
Received 30 January 2026
Accepted for publication 15 March 2026
Published 25 March 2026 Volume 2026:19 597686
DOI https://doi.org/10.2147/JIR.S597686
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ujjwol Risal
Hong-chen Ren,1,* Yu-chen Wang,1,* Jing-bo Cheng,1 Hai-cheng Tao,2 Hui Feng,3 Ming-li Feng1
1Department of Orthopedics, Xuanwu Hospital, Capital Medical University, Beijing, 100053, People’s Republic of China; 2Department of Emergency Medicine, Xuanwu Hospital, Capital Medical University, Beijing, 100053, People’s Republic of China; 3Department of Orthopedics, Xiong’an Xuanwu Hospital, Xiong’an New Area, 071702, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ming-li Feng, Email [email protected]
Background: Osteoarthritis (OA) is a common disabling joint disorder traditionally attributed to mechanical wear. Emerging evidence shows that metabolic syndrome (MetS) and its components—obesity, hypertension, hyperglycemia, and dyslipidemia—are closely associated with OA onset and progression, suggesting that OA also has a metabolic-inflammatory nature.
Main Text: This review highlights mechanisms linking each MetS component to OA. Obesity contributes not only by increasing joint load but also through adipokines such as leptin, resistin, and visfatin, which activate inflammatory pathways and promote cartilage degradation and synovitis. Hypertension may worsen OA via joint ischemia, oxidative stress, and renin–angiotensin system activation. Hyperglycemia damages cartilage and ligaments by promoting advanced glycation end product accumulation and oxidative stress. Dyslipidemia influences OA through cholesterol deposition and inflammatory responses. Metabolic inflammation and immunometabolic reprogramming further drive OA progression.
Conclusion: MetS and OA are interconnected through mechanical stress, adipokine activity, inflammatory signaling, and metabolic dysregulation. Future studies should clarify how MetS affects pain, subchondral bone remodeling, and other OA phenotypes, aiding the development of individualized, metabolism-targeted strategies for early intervention and comprehensive OA management.
Keywords: inflammatory factors, metabolic syndrome, metabolomics, osteoarthritis
Review Criteria
Using the keywords “osteoarthritis”, together with “metabolic abnormalities”, “metabolic syndrome”, “obesity”, “hypertension”, “hyperglycemia”, “diabetes”, “insulin resistance”, “dyslipidemia”, “inflammatory factors”, and “immunometabolism”, we conducted independent searches in the PubMed database. We focused on research evidence published within the past five years and restricted the search to studies written in English. Duplicate records, studies with low relevance or poor quality, articles with unclear viewpoints, and literature that was not accessible for full-text download were excluded.
Background
Osteoarthritis (OA) is the most common form of arthritis and is characterized as a chronic degenerative disease affecting the joints. Its primary clinical manifestation is joint pain, which ultimately leads to structural abnormalities and loss of joint function, making OA a major cause of disability.1 Pathological changes typically include partial or full-thickness loss of articular cartilage, synovial inflammation, osteophyte formation, meniscal extrusion and tearing, bone marrow lesions, and narrowing of the joint space (Figure 1).2,3 As there are currently no effective therapies capable of halting OA progression, patients with end-stage disease often require joint replacement surgery, imposing a substantial global medical and economic burden.4 As of 2024, OA remains the most prevalent musculoskeletal disorder, with its global burden continuing to rise; approximately 7.6% of the world’s population is affected, and this figure is projected to increase by 60–100% by 2050. Notably, among all disabling conditions in individuals aged over 70 years, OA ranks seventh worldwide and predominantly affects the knee joint.5 In traditional concepts, the etiology of OA was primarily attributed to mechanical wear and other external forces, with obesity considered the major contributing factor. However, the most recent perspectives regard OA as a highly complex mechanical–inflammatory disease characterized by heterogeneity, multifactorial influences, multidimensional interactions, and multiple sources and origins. Accordingly, OA can be classified into several distinct phenotypes and endotypes, among which metabolic OA represents an important subtype.6,7 Moreover, studies have shown that hand joints, as non–weight-bearing regions, can still develop hand osteoarthritis (HOA). After excluding the direct confounding mechanical effects of obesity, HOA was found to be significantly associated with metabolic syndrome (MetS).8–10 These findings highlight the critical role of metabolic abnormalities in the onset and progression of OA.
 |
Figure 1 Native knees and osteoarthritic knees exhibit significant structural and functional differences. |
Table 1 summarizes the commonly used definitions of MetS in recent years.11–13 MetS is regarded as a clinical syndrome comprising multiple metabolic abnormalities, including central obesity, hypertension, hyperglycemia, and dysregulated lipid metabolism.14 It is also considered a major risk factor for cardiovascular diseases.15 In clinical practice, we frequently observe that many patients with OA requiring surgical intervention present with comorbid MetS. Joint pain is the predominant symptom of OA, and current evidence suggests that OA-related pain mainly originates from synovitis, bone marrow lesions, osteophyte formation, meniscal degeneration or tears, and the infrapatellar fat pad (IFP).16–18 Moreover, OA is often accompanied by hyperalgesia.19 The systemic and local inflammatory state induced by MetS is closely intertwined with these pain-generating mechanisms in OA.20,21 Furthermore, the presence of MetS is associated with an earlier onset of OA and more severe symptoms. Therefore, elucidating the relationship between MetS and OA holds substantial clinical and scientific significance.
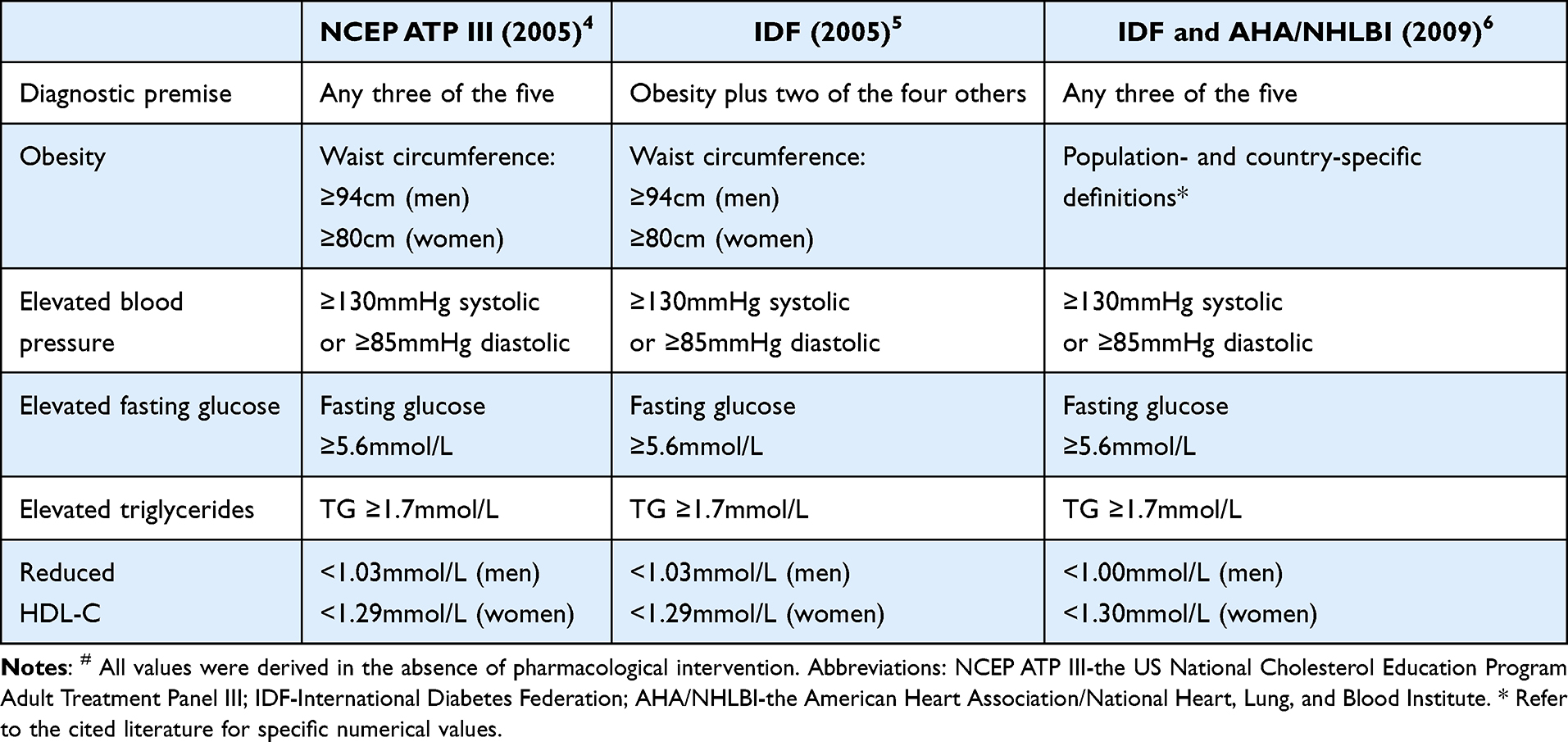 |
Table 1 Definitions of Metabolic Syndrome# |
In this review, we primarily discuss the relationships between various metabolic abnormalities within MetS and the pathological changes observed in different joint tissues affected by OA. In addition, we address the interactions between inflammatory and immune factors and the development of OA.
Obesity and Osteoarthritis
Obesity—particularly abdominal obesity—plays a critical role in the onset and progression of OA22 and is widely recognized as a major risk factor. Obesity directly increases the mechanical load on weight-bearing joints, exacerbating mechanical wear and thereby promoting the development and progression of OA.23 For example, a high body mass index (BMI) is significantly positively correlated with the incidence of OA in weight-bearing joints such as the hip and knee, and is also associated with increased pain severity.1 At the same time, obesity reflects an expansion of adipose tissue volume. Numerous studies have demonstrated that adipokines released by adipose tissue contribute to OA progression24,25 and exert detrimental effects on the synovium, bone, and other joint tissues.26 In the following sections, we discuss the relationship between obesity and OA from both mechanical and biological perspectives.
Mechanical Factors
Studies have shown that moderate mechanical stress—such as jogging or swimming—can stimulate chondrocytes to secrete collagen, thereby prolonging joint longevity. In contrast, excessive mechanical loading in individuals with obesity generates high-intensity stress that promotes chondrocyte catabolism and induces a senescence-associated secretory phenotype (SASP).27,28 Moreover, excessive mechanical stress can directly cause cartilage injury.29,30 Recent evidence further demonstrates that in both in vivo and in vitro settings, mechanical overload leads to downregulation of FBXW7 (a ubiquitin ligase) in chondrocytes, which subsequently promotes chondrocyte senescence and apoptosis through the MKK7–JNK signaling pathway.30 However, the mechanisms underlying the FBXW7 downregulation induced by excessive mechanical stimulation remain unclear.
In addition, an increasing number of studies have identified numerous mechanosensitive ion channels on the chondrocyte surface that respond to external mechanical stress and help maintain the stability of their physiological structure.31–33 Consequently, mechanical stimulation can influence the metabolic microenvironment of chondrocytes. For example, excessive mechanical loading has been shown to induce GPX4-downregulation–related ferroptosis in chondrocytes. Mechanical overload activates the mechanosensitive Piezo1 channel on the chondrocyte membrane, leading to increased Ca2⁺ influx and accelerated ferroptosis. It can also activate the NF-κB signaling pathway, promoting the senescence of human nucleus pulposus cells (hNPCs) and contributing to spinal degeneration. Interestingly, supplementing overloaded chondrocytes with ferroptosis-regulating factors such as Fsp1 and CoQ10 can rescue cells from ferroptosis and oxidative damage.34,35 For example, excessive mechanical loading reduces the expression of PDZK1 (a PDZ domain–containing protein that functions as a mechanosensitive Na⁺/H⁺ exchange channel) on the surface of chondrocytes. This reduction directly impairs the mitochondrial oxidative respiratory chain, leading to mitochondrial dysfunction, accumulation of reactive oxygen species (ROS), and subsequent cellular senescence. Notably, overexpression of PDZK1, exogenous supplementation with the mitochondria-targeted coenzyme Q analogue (MitoQ), or treatment with β-hydroxybutyrate (BHB) can alleviate OA symptoms.36 These findings indicate that excessive mechanical stress negatively affects chondrocyte metabolism through mechanogated channels. Age-related degeneration of articular cartilage leads to joint space narrowing and alterations in limb alignment, followed by the formation of periarticular osteophytes. Excessive osteophyte growth can further trigger synovitis and meniscal damage, thereby exacerbating knee pain.18,37 In addition, hypertrophic osteophytes in the intercondylar notch may exert a cutting effect on the anterior cruciate ligament (ACL), resulting in knee instability.38,39 Figure 2 summarizes these mechanistic processes.
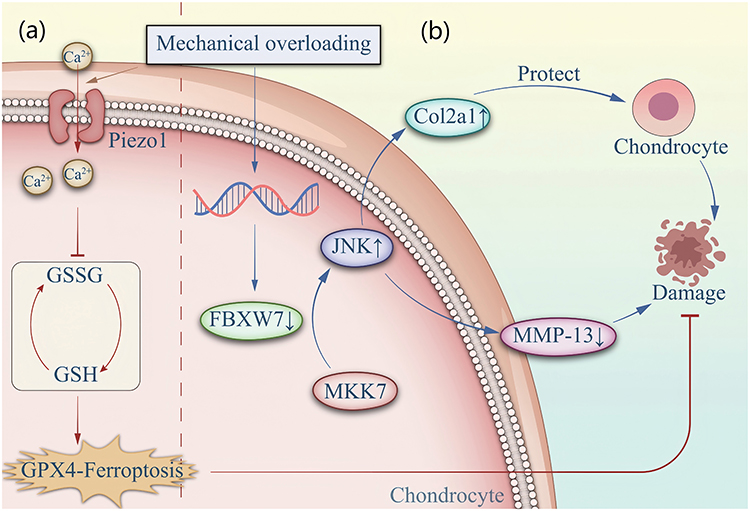 |
Figure 2 (a) Mechanical overloading stimulates an increase in intracellular Ca2+ influx, leading to a decrease in intracellular GSH content and a concurrent reduction in GPX4 levels, which induces ferroptosis. (b) Under normal conditions, FBXW7 inhibits the activation of the MKK7-JNK signaling pathway, thereby protecting chondrocytes. However, mechanical overloading downregulates FBXW7 in chondrocytes, leading to cellular senescence. |
Biological Factors
In addition to increasing the mechanical load on weight-bearing joints, obesity is characterized most prominently by an overall expansion of adipose tissue mass. The role of adipose tissue in OA was previously underappreciated; however, with advances in clinical research, it has become clear that adipose tissue produces numerous adipokines—such as leptin, resistin, visfatin, and adiponectin.40–42 These adipokines contribute to systemic or local inflammatory states and play a crucial role in the initiation and progression of OA.43–46 Adipose tissue is now widely regarded as a specialized endocrine organ.47 Within human joints, articular cartilage consists of chondrocytes and the extracellular matrix (ECM). Early degenerative changes in OA are characterized by the degradation of collagen and proteoglycans within the ECM, and adipokines are key triggers of this matrix breakdown.2,43 In addition, the IFP is an important contributor to knee pain in patients with OA. Thrombospondin-4 (TSP-4), released by the IFP, is closely associated with synovitis and synovial fibrosis.48–50 Both clinical studies and animal experiments have demonstrated that partial resection of the IFP can significantly reduce joint inflammation and pain, while also exerting protective effects on joint stability and cartilage longevity.51,52 The following sections will discuss in detail the relationships between adipokines and various joint tissues involved in OA.
Leptin
Leptin is primarily produced by adipose tissue,53 and its overall circulating level is largely associated with the amount of white adipose tissue (WAT).54 Generally, women exhibit higher leptin levels than men;55 however, intra-articular leptin is mainly derived from the IFP.56 Numerous studies have demonstrated that leptin can act synergistically with various chemokines to accelerate the degradation of the ECM in chondrocytes. For example, in primary human articular chondrocytes (HACs), leptin binds to its receptor and activates signaling pathways such as JAK–STAT, MAPK, and PI3K–Akt to promote the synthesis of MMP-1 and MMP-13. It can also act in synergy with IL-1 to further enhance ECM breakdown in cartilage tissue.57,58 Additionally, leptin promotes the expression of histone deacetylases (HDAC3/4), thereby activating the TGF-β/Smad pathway and enhancing the production of MMP-1, MMP-13, and ADAMTS-4.59 Based on these characteristics of leptin, Koskinen-Kolasa et al identified a potential therapeutic opportunity for OA: suppressor of cytokine signaling-3 (SOCS-3) can inhibit these signaling pathways and counteract leptin-induced protease release, ultimately alleviating OA-related symptoms.60 Furthermore, MRI findings in OA patients show that serum leptin levels are positively correlated with osteophyte size, infrapatellar synovitis, and joint effusion.61
Resistin
Resistin is a dimeric protein that can induce insulin resistance in both humans and mice.62–64 In contrast to leptin, the concentration of resistin in serum is much higher than in synovial fluid, and its levels are significantly elevated in patients with OA.65 Compared with articular cartilage, resistin exhibits a stronger catabolic effect on the meniscus, and among these adipokines, it exerts the most detrimental influence on meniscal tissue by depleting sulfated glycosaminoglycans (sGAG).66 In articular cartilage, resistin promotes matrix degradation by upregulating pro-inflammatory cytokines and chemokines, thereby activating signaling pathways such as cAMP/PKA, p38-MAPK, C/EBP-β, and NF-κB, leading to enhanced release of matrix metalloproteinases and subsequent cartilage destruction.67 However, in human OA synovial fibroblasts (OASFs), resistin can activate the MEK and ERK pathways to suppress the levels of inflammatory mediators such as TNF-α and IL-1β, suggesting that resistin may exert certain protective effects on the synovium.68
Visfatin
Visfatin was initially identified in the liver, skeletal muscle, and bone marrow, and was previously known as pre-B colony–enhancing factor (PBEF). It exerts pro-inflammatory and catabolic effects on cartilage.69 Studies have shown that visfatin increases the expression of intercellular adhesion molecule-1 (ICAM-1) in synovial cells. In OA synovial tissue, local infiltration of monocytes into the periarticular synovium is considered an important contributor to synovial pathology, and this process is mediated through ICAM-1–dependent chemotaxis. Therefore, visfatin facilitates monocyte infiltration in OA synovium.70 Furthermore, visfatin promotes OA progression by stimulating human chondrocytes to produce PGE2, MMP-3, and MMP-13, thereby inducing a pro-degradative and pro-inflammatory chondrocyte phenotype.71–73 The therapeutic potential of targeting visfatin in OA has now been widely recognized.
Adiponectin
Adiponectin is also primarily produced by white adipose tissue. In the circulation, native adiponectin mainly exists in two forms—globular adiponectin and full-length adiponectin74 and exerts its biological functions through two different receptors. AdipoR1 is predominantly expressed in skeletal muscle and cartilage, whereas AdipoR2 is mainly found in the liver.75 The role of adiponectin in the pathogenesis and progression of OA remains controversial, with evidence supporting both protective and deleterious effects.76 Some studies have reported that globular adiponectin (gAPN) protects rat chondrocytes from H2O2-induced apoptosis via the AMPK/mTOR signaling pathway. Low concentrations of gAPN antagonize H2O2-induced cytotoxicity in a dose-dependent manner; however, at higher concentrations, gAPN exhibits cytotoxic effects.74 Consistent with these findings, a meta-analysis has also indicated a positive association between adiponectin levels and the incidence of OA.77 The adiponectin receptor agonist AdipoRon has been shown to activate the AMPK/mTOR signaling pathway to regulate autophagy and protect chondrocytes from calcification,78 and it can also slow intervertebral disc (IVD) degeneration.79 In addition, adiponectin promotes the osteogenic differentiation of human bone marrow–derived stromal cells (hBMSC) in vitro by suppressing the expression of phosphatase and tensin (PTEN) homolog deleted on chromosome ten.80
Hypertension and OA
Hypertension is a common comorbidity of OA, yet its association with OA has only gained increasing attention in recent years. An analysis of nearly two decades of nationally representative data from the U.S. National Health and Nutrition Examination Survey (NHANES) confirmed a significant relationship between hypertension and arthritis. Notably, this association remained statistically significant even after adjusting for sex, race, age, socioeconomic factors, excessive sodium intake, physical activity, obesity, smoking history, diabetes, and other comorbidities. In addition, elevations in systolic blood pressure and pulse pressure have been correlated with an increased incidence of radiographic osteoarthritis (ROA).81
Hypertension can exert varying degrees of influence on intra-articular tissues, particularly being closely associated with cartilage degeneration and synovitis, with this association appearing more pronounced in female OA populations.82 This phenomenon may be attributed to multifaceted physiological mechanisms. Primarily, the decline in estrogen levels following menopause diminishes its protective effects on both the cardiovascular system and joint tissues, rendering females more susceptible to hypertension-induced systemic low-grade inflammation, characterized by elevated levels of IL-6 and TNF-α. Furthermore, evidence suggests that hypertension-related microcirculatory disturbances in multiple joints are more prevalent in women, which exacerbates subchondral ischemia and subsequent cartilage degradation. Recently, a large-scale cohort study demonstrated that hypertensive women exhibit significantly higher expression levels of synovial inflammatory markers compared to their male counterparts, suggesting that hypertension may accelerate the progression of synovitis through sex-specific inflammatory pathways.83–85 During OA progression, hypertension may lead to subchondral ischemia, thereby damaging cartilage tissue, which is currently a widely accepted mechanism.86–88 Chronic hypertension impairs the microcirculation within the subchondral bone, reducing the delivery of essential nutrients and oxygen to the overlying articular cartilage, thus accelerating its degeneration. In recent years, however, chronic low-grade inflammation has emerged as another important factor mediating OA development in hypertensive patients. Elevated circulating inflammatory mediators such as IL-6, TNF-α, and CRP have been observed in individuals with hypertension;89 these factors can enter the joint cavity through the bloodstream, activating synovial macrophages to release inflammatory mediators and subsequently inducing synovial inflammation and cartilage destruction.30,90 Animal studies have demonstrated that hypertension is associated with OA progression in rats. Activation of the renin–angiotensin system (RAS) exacerbates cartilage damage in hypertensive mice, primarily via stimulation of angiotensin type 1 receptors (AT1R) on chondrocytes. This suggests that modulation of the RAS pathway may represent a potential therapeutic target for OA patients with hypertension.82,91 Furthermore, hypertension-related endothelial dysfunction can result in localized ischemia–reperfusion injury, further triggering oxidative stress and inflammatory cascades within the joint.92 Antihypertensive medications may mitigate OA progression; for instance, traditional medicine–derived agents such as lyophilized methanolic extract from flowering buds of Capparis spinosa (LECS) have shown cartilage-protective effects. Experimental evidence also indicates that calcium channel blockers can reduce chondrocyte apoptosis by inhibiting calcium influx.93,94 Nevertheless, current research on the relationship between hypertension and OA-related meniscal injury, infrapatellar fat pad inflammation, and osteophyte formation remains limited, despite these being important contributors to OA-associated pain and restricted joint mobility.
Hyperglycemia and OA
In recent years, the association between hyperglycemia and OA has become a research hotspot. However, regarding the study outcomes, there is still no definitive evidence that hyperglycemia is an independent risk factor for OA. Some studies have suggested that there is no significant association between diabetes and OA, which may be attributed to the confounding effect of BMI, as BMI significantly increases the risk of diabetes.95–97 Recent Mendelian randomization studies and large-scale cohort analyses have further demonstrated that after rigorous adjustment for BMI, the direct causal link between type 2 diabetes and OA development is considerably attenuated, highlighting the pivotal role of adiposity in this relationship. Many previous studies did not adequately adjust for obesity, which may act as a bridge linking diabetes mellitus (DM) and OA, or indicate that diabetes itself is not a risk factor for OA.98,99 Nevertheless, from the perspective of OA pathogenesis, recent studies have provided several important insights into hyperglycemia-induced OA. Diabetes and chronic hyperglycemia have been shown to promote OA development, and this association appears to be independent of BMI and age. In a prospective study involving 10,730 participants without knee OA, HbA1c >7.7% and fasting blood glucose >10.3 mmol/L were significantly positively associated with the risk of knee OA. Hyperglycemia was also found to reduce quadriceps muscle strength, leading to joint instability and contributing to symptomatic OA.100 Mechanistically, hyperglycemia-induced oxidative stress, accumulation of inflammatory mediators, and increased advanced glycation end products (AGEs) can negatively affect key joint tissues, including articular cartilage, bone, synovium, and ligaments.101–103
For the knee joint, the synovium plays a crucial role in cartilage metabolism. Immunohistochemical comparisons between synovia from diabetic OA patients and non-diabetic OA patients revealed that levels of MMP-13, ADAMTS-5, and inflammatory mediators were significantly elevated in the diabetic OA group. This suggests that hyperglycemia can disrupt normal cartilage structure by stimulating synovial cells to release proteases and inflammatory factors. In addition, animal studies have shown that hyperglycemia can activate the HIF-1α/GLUT1 pathway in synovial cells, promoting the accumulation of AGEs and thereby damaging cartilage.104 Prolonged glucose overload allows glucose to infiltrate the joint cavity, altering the cartilage microenvironment and impairing the optimal morphology of chondrocytes. This is manifested as tissue loosening, including reduced chondrocyte volume and increased intercellular space, along with decreased ECM density. Consequently, the cartilage’s tolerance to mechanical load diminishes, potentially serving as a trigger for OA development.105 At the microscopic level, hyperglycemia excessively activates the p38-MAPK signaling pathway, leading to elevated proteases and inflammatory mediators and resulting in ECM degradation of chondrocytes. This also indicates that the p38-MAPK pathway may serve as a potential target for disease intervention.106
Moreover, a high-glucose environment reduces the expression of peroxisome proliferator-activated receptor γ (PPARγ) and type II collagen in chondrocytes, both of which represent potential pathogenic mechanisms in hyperglycemic OA. Therefore, pioglitazone, a PPARγ agonist, can not only lower blood glucose but also mitigate the deleterious effects of hyperglycemia on chondrocytes.107 Oleanolic acid (OLA) exerts a similar but slightly different effect by activating the PPARγ/SOD2 signaling pathway, thereby protecting mitochondrial function in chondrocytes, reducing inflammatory mediator levels, and promoting type II collagen production.108
Hyperglycemia also adversely affects ligaments. In OA, ligament contracture and tears are common, particularly ACL injuries, which contribute to knee instability. Animal experiments have confirmed that a hyperglycemic environment promotes fibrosis of the medial collateral ligament (MCL), reducing its elasticity and hindering self-repair of injured ligaments. Interestingly, melatonin can counteract the detrimental effects of hyperglycemia on ligaments;109 however, whether melatonin provides similar benefits for human joint ligaments requires further investigation.
Dyslipidemia and OA
Currently, most evidence, including recent meta-analyses and mechanistic explorations, indicates a significant association between lipid metabolism disorders (especially hypercholesterolemia) and OA.110,111 However, a few studies using Mendelian randomization have presented equivocal findings regarding the direct causal links, suggesting a more complex interplay.112,113 The lipid abnormalities commonly referred to include elevated total cholesterol (TCHO), decreased high-density lipoprotein cholesterol (HDL), and increased low-density lipoprotein cholesterol (LDL).114
Multiple studies have indicated that elevated cholesterol levels are closely associated with the onset and progression of OA.115–118 The key protein responsible for cholesterol efflux in OA chondrocytes—apolipoprotein A1 binding protein (A1BP)—is significantly downregulated, leading to markedly increased intracellular cholesterol levels, which in turn results in cholesterol accumulation and inhibition of ECM synthesis. Upregulation of A1BP can reverse these effects.119,120 Animal studies have further confirmed that intracellular cholesterol accumulation in chondrocytes markedly reduces the expression of cholesterol metabolism–related genes, such as low-density lipoprotein receptor–related protein 3 (LRP3), thereby promoting protease production and damaging the chondrocyte ECM. This mechanism has been referred to as the cholesterol–LRP3–SDC4 metabolic axis.121 Given the role of cholesterol in OA, several therapeutic strategies have been proposed. For instance, cholesterol-lowering therapy combined with IL-1β inhibitors can reduce synovial hyperplasia and cartilage degeneration in mice.122 Adenoviral-mediated upregulation of circRNA, specifically circARPC1B, significantly delays OA progression induced by a high-cholesterol diet, as circRNAs play a critical role in maintaining ECM homeostasis.123 In addition, lipid-lowering drugs such as atorvastatin and simvastatin can ameliorate the metabolic impact of cholesterol on chondrocytes and slow OA progression,124,125 representing a potential breakthrough for symptom intervention in OA patients with hyperlipidemia.
Regarding HOA, studies have indicated that hyperlipidemia may serve as an independent risk factor for HOA,126 and lipid abnormalities are significantly associated with both the onset and progression of HOA, particularly elevated triglyceride levels.127 Concerning HDL, experimental evidence has shown that direct HDL treatment can downregulate the expression of matrix metalloproteinases (MMP3/9/12/13) and ADAMTS4/5, thereby exerting a protective effect on OA cartilage.120
Metabolic Inflammation and OA
With the deepening investigation into the relationship between inflammatory factors and OA in recent years, OA is increasingly recognized not merely as a wear-and-tear disorder but as a complex, multifactorial, chronic degenerative disease mediated by inflammatory cytokines. Inflammatory mediators can damage multiple non–mechanically loaded joint tissues, such as the synovium and infrapatellar fat pad, initiating inflammatory cascades and exacerbating OA symptoms.128,129 Moreover, low-grade systemic inflammation is consistently present in patients with OA. Locally within the joint, numerous studies have demonstrated that OA is frequently accompanied by infiltration of inflammatory cells within the synovium—including macrophages, T cells, mast cells, and B cells—whose secreted cytokines contribute substantially to OA onset and progression.130–132 Synovial inflammation is also strongly associated with knee pain.133,134
Regarding systemic inflammation, there is no doubt that each component of MetS contributes to elevated levels of circulating inflammatory mediators, exerting detrimental effects on joint tissues. Metabolic dysregulation also disrupts the balance between M1 and M2 macrophage phenotypes, leading to chronic inflammation, which serves as an important driver of synovitis, cartilage degeneration, and osteophyte formation in OA.129 Subsequently, as inflammatory cytokines induce joint-tissue damage, the accumulation of senescent cells further exacerbates oxidative stress,135 creating a vicious cycle of persistent local inflammation.
Immunometabolism and OA
Immunometabolism is an emerging research field that focuses on metabolic pathway alterations within immune cells and their effects on the survival and function of surrounding cells. Although immunometabolism is not currently classified as a component of MetS, several elements of MetS—particularly obesity and hypertension—are closely associated with metabolic reprogramming in immune cells such as macrophages.136,137 Dysregulation of immune-cell metabolism can lead to persistent inflammatory responses. Immunometabolism highlights the pivotal role of metabolic reprogramming within the immune system in the pathogenesis and progression of chronic inflammatory diseases.
To date, six major metabolic pathways have been identified as contributors to immunometabolism: glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway (PPP), fatty acid oxidation, fatty acid synthesis, and amino acid metabolism.138,139 Under adverse conditions, mammalian metabolism typically shifts from a low-level resting state to a highly active one. For instance, in the joints of patients with OA, the metabolic behavior of articular chondrocytes, subchondral bone cells, and synovial cells undergoes significant changes, and these cells interact with macrophages in the synovium and the immune system to influence disease progression.140 A metabolic shift toward glycolysis is essential for activating inflammatory pathways during immune responses and in chronic inflammatory diseases such as OA and rheumatoid arthritis (RA).141 Currently, research on immunometabolism in OA remains in its early stages, and more extensive and in-depth studies are needed.
Conclusion and Future Perspectives
In this review, we systematically examined recent studies concerning the relationship between MetS and OA, highlighting the complex and extensive connections between these two conditions. Although this review provides a detailed overview of the latest findings on the involvement of MetS in OA, many important questions remain to be explored. As a complex disorder involving the degeneration of multiple intraarticular tissues, OA must be viewed in its entirety. Cartilage aging and cartilage loss are indeed important components of disease progression, but they do not represent the whole picture. Notably, the primary symptom of OA, pain, is in most cases not caused by cartilage wear. Instead, it arises from synovial inflammation, subchondral bone remodeling, bone marrow lesions, osteophyte formation, and capsular contracture. However, current research places disproportionate emphasis on the roles of MetS in cartilage degeneration and synovitis, while studies investigating the effects of MetS on subchondral bone remodeling, bone marrow lesions, osteophyte formation, meniscal degeneration and tearing, and ligament injury are very limited. These factors are critical contributors to pain and functional impairment in patients with OA. Since the primary goal of all current OA treatments is still pain relief, it is clear that significant gaps remain in research focused on symptomatic OA. For patients with OA, pain is the predominant symptom and the problem they most urgently wish to resolve. Relief of pain is also one of the primary indicators for evaluating the effectiveness of OA treatments. In addition, many current theories regarding MetS-OA are derived from animal studies, particularly those conducted in rat models. Although the scientific value of animal research is undeniable, substantial and reliable clinical evidence is still lacking before these theories can be fully applied to human OA. For instance, whether pharmacologic interventions that alleviate metabolic abnormalities and improve OA symptoms in rodent models can be effectively translated to human patients remains uncertain, and many questions require further verification. Furthermore, although the signaling pathways through which individual components of MetS contribute to OA have been extensively studied, very few therapeutic agents have been developed to halt OA progression based on these pathways. This may be due to the fact that many of these molecular mechanisms have only been uncovered in recent years. With continued research, it is reasonable to anticipate that targeted interventions will gradually be developed and eventually integrated into clinical practice. Both MetS and OA are highly prevalent in older adults, and MetS is undoubtedly a major risk factor for the development of OA. However, whether OA itself may promote the onset of MetS has not yet been systematically investigated. Theoretically, OA-related knee pain can lead to reduced physical activity, which may in turn contribute to the development of MetS.
With continued advancements in understanding the etiology of OA, our knowledge of this disease has reached an unprecedented level. The strategies available to halt OA progression are now more diverse and effective than ever before. Future research should build upon the existing foundation and address the current gaps through targeted investigations. Inflammatory mediators serve as a central link between MetS and the progression of OA, and the relationships between metabolic abnormalities and damage to different joint tissues are intricate and highly complex (Figure 3). However, more systematic research in the future will undoubtedly clarify these mechanisms. It is conceivable that individualized treatment approaches based on specific metabolic abnormalities in patients with OA may eventually become possible, which would represent a transformative advance in the management of this disease.
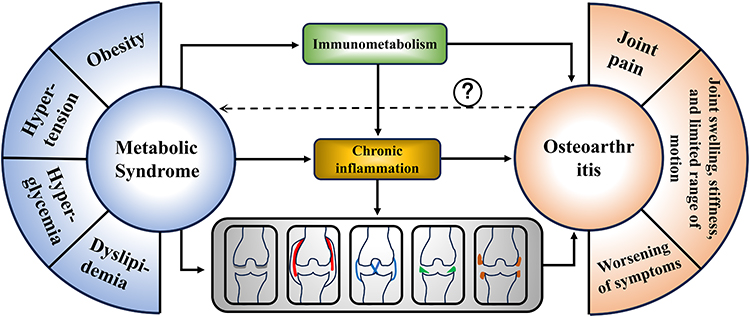 |
Figure 3 MetS is a heterogeneous condition characterized by obesity, hypertension, hyperglycemia, elevated triglycerides, and reduced high-density lipoprotein cholesterol; the presence of three or more of these components establishes the diagnosis. In OA, MetS is associated to varying degrees with aging-related degeneration across multiple intra-articular tissues, and low-grade chronic inflammation is a key driver of OA. Current evidence indicates that MetS is most strongly linked to OA-related cartilage and synovial pathology, whereas whether OA can in turn promote metabolic syndrome remains inconclusive (Dashed Arrow). |
Abbreviations
A1BP, A1 binding protein; ACL, anterior cruciate ligament; AGEs, advanced glycation end products; AT1R, angiotensin type 1 receptors; BHB, β-hydroxybutyrate; BMI, body mass index; DM, diabetes mellitus; ECM, extracellular matrix; gAPN, globular adiponectin; HACs, human articular chondrocytes; hBMSC, human bone marrow–derived stromal cells; HDAC, histone deacetylases; HDL, high-density lipoprotein cholesterol; hNPCs, human nucleus pulposus cells; HOA, hand osteoarthritis; ICAM-1, intercellular adhesion molecule-1; IFP, infrapatellar fat pad; IVD, intervertebral disc; LDL, low-density lipoprotein cholesterol; LECS, lyophilized methanolic extract from flowering buds of Capparis spinosa; LRP3, lipoprotein receptor–related protein 3; MCL, medial collateral ligament; MetS, Metabolic syndrome; MitoQ, mitochondria-targeted coenzyme Q analogue; NHANES, National Health and Nutrition Examination Survey; OA, osteoarthritis; OASFs, osteoarthritis synovial fibroblasts; OLA, Oleanolic acid; PBEF, pre-B colony–enhancing factor; PPARγ, peroxisome proliferator-activated receptor γ; PPP, pentose phosphate pathway; PTEN, phosphatase and tensin; RA, rheumatoid arthritis; RAS, renin–angiotensin system; ROA, radiographic osteoarthritis; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; sGAG, sulfated glycosaminoglycans; SOCS-3, suppressor of cytokine signaling-3; TCA, tricarboxylic acid; TCHO, total cholesterol; TSP-4, Thrombospondin-4; WAT, white adipose tissue.
Data Sharing Statement
No new data was generated for this article.
Author Contributions
Yuchen Wang is a co–first author of this article. Hongchen Ren; Conceptualization, Investigation, Writing – original draft. Yuchen Wang, Jingbo Cheng, Haicheng Tao and Hui Feng; Conceptualization, Data curation, Validation. Mingli Feng; Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology. All authors have made significant contributions to the work of the report and provided critical feedback on the revisions made to the article. Finally, all authors approved the upcoming version. They have reached an agreement on the journal to which the article will be submitted and have agreed to take responsibility for all aspects of the work.
Funding
This research received funding from Capital Health Development Fund (2026-2-2015), Beijing Medical and Health Collaborative High-Quality Traditional Chinese Medicine Research and Development Project (zyygzl-2026-007) and Translational Research Project of Xuanwu Hospital, Capital Medical University (CXZH202403).
Disclosure
The authors declare that they have no competing interests.
References
1. Tang S, Zhang C, Oo WM, et al. Osteoarthritis. Nat Rev Dis Primers. 2025;11(1):10. doi:10.1038/s41572-025-00594-6
2. Tong L, Yu H, Huang X, et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022;10(1):60. doi:10.1038/s41413-022-00226-9
3. Jenei-Lanzl Z, Zaucke F. Osteoarthritis year in review 2024: biology. Osteoarthritis Cartilage. 2025;33(1):58–15. doi:10.1016/j.joca.2024.10.008
4. Yao Q, Wu X, Tao C, et al. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct Target Ther. 2023;8(1):56. doi:10.1038/s41392-023-01330-w
5. Courties A, Kouki I, Soliman N, et al. Osteoarthritis year in review 2024: epidemiology and therapy. Osteoarthritis Cartilage. 2024;32(11):1397–1404. doi:10.1016/j.joca.2024.07.014
6. Mobasheri A, Loeser R. Clinical phenotypes, molecular endotypes and theratypes in OA therapeutic development. Nat Rev Rheumatol. 2024;20(9):525–526. doi:10.1038/s41584-024-01126-4
7. Fan X, Sun AR, Young RSE, et al. Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications. Bone Res. 2024;12(1):7. doi:10.1038/s41413-023-00304-6
8. Sanchez-Santos MT, Judge A, Gulati M, et al. Association of metabolic syndrome with knee and hand osteoarthritis: a community-based study of women. Semin Arthritis Rheum. 2019;48(5):791–798. doi:10.1016/j.semarthrit.2018.07.007
9. Visser AW, de Mutsert R, le Cessie S, et al. The relative contribution of mechanical stress and systemic processes in different types of osteoarthritis: the NEO study. Ann Rheum Dis. 2015;74(10):1842–1847. doi:10.1136/annrheumdis-2013-205012
10. Tomi AL, Sellam J, Lacombe K, et al. Increased prevalence and severity of radiographic hand osteoarthritis in patients with HIV-1 infection associated with metabolic syndrome: data from the cross-sectional METAFIB-OA study. Ann Rheum Dis. 2016;75(12):2101–2107. doi:10.1136/annrheumdis-2016-209262
11. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement: executive summary. Crit Pathw Cardiol. 2005;4(4):198–203. doi:10.1097/00132577-200512000-00018
12. Zimmet P, Magliano D, Matsuzawa Y, et al. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. 2005;12(6):295–300. doi:10.5551/jat.12.295
13. Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. doi:10.1161/circulationaha.109.192644
14. Romeo S, Vidal-Puig A, Husain M, et al. Clinical staging to guide management of metabolic disorders and their sequelae: a European Atherosclerosis Society consensus statement. Eur Heart J. 2025;46(38):3685–3713. doi:10.1093/eurheartj/ehaf314
15. Fahed G, Aoun L, Bou Zerdan M, et al. Metabolic syndrome: updates on pathophysiology and management in 2021. Int J Mol Sci. 2022;23(2):786. doi:10.3390/ijms23020786
16. Duong V, Oo WM, Ding C, et al. Evaluation and treatment of knee pain: a review. JAMA. 2023;330(16):1568–1580. doi:10.1001/jama.2023.19675
17. O’Neill TW, Felson DT. Mechanisms of osteoarthritis (OA) pain. Curr Osteoporos Rep. 2018;16(5):611–616. doi:10.1007/s11914-018-0477-1
18. Roelofs AJ, Kania K, Rafipay AJ, et al. Identification of the skeletal progenitor cells forming osteophytes in osteoarthritis. Ann Rheum Dis. 2020;79(12):1625–1634. doi:10.1136/annrheumdis-2020-218350
19. Courtney CA, O’Hearn MA, Hornby TG. Neuromuscular function in painful knee osteoarthritis. Curr Pain Headache Rep. 2012;16(6):518–524. doi:10.1007/s11916-012-0299-2
20. Singh A, Fraser B, Venn A, et al. Trajectory of metabolic syndrome and its association with knee pain in middle-aged adults. Diabetes Metab Syndr. 2023;17(12):102916. doi:10.1016/j.dsx.2023.102916
21. Valdes AM. Metabolic syndrome and osteoarthritis pain: common molecular mechanisms and potential therapeutic implications. Osteoarthritis Cartilage. 2020;28(1):7–9. doi:10.1016/j.joca.2019.06.015
22. Lyu L, Cai Y, Xiao M, et al. Causal relationships of general and abdominal adiposity on osteoarthritis: a two-sample mendelian randomization study. J Clin Med. 2022;12(1):320. doi:10.3390/jcm12010320
23. Voinier D, Neogi T, Stefanik JJ, et al. Using cumulative load to explain how body mass index and daily walking relate to worsening knee cartilage damage over two years: the MOST study. Arthritis Rheumatol. 2020;72(6):957–965. doi:10.1002/art.41181
24. Francisco V, Pérez T, Pino J, et al. Biomechanics, obesity, and osteoarthritis. The role of adipokines: when the levee breaks. J Orthop Res. 2018;36(2):594–604. doi:10.1002/jor.23788
25. Neumann E, Junker S, Schett G, et al. Adipokines in bone disease. Nat Rev Rheumatol. 2016;12(5):296–302. doi:10.1038/nrrheum.2016.49
26. Chen HT, Tsou HK, Chen JC, et al. Adiponectin enhances intercellular adhesion molecule-1 expression and promotes monocyte adhesion in human synovial fibroblasts. PLoS One. 2014;9(3):e92741. doi:10.1371/journal.pone.0092741
27. Miller RH. Joint loading in runners does not initiate knee osteoarthritis. Exerc Sport Sci Rev. 2017;45(2):87–95. doi:10.1249/jes.0000000000000105
28. Davis S, Roldo M, Blunn G, et al. Influence of the mechanical environment on the regeneration of osteochondral defects. Front Bioeng Biotechnol. 2021;9:603408. doi:10.3389/fbioe.2021.603408
29. Moon PM, Shao ZY, Wambiekele G, et al. Global deletion of Pannexin 3 resulting in accelerated development of aging-induced osteoarthritis in mice. Arthritis Rheumatol. 2021;73(7):1178–1188. doi:10.1002/art.41651
30. Zhang H, Shao Y, Yao Z, et al. Mechanical overloading promotes chondrocyte senescence and osteoarthritis development through downregulating FBXW7. Ann Rheum Dis. 2022;81(5):676–686. doi:10.1136/annrheumdis-2021-221513
31. Fu W, Vasylyev D, Bi Y, et al. Nav1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature. 2024;625(7995):557–565. doi:10.1038/s41586-023-06888-7
32. Lee W, Leddy HA, Chen Y, et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc Natl Acad Sci U S A. 2014;111(47):E5114–5122. doi:10.1073/pnas.1414298111
33. Xu X, Liu S, Liu H, et al. Piezo channels: awesome mechanosensitive structures in cellular mechanotransduction and their role in bone. Int J Mol Sci. 2021;22(12):6429. doi:10.3390/ijms22126429
34. Wang S, Li W, Zhang P, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75. doi:10.1016/j.jare.2022.01.004
35. Wu J, Chen Y, Liao Z, et al. Self-amplifying loop of NF-κB and periostin initiated by PIEZO1 accelerates mechano-induced senescence of nucleus pulposus cells and intervertebral disc degeneration. Mol Ther. 2022;30(10):3241–3256. doi:10.1016/j.ymthe.2022.05.021
36. Shao Y, Zhang H, Guan H, et al. PDZK1 protects against mechanical overload-induced chondrocyte senescence and osteoarthritis by targeting mitochondrial function. Bone Res. 2024;12(1):41. doi:10.1038/s41413-024-00344-6
37. Fan T, Ruan G, Antony B, et al. The interactions between MRI-detected osteophytes and bone marrow lesions or effusion-synovitis on knee symptom progression: an exploratory study. Osteoarthritis Cartilage. 2021;29(9):1296–1305. doi:10.1016/j.joca.2021.06.008
38. Nakamura Y, Ogawa H, Sohmiya K, et al. Relationship between histological changes of the anterior cruciate ligament and knee function in osteoarthritis patients. Orthop Traumatol Surg Res. 2022;108(8):103341. doi:10.1016/j.otsr.2022.103341
39. Panzer S, Augat P, Atzwanger J, et al. 3-T MRI assessment of osteophyte formation in patients with unilateral anterior cruciate ligament injury and reconstruction. Skeletal Radiol. 2012;41(12):1597–1604. doi:10.1007/s00256-012-1445-y
40. Collins KH, Lenz KL, Pollitt EN, et al. Adipose tissue is a critical regulator of osteoarthritis. Proc Natl Acad Sci U S A. 2021;118(1). doi:10.1073/pnas.2021096118
41. Chang J, Liao Z, Lu M, et al. Systemic and local adipose tissue in knee osteoarthritis. Osteoarthritis Cartilage. 2018;26(7):864–871. doi:10.1016/j.joca.2018.03.004
42. Binvignat M, Sellam J, Berenbaum F, et al. The role of obesity and adipose tissue dysfunction in osteoarthritis pain. Nat Rev Rheumatol. 2024;20(9):565–584. doi:10.1038/s41584-024-01143-3
43. Taylor EB. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin Sci. 2021;135(6):731–752. doi:10.1042/cs20200895
44. Xie C, Chen Q. Adipokines: new therapeutic target for osteoarthritis? Curr Rheumatol Rep. 2019;21(12):71. doi:10.1007/s11926-019-0868-z
45. Deng T, Lyon CJ, Bergin S, et al. Obesity, inflammation, and cancer. Annu Rev Pathol. 2016;11:421–449. doi:10.1146/annurev-pathol-012615-044359
46. Kontny E, Plebanczyk M, Lisowska B, et al. Comparison of rheumatoid articular adipose and synovial tissue reactivity to proinflammatory stimuli: contribution to adipocytokine network. Ann Rheum Dis. 2012;71(2):262–267. doi:10.1136/annrheumdis-2011-200123
47. Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. 2019;129(10):3990–4000. doi:10.1172/jci129187
48. Braun S, Pollinger P, Sohn R, et al. Expression of thrombospondin-4 in the infrapatellar fat pad and synovial fluid - potential contribution to osteoarthritis pain. Arthritis Res Ther. 2025;27(1):170. doi:10.1186/s13075-025-03615-7
49. Sriwatananukulkit O, Desclaux S, Tawonsawatruk T, et al. Effectiveness of losartan on infrapatellar fat pad/synovial fibrosis and pain behavior in the monoiodoacetate-induced rat model of osteoarthritis pain. Biomed Pharmacother. 2023;158:114121. doi:10.1016/j.biopha.2022.114121
50. Inomata K, Tsuji K, Onuma H, et al. Time course analyses of structural changes in the infrapatellar fat pad and synovial membrane during inflammation-induced persistent pain development in rat knee joint. BMC Musculoskelet Disord. 2019;20(1):8. doi:10.1186/s12891-018-2391-1
51. Li Y, Lu P, Yao H, et al. Observation of the effects of infrapatellar fat pad excision on the inflammatory progression of knee osteoarthritis in mice. J Inflamm Res. 2025;18:6653–6672. doi:10.2147/jir.S517314
52. Liu Y, Gao Q. Partial excision of infrapatellar fat pad for the treatment of knee osteoarthritis. J Orthop Surg Res. 2024;19(1):631. doi:10.1186/s13018-024-05114-y
53. Yadav A, Kataria MA, Saini V, et al. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta. 2013;417:80–84. doi:10.1016/j.cca.2012.12.007
54. Solsona-Vilarrasa E, Vousden KH. Obesity, white adipose tissue and cancer. Febs J. 2025;292(9):2189–2207. doi:10.1111/febs.17312
55. Pérez-Pérez A, Vilariño-García T, Fernández-Riejos P, Martín-González J, Segura-Egea JJ, Sánchez-Margalet V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017;35:71–84. doi:10.1016/j.cytogfr.2017.03.001
56. Liu Z, Xie W, Li H, et al. Novel perspectives on leptin in osteoarthritis: focus on aging. Genes Dis. 2024;11(6):101159. doi:10.1016/j.gendis.2023.101159
57. Scotece M, Mobasheri A. Leptin in osteoarthritis: focus on articular cartilage and chondrocytes. Life Sci. 2015;140:75–78. doi:10.1016/j.lfs.2015.05.025
58. Hui W, Litherland GJ, Elias MS, et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Ann Rheum Dis. 2012;71(3):455–462. doi:10.1136/annrheumdis-2011-200372
59. Su YP, Chen CN, Huang KC, et al. Leptin induces MMP1/13 and ADAMTS 4 expressions through bone morphogenetic protein-2 autocrine effect in human chondrocytes. J Cell Biochem. 2018;119(4):3716–3724. doi:10.1002/jcb.26593
60. Koskinen-Kolasa A, Vuolteenaho K, Korhonen R, et al. Catabolic and proinflammatory effects of leptin in chondrocytes are regulated by suppressor of cytokine signaling-3. Arthritis Res Ther. 2016;18(1):215. doi:10.1186/s13075-016-1112-0
61. Chong TKY, Tan JR, Ma CA, et al. Association of adipokines with severity of knee osteoarthritis assessed clinically and on magnetic resonance imaging. Osteoarthr Cartil Open. 2023;5(4):100405. doi:10.1016/j.ocarto.2023.100405
62. Jiang Y, Lu L, Hu Y, et al. Resistin induces hypertension and insulin resistance in mice via a TLR4-dependent pathway. Sci Rep. 2016;6:22193. doi:10.1038/srep22193
63. Kim SJ, Nian C, McIntosh CH. Resistin knockout mice exhibit impaired adipocyte glucose-dependent insulinotropic polypeptide receptor (GIPR) expression. Diabetes. 2013;62(2):471–477. doi:10.2337/db12-0257
64. Yang HM, Kim J, Kim BK, et al. Resistin regulates inflammation and insulin resistance in humans via the endocannabinoid system. Research. 2024;7:0326. doi:10.34133/research.0326
65. Perruccio AV, Mahomed NN, Chandran V, et al. Plasma adipokine levels and their association with overall burden of painful joints among individuals with Hip and knee osteoarthritis. J Rheumatol. 2014;41(2):334–337. doi:10.3899/jrheum.130709
66. Nishimuta JF, Levenston ME. Meniscus is more susceptible than cartilage to catabolic and anti-anabolic effects of adipokines. Osteoarthritis Cartilage. 2015;23(9):1551–1562. doi:10.1016/j.joca.2015.04.014
67. Zhao CW, Gao YH, Song WX, et al. An update on the emerging role of resistin on the pathogenesis of osteoarthritis. Mediators Inflamm. 2019;2019:1532164. doi:10.1155/2019/1532164
68. Chen WC, Lu YC, Kuo SJ, et al. Resistin enhances IL-1β and TNF-α expression in human osteoarthritis synovial fibroblasts by inhibiting miR-149 expression via the MEK and ERK pathways. FASEB J. 2020;34(10):13671–13684. doi:10.1096/fj.202001071R
69. Franco-Trepat E, Guillán-Fresco M, Alonso-Pérez A, et al. Visfatin connection: present and future in osteoarthritis and osteoporosis. J Clin Med. 2019;8(8):1178. doi:10.3390/jcm8081178
70. Law YY, Lin YM, Liu SC, et al. Visfatin increases ICAM-1 expression and monocyte adhesion in human osteoarthritis synovial fibroblasts by reducing miR-320a expression. Aging. 2020;12(18):18635–18648. doi:10.18632/aging.103889
71. Zhang Q, Zhao YX, Li LF, et al. Metabolism-related adipokines and metabolic diseases: their role in osteoarthritis. J Inflamm Res. 2025;18:1207–1233. doi:10.2147/JIR.S499835
72. Yang S, Ryu JH, Oh H, et al. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann Rheum Dis. 2015;74(3):595–602. doi:10.1136/annrheumdis-2013-204355
73. Cheleschi S, Tenti S, Barbarino M, et al. Exploring the crosstalk between hydrostatic pressure and adipokines: an in vitro study on human osteoarthritic chondrocytes. Int J Mol Sci. 2021;22(5):2745. doi:10.3390/ijms22052745
74. Hu J, Cui W, Ding W, et al. Globular adiponectin attenuated H2O2-induced apoptosis in rat chondrocytes by inducing autophagy through the AMPK/ mTOR pathway [published correction appears in Cell Physiol Biochem. 2022 Oct 31;56(5):602-604. doi: 10.33594/000000579]. Cell Physiol Biochem. 2017;43(1):367–382. doi:10.1159/000480416
75. Okada-Iwabu M, Yamauchi T, Iwabu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503(7477):493–499. doi:10.1038/nature12656
76. Ilia I, Nitusca D, Marian C. Adiponectin in osteoarthritis: pathophysiology, relationship with obesity and presumptive diagnostic biomarker potential. Diagnostics. 2022;12(2):455. doi:10.3390/diagnostics12020455
77. Tang Q, Hu ZC, Shen LY, et al. Association of osteoarthritis and circulating adiponectin levels: a systematic review and meta-analysis. Lipids Health Dis. 2018;17(1):189. doi:10.1186/s12944-018-0838-x
78. Duan ZX, Tu C, Liu Q, et al. Adiponectin receptor agonist AdipoRon attenuates calcification of osteoarthritis chondrocytes by promoting autophagy. J Cell Biochem. 2020;121(5–6):3333–3344. doi:10.1002/jcb.29605
79. Ohnishi H, Zhang Z, Yurube T, et al. Anti-inflammatory effects of adiponectin receptor agonist adiporon against intervertebral disc degeneration. Int J Mol Sci. 2023;24(10):8566. doi:10.3390/ijms24108566
80. Liu X, Chen T, Wu Y, et al. Role and mechanism of PTEN in adiponectin-induced osteogenesis in human bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2017;483(1):712–717. doi:10.1016/j.bbrc.2016.12.076
81. Lo GH, McAlindon TE, Katz JN, et al. Systolic and pulse pressure associate with incident knee osteoarthritis: data from the osteoarthritis initiative. Clin Rheumatol. 2017;36(9):2121–2128. doi:10.1007/s10067-017-3656-z
82. Yeater TD, Griffith JL, Cruz CJ, et al. Hypertension contributes to exacerbated osteoarthritis pathophysiology in rats in a sex-dependent manner. Arthritis Res Ther. 2023;25(1):7. doi:10.1186/s13075-022-02966-9
83. Zhang Y, Cai W, Han G, et al. Sex differences in the association between hypertension and osteoarthritis: a systematic review and meta-analysis. Arthritis Res Ther. 2021;23(1):118. doi:10.1186/s13075-021-02503-z
84. Arkill KP, Winlove CP. Solute transport in the deep and calcified zones of articular cartilage. Osteoarthr Cartil. 2008;16(6):708–714. doi:10.1016/j.joca.2007.10.001
85. Wang M, Qin T, Jin Y, et al. Impact of vascular hypertension on synovial inflammation and cartilage integrity: a gender-stratified analysis. J Orthop Trans. 2023;39:45–56. doi:10.1016/j.jot.2023.02.001
86. Findlay DM, Kuliwaba JS. Bone-cartilage crosstalk: a conversation for understanding osteoarthritis. Bone Res. 2016;4:16028. doi:10.1038/boneres.2016.28
87. Ching K, Houard X, Berenbaum F, Wen C. Hypertension meets osteoarthritis - revisiting the vascular aetiology hypothesis. Nat Rev Rheumatol. 2021;17(9):533–549. doi:10.1038/s41584-021-00650-x
88. Ni R, Guo XE, Yan C, Wen C. Hemodynamic stress shapes subchondral bone in osteoarthritis: an emerging hypothesis. J Orthop Translat. 2021;32:85–90. doi:10.1016/j.jot.2021.11.007
89. Jian ZW, Zhang XM, Huang GS. Clinical value of the platelet and inflammatory factor activation in vascular endothelial injury in essential hypertension. Clin Hemorheol Microcirc. 2023;83(2):171–180. doi:10.3233/ch-221638
90. Zhou H, Shen X, Yan C, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res Ther. 2022;13(1):322. doi:10.1186/s13287-022-03005-9
91. Yamagishi K, Tsukamoto I, Nakamura F, et al. Activation of the renin-angiotensin system in mice aggravates mechanical loading-induced knee osteoarthritis. Eur J Histochem. 2018;62(3):2930. doi:10.4081/ejh.2018.2930
92. Griendling KK, Camargo LL, Rios FJ, et al. Oxidative Stress and Hypertension. Circ Res. 2021;128(7):993–1020. doi:10.1161/circresaha.121.318063
93. Visser AW, Kloppenburg M, de Mutsert R. Cardiovascular drugs and osteoarthritis: effects of targeting ion channels. Cells. 2021;10(10):2572. doi:10.3390/cells10102572
94. Smith SM, Cooper-DeHoff RM. Fixed-dose combination amlodipine/celecoxib (consensi) for hypertension and osteoarthritis. Am J Med. 2019;132(2):172–174. doi:10.1016/j.amjmed.2018.08.027
95. Wang J, Peng L, Yang M, et al. Is there a genetic relationship between blood glucose and osteoarthritis? A mendelian randomization study. Diabetol Metab Syndr. 2024;16(1):274. doi:10.1186/s13098-024-01517-3
96. Kim JS, Choi JH, Shin SR, Han AL. Association between insulin resistance indices and prevalence of knee osteoarthritis using the Korean National health and examination survey. Sci Rep. 2025;15(1):18195. doi:10.1038/s41598-025-91526-7
97. Zhang B, Liu D. Integrated bioinformatic analysis of the molecular mechanisms between type 2 diabetes mellitus and osteoarthritis. Medicine. 2024;103(35):e39469. doi:10.1097/MD.0000000000039469
98. Dawson LP, Fairley JL, Papandony MC, et al. Is abnormal glucose tolerance or diabetes a risk factor for knee, hip, or hand osteoarthritis? A systematic review. Semin Arthritis Rheum. 2018;48(2):176–189. doi:10.1016/j.semarthrit.2018.02.008
99. Khor A, Ma CA, Hong C, et al. Diabetes mellitus is not a risk factor for osteoarthritis. RMD Open. 2020;6(1):e001030. doi:10.1136/rmdopen-2019-001030
100. Zheng J, Huang X, Huang J, et al. Association of diabetes mellitus status and hyperglycemia with symptomatic knee osteoarthritis. Arthritis Care Res. 2023;75(3):509–518. doi:10.1002/acr.24872
101. Luc K, Schramm-Luc A, Guzik TJ, et al. Oxidative stress and inflammatory markers in prediabetes and diabetes. J Physiol Pharmacol. 2019;70(6). doi:10.26402/jpp.2019.6.01
102. Yang F, Zhu D, Wang Z, et al. Role of advanced glycation end products in intervertebral disc degeneration: mechanism and therapeutic potential. Oxid Med Cell Longev. 2022;2022:7299005. doi:10.1155/2022/7299005
103. Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules. 2022;12(4):542. doi:10.3390/biom12040542
104. Li Q, Wen Y, Wang L, et al. Author correction: hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like synoviocytes promotes knee osteoarthritis. Exp Mol Med. 2022;54(6):862–865. doi:10.1038/s12276-022-00788-y
105. Njoto I, Soekanto A, Ernawati E, et al. Chondrocyte intracellular matrix strain fields of articular cartilage surface in hyperglycemia model of rat: cellular morphological study. Med Arch. 2018;72(5):348–351. doi:10.5455/medarh.2018.72.348-351
106. Cheng X, Ni B, Zhang F, et al. High glucose-induced oxidative stress mediates apoptosis and extracellular matrix metabolic imbalances possibly via p38 MAPK activation in rat nucleus pulposus cells. J Diabetes Res. 2016;2016:3765173. doi:10.1155/2016/3765173
107. Chen YJ, Chan DC, Lan KC, et al. PPARγ is involved in the hyperglycemia-induced inflammatory responses and collagen degradation in human chondrocytes and diabetic mouse cartilages. J Orthop Res. 2015;33(3):373–381. doi:10.1002/jor.22770
108. Kang X, Yang Z, Sheng J, et al. Oleanolic acid prevents cartilage degeneration in diabetic mice via PPARγ associated mitochondrial stabilization. Biochem Biophys Res Commun. 2017;490(3):834–840. doi:10.1016/j.bbrc.2017.06.127
109. Adamska O, Wnuk A, Kamińska A, et al. Melatonin supplementation counteracts fiber loss in knee ligaments of diabetes-induced rats. Front Pharmacol. 2024;15(1399719). doi:10.3389/fphar.2024.1399719
110. Schwager JL, Nevitt MC, Torner J, et al. Association of serum low-density lipoprotein, high-density lipoprotein, and total cholesterol with development of knee osteoarthritis. Arthritis Care Res. 2022;74(2):274–280. doi:10.1002/acr.24455
111. Hsueh TY, Chen YP. The association between dyslipidemia and hand osteoarthritis: a systematic review and meta-analysis. Clin Rheumatol. 2025;44(4):1439–1448. doi:10.1007/s10067-025-07384-1
112. Zhu XW, Zheng X, Wang L, et al. Evaluation of the causal relationship between 28 circulating biomarkers and osteoarthritis: a bidirectional Mendelian randomization study. Bone Joint Res. 2025;14(3):259–269. doi:10.1302/2046-3758.143.BJR-2024-0207.R1
113. Ma W, Chen H, Zhang Z, Xiong Y. Association of lipid-lowering drugs with osteoarthritis outcomes from a drug-target Mendelian randomization study. PLoS One. 2024;19(2):e0293960. doi:10.1371/journal.pone.0293960
114. Vekic J, Zeljkovic A, Stefanovic A, et al. Obesity and dyslipidemia. Metabolism. 2019;92:71–81. doi:10.1016/j.metabol.2018.11.005
115. Song Y, Liu J, Zhao K, et al. Cholesterol-induced toxicity: an integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021;33(10):1911–1925. doi:10.1016/j.cmet.2021.09.001
116. Little CB. Cholesterol, systemic inflammation, interleukin-1β, and osteoarthritis risk - aligning animal models with specific patient endotypes provides novel insights. Osteoarthritis Cartilage. 2023;31(3):298–299. doi:10.1016/j.joca.2022.11.009
117. Papathanasiou I, Anastasopoulou L, Tsezou A. Cholesterol metabolism related genes in osteoarthritis. Bone. 2021;152:116076. doi:10.1016/j.bone.2021.116076
118. Xiong J, Long J, Chen X, et al. Dyslipidemia might be associated with an increased risk of osteoarthritis. Biomed Res Int. 2020;2020:3105248. doi:10.1155/2020/3105248
119. Lee G, Yang J, Kim SJ, et al. Enhancement of intracellular cholesterol efflux in chondrocytes leading to alleviation of osteoarthritis progression. Arthritis Rheumatol. 2025;77(2):151–162. doi:10.1002/art.42984
120. Yan L, Ge H, Xu Q, et al. Dyslipidemia induced inflammation mediated the association between obesity and osteoarthritis: a population-based study. BMC Public Health. 2024;24(1):3155. doi:10.1186/s12889-024-20616-4
121. Cao C, Shi Y, Zhang X, et al. Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4. Nat Commun. 2022;13(1):7139. doi:10.1038/s41467-022-34830-4
122. Van Gemert Y, Kruisbergen NNL, Blom AB, et al. IL-1β inhibition combined with cholesterol-lowering therapies decreases synovial lining thickness and spontaneous cartilage degeneration in a humanized dyslipidemia mouse model. Osteoarthritis Cartilage. 2023;31(3):340–350. doi:10.1016/j.joca.2022.09.014
123. Li J, Li X, Zhou S, et al. Circular RNA circARPC1B functions as a stabilisation enhancer of Vimentin to prevent high cholesterol-induced articular cartilage degeneration. Clin Transl Med. 2023;13(9):e1415. doi:10.1002/ctm2.1415
124. Gierman LM, Kühnast S, Koudijs A, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice--a translational model for atherosclerosis. Ann Rheum Dis. 2014;73(5):921–927. doi:10.1136/annrheumdis-2013-203248
125. Robby AI, Kim EH, Huh KM, et al. Mevalonate pathway-triggered phase transition of injectable hydrogel for cholesterol-downregulated therapy of osteoarthritis. Bioact Mater. 2025;51:876–888. doi:10.1016/j.bioactmat.2025.06.047
126. Frey N, Hügle T, Jick SS, et al. Hyperlipidaemia and incident osteoarthritis of the hand: a population-based case-control study. Osteoarthritis Cartilage. 2017;25(7):1040–1045. doi:10.1016/j.joca.2017.01.014
127. Baloun J, Švec X, Cerezo LA, et al. The association between radiographic progression, functional impairment, markers of dyslipidaemia and inflammation in patients with hand osteoarthritis: a five-year longitudinal study. Clin Exp Rheumatol. 2025;43(3):486–496. doi:10.55563/clinexprheumatol/9b9fnm
128. Han Z, Wang K, Ding S, et al. Cross-talk of inflammation and cellular senescence: a new insight into the occurrence and progression of osteoarthritis. Bone Res. 2024;12(1):69. doi:10.1038/s41413-024-00375-z
129. Warmink K, Vinod P, Korthagen NM, et al. Macrophage-driven inflammation in metabolic osteoarthritis: implications for biomarker and therapy development. Int J Mol Sci. 2023;24(7):6112. doi:10.3390/ijms24076112
130. Li YS, Luo W, Zhu SA, et al. T cells in osteoarthritis: alterations and beyond. Front Immunol. 2017;8(356). doi:10.3389/fimmu.2017.00356
131. Rosshirt N, Trauth R, Platzer H, et al. Proinflammatory T cell polarization is already present in patients with early knee osteoarthritis. Arthritis Res Ther. 2021;23(1):37. doi:10.1186/s13075-020-02410-w
132. Xie X, Doody GM, Shuweihdi F, et al. B-cell capacity for expansion and differentiation into plasma cells are altered in osteoarthritis. Osteoarthritis Cartilage. 2023;31(9):1176–1188. doi:10.1016/j.joca.2023.03.017
133. Gómez-Aristizábal A, Gandhi R, Mahomed NN, et al. Synovial fluid monocyte/macrophage subsets and their correlation to patient-reported outcomes in osteoarthritic patients: a cohort study. Arthritis Res Ther. 2019;21(1):26. doi:10.1186/s13075-018-1798-2
134. Sarmanova A, Hall M, Fernandes GS, et al. Association between ultrasound-detected synovitis and knee pain: a population-based case-control study with both cross-sectional and follow-up data. Arthritis Res Ther. 2017;19(1):281. doi:10.1186/s13075-017-1486-7
135. Motta F, Barone E, Sica A, et al. Inflammaging and osteoarthritis. Clin Rev Allergy Immunol. 2023;64(2):222–238. doi:10.1007/s12016-022-08941-1
136. Mouton AJ, Li X, Hall ME, et al. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res. 2020;126(6):789–806. doi:10.1161/circresaha.119.312321
137. Makowski L, Chaib M, Rathmell JC. Immunometabolism: from basic mechanisms to translation. Immunol Rev. 2020;295(1):5–14. doi:10.1111/imr.12858
138. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi:10.1038/nri.2016.70
139. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–643. doi:10.1016/j.immuni.2013.04.005
140. Henry ÓC, O’Neill LAJ. Metabolic reprogramming in stromal and immune cells in rheumatoid arthritis and osteoarthritis: therapeutic possibilities. Eur J Immunol. 2025;55(4):e202451381. doi:10.1002/eji.202451381
141. Mobasheri A, Rayman MP, Gualillo O, et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13(5):302–311. doi:10.1038/nrrheum.2017.50
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Skin Metabolic Signatures of Psoriasis and Psoriasis Concurrent with Metabolic Syndrome
Yan L, Wang W, Dong M, Wang R, Li C
Journal of Inflammation Research 2025, 18:505-517
Published Date: 10 January 2025