Back to Journals » Nature and Science of Sleep » Volume 13
Reviewing the Clinical Implications of Treating Narcolepsy as an Autoimmune Disorder
Authors Giannoccaro MP, Liguori R ![]() , Plazzi G
, Plazzi G ![]() , Pizza F
, Pizza F
Received 16 December 2020
Accepted for publication 19 March 2021
Published 11 May 2021 Volume 2021:13 Pages 557—577
DOI https://doi.org/10.2147/NSS.S275931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sarah L Appleton
Maria Pia Giannoccaro,1,2 Rocco Liguori,1,2 Giuseppe Plazzi,1,3 Fabio Pizza1,2
1IRCCS Istituto delle Scienze Neurologiche di Bologna, Ospedale Bellaria, Bologna, Italy; 2Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy; 3Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy
Correspondence: Giuseppe Plazzi
IRCCS Istituto delle Scienze Neurologiche di Bologna, Ospedale Bellaria, via Altura 3, Bologna, 40139, Italy
Tel +39 051 4966926
Fax +39 051 4966176
Email [email protected]
Abstract: Narcolepsy type 1 (NT1) is a lifelong sleep disorder, primarily characterized clinically by excessive daytime sleepiness and cataplexy and pathologically by the loss of hypocretinergic neurons in the lateral hypothalamus. Despite being a rare disorder, the NT1-related burden for patients and society is relevant due to the early onset and chronic nature of this condition. Although the etiology of narcolepsy is still unknown, mounting evidence supports a central role of autoimmunity. To date, no cure is available for this disorder and current treatment is symptomatic. Based on the hypothesis of the autoimmune etiology of this disease, immunotherapy could possibly represent a valid therapeutic option. However, contrasting and limited results have been provided so far. This review discusses the evidence supporting the use of immunotherapy in narcolepsy, the outcomes obtained so far, current issues and future directions.
Keywords: narcolepsy type 1, immunotherapy, immunomodulation, intravenous immunoglobulin, steroid, monoclonal antibodies
Introduction
Narcolepsy is a chronic sleep disorder, primarily associated with excessive daytime sleepiness (EDS) and cataplexy, a sudden and transient loss of muscle tone triggered mainly by intense, usually positive, emotions, during wakefulness. Other symptoms, including sleep-related hallucinations, sleep paralyses, and fragmented nocturnal sleep point to an intrinsic REM sleep dysfunction (ICSD3).1 In most cases, symptom onset is in the first two decades of life, with up to 65% of the cases presenting before the age of 20 years.2,3
According to the American Academy of Sleep Medicine (AASM),1 two distinct subtypes are identified, Narcolepsy type 1 (NT1) and Narcolepsy type 2 (NT2). NT1 results from the loss of hypothalamic hypocretin (orexin)-producing neurons as documented by reduced or undetectable levels of hypocretin-1 (hcrt-1) in the cerebrospinal fluid (CSF) and is clinically marked by cataplexy, whereas NT2 is characterized by normal CSF hcrt-1 concentration and absence of cataplexy. The CSF hcrt-1 deficiency observed in NT1 is due to the destruction of a small group of hypocretin secreting neurons in the lateral hypothalamus.4 In NT2, a less severe loss of these neurons or an altered hypocretin receptor signalling5,6 has been postulated. About 10% of NT2 cases transform into the NT1 phenotype, indicating disease progression over time, at least in some cases.7–10
Narcolepsy is classified as a rare disorder with a prevalence of 20–50/100,000 individuals worldwide11,12 but is however poorly and lately recognized13,14 and burdened by a high socioeconomic impact. Indeed, narcolepsy patients have lower education and higher unemployment rate compared to the general population, resulting in reduced incomes and lowered life standards.15–18 Moreover, they present higher frequency of other medical/psychiatric comorbidities and concurrent medication usage, and reduced rates of marriage/cohabitation. Despite the availability of several symptomatic treatments,4,19 complete control of symptoms is only rarely achieved.20,21
The necessity to find a cure for this lifelong and disabling condition has driven the investigation of new treatments targeting the underlying mechanisms of the disease. In this review, we will discuss the implications of treating narcolepsy as an autoimmune disorder, the therapeutic approaches used so far and their outcomes as well as the future directions.
Evidence of Autoimmune Etiology in Narcolepsy
Autoimmune disorders are pathological conditions characterized by an aberrant immune response against “self-antigens” due to the loss of tolerance, which leads to inflammation, cell injury or dysfunction and clinical manifestations. Formal demonstration of the autoimmune nature of a disease requires several pieces of evidence.22 Direct evidence is provided by the passive transfer of pathology by antibodies or T-cells from an affected individual to laboratory animals or to cells in culture. Indirect evidence comes from the simulation of disease in animal models either by active immunization or by manipulation of the immune system, or by isolation of self-reactive T-cells/autoantibodies from the organ targeted by the autoimmune attack. Finally, circumstantial evidence derives from different clinical observations such as: a) presence of genetic susceptibility (ie, recurrence in the same family and human leukocyte antigen (HLA) association); b) presence of antibodies in relation with a specific clinical phenotype; and c) response to immunotherapy.23
The loss of the hypocretin secreting cells represents the core feature of NT1. Nevertheless, the pathological mechanisms leading to the highly selective destruction of these hypothalamic cells, with sparing of the neighboring melanin-concentrating hormone neurons, are still unknown. However, the specificity of this loss itself, the strong association with the HLA DQB1*06:0224 and other genetically determined features of the immune system pointed towards the hypothesis of the autoimmune etiology of narcolepsy. This hypothesis was further supported by circumstantial evidence coming from epidemiological studies showing an association between NT1 and infections, which can provoke autoimmune reactions through different mechanisms such as bystander activation, molecular mimicry, superantigens and epitope spreading.25 A questionnaire-based study revealed an increased frequency of narcolepsy among subjects diagnosed with strep throat before the age of 2126 and elevated streptococcal antibodies levels were found in patients’ sera taken within 3 years from disease onset compared to age-matched controls.27 Lately, a link between narcolepsy and the influenza A virus subtype H1N1 (A/H1N1) was observed. In China, the incidence of narcolepsy increased three-fold within 6 months of the peak of the A/H1N1 influenza pandemic.28 Furthermore, an increase in the incidence of narcolepsy was observed in subjects vaccinated with the H1N1 Pandemrix® vaccine.29–33 These observations led to speculate that the autoimmune destruction of the hypocretin neurons could be triggered by molecular mimicry with H1N1 flu antigens. Indeed, a study reported the presence of hypocretin-specific CD4+ T-cells cross-reactive to H1N1 hemagglutinin (HA) protein in narcolepsy patients,34 although this finding was not replicated.35
Indeed, HLA class II molecules, such as DQB1*06:02, are responsible for the presentation of antigenic peptides to CD4+ T-cells, possibly implying a prominent role of T-cells in the pathogenesis of narcolepsy. Subsequently, the discovery that polymorphisms in other genes involved in the immune response, such as the T-cell receptor alpha (TCRalpha)36 and the purinergic receptor subtype P2RY11 (P2RY11)37 among others,38–40 are associated with an increased risk of developing the disease, further implicated the T-cells in its pathogenesis. Indeed, recently an increased hypocretin-specific CD4+ T-cell response was found in blood samples from 19 NT1 patients compared to controls.41 However, unexpectedly, the majority of these cells were mainly HLA-DR- and not HLA-DQ6- restricted.41 Another study showed the presence, in NT1 patients, of DQ0602-restricted CD4+ T-cells cross-reactive to hypocretin and the HA protein of the pandemic 2009/2010 A/H1N1 influenza virus.34 Higher frequency of hypocretin responsive CD4+ and CD8+ T-cells was also observed in NT1 children.42 However, it remains unclear if43 and how CD4+ T-cells are involved in the destruction of the hypocretin neurons. Indeed, one critical observation is that neuronal cells do not express HLA class II molecules but only class I, which are recognized by CD8+ and not CD4+ T lymphocytes. Nevertheless, a role of CD8+ T-cells is supported by the association with several HLA class I alleles44,45 and by the finding, in nine NT1 patients, of mutations in P2RY11, encoding for a receptor highly expressed in cytotoxic CD8+ T lymphocytes.46 The potential ability of CD8+ T-cells to destroy the hypocretinergic neurons has been further supported by the serendipitous pathological observation of extensive CD8+ T-cell infiltrates and gliosis in the hypothalamus of a patient with NT1 secondary to Ma2 antibody-mediated encephalitis.47 A similar proof of concept was demonstrated in a transgenic mice animal model where cytotoxic CD8+, but not CD4+, HA-reactive T-cells were able to destroy hypocretin neurons expressing HA as a neo-antigen.48 Interestingly, a higher frequency of autoreactive CD8+ T-cells was recently documented in NT1 patients’ blood samples compared to controls.49 Moreover, CD8+ T-cells were identified in the CSF of patient with recent onset NT2 who later on progressed into NT1 with hypocretin deficiency.41 These observations suggest a direct involvement of CD8+ T-cells in the destruction of hypocretin neurons. Finally, it has recently been observed that NT1 patients display effector CD4+ T-cells with an unconventional profile which might have cytotoxic activity.50
Although these evidence point to a T-cell-mediated process associated with narcolepsy, these findings are mainly based on blood sample studies, and the primary role of these cells is not clear yet.
Since B cells are usually involved in CD4+ T-cell-mediated responses,51 several studies investigated the presence of neuronal autoantibodies. Screening for antibodies against specific antigens including the hypocretin precursor-peptide,52 hypocretin 1 and 2 and their receptors (HCRTR1 and 2)53 and other neuronal antigens in narcoleptic patients54–57 produced negative or indecisive results. Antibodies directed against Tribbles homologue 2 (TRIB2) were detected in 14% of narcoleptic patients but also in a small percentage (5%) of control sera,58 a finding replicated also in other cohorts59,60 but not in post-H1N1 cases.61 TRIB2 is highly expressed in hypocretinergic neurons, but also in other cells types; moreover, it is an intracellular antigen, and as such is unlikely to play a primary role in the destruction of hypocretin-producing cells. Subsequently, HCRTR2 antibodies were found in 85% of post-Pandemrix® narcolepsy cases as well as in controls (35%),62 but these findings have not been reproduced in idiopathic cases.63,64 Several studies showed the presence of antibodies directed against different neuronal targets in NT1 patients, but usually only in a small percentage of cases,65 suggesting that the humoral response is not primarily relevant in the pathogenesis of the disease.
Therefore, despite significant progresses, most evidence supporting the autoimmune etiology of narcolepsy is still circumstantial. According to Witebsky’s postulate,23 definitive proof such as the passive transfer of the disease to healthy individuals by autoreactive T-cells and/or autoantibodies or the active immunization with autoantigen able to induce the disease in animal models, are still missing.22
Immunotherapies
Soon after the autoimmune etiology of narcolepsy was hypothesized, the first attempts to arrest the pathogenic process using immunotherapy were made. These early attempts were based on the same approaches used for the treatment of classical neurological autoimmune disorders, and included corticosteroids, plasmapheresis (PLEX) and intravenous immunoglobulins (IVIG). These therapies indeed exert their action at multiple levels and could possibly be effective independently from the primary involvement of a humoral or cellular immune response. More recently, therapies specifically targeting B- and T- cells were attempted in limited cases, as discussed below.
Immunotherapies with Pleiotropic Effects
Corticosteroids exert a broad range of effects on the immune system, from inflammatory cytokines synthesis inhibition to impairment of function and survival of multiple types of immune cells, including neutrophils, monocytes, macrophages and B and T lymphocytes.66 Because of these wide effects, corticosteroids are widely employed to treat a variety of neurological inflammatory and autoimmune disorders and were therefore the first immunomodulatory treatment used in NT1 (Table 1). An 8-year-old boy was treated two months after the acute onset of NT1 with prednisone (1 mg/kg/day) for 3 weeks. However, no clinical improvement or modification of sleep parameters was observed.67 Similar negative results were reported in a 29-year-old woman who received intravenous methylprednisolone (IVMP) for a transverse myelitis of possible autoimmune etiology nine years after the onset of NT1.68 Conversely, Coelho et al69 described two men with a longstanding NT1 history, who reported the disappearance of EDS, and in one case also of cataplexy, upon prednisone treatment (40 mg/day) for other inflammatory conditions (inflammatory intestinal disease and asthma). However, these cases were not corroborated by CSF hypocretin-1 levels measurements nor the improvement was confirmed by repeated sleep studies after treatment and, in consideration of the long interval between disease onset and treatment, it is likely that the arousing effects of the steroids played a major role in the control of EDS.
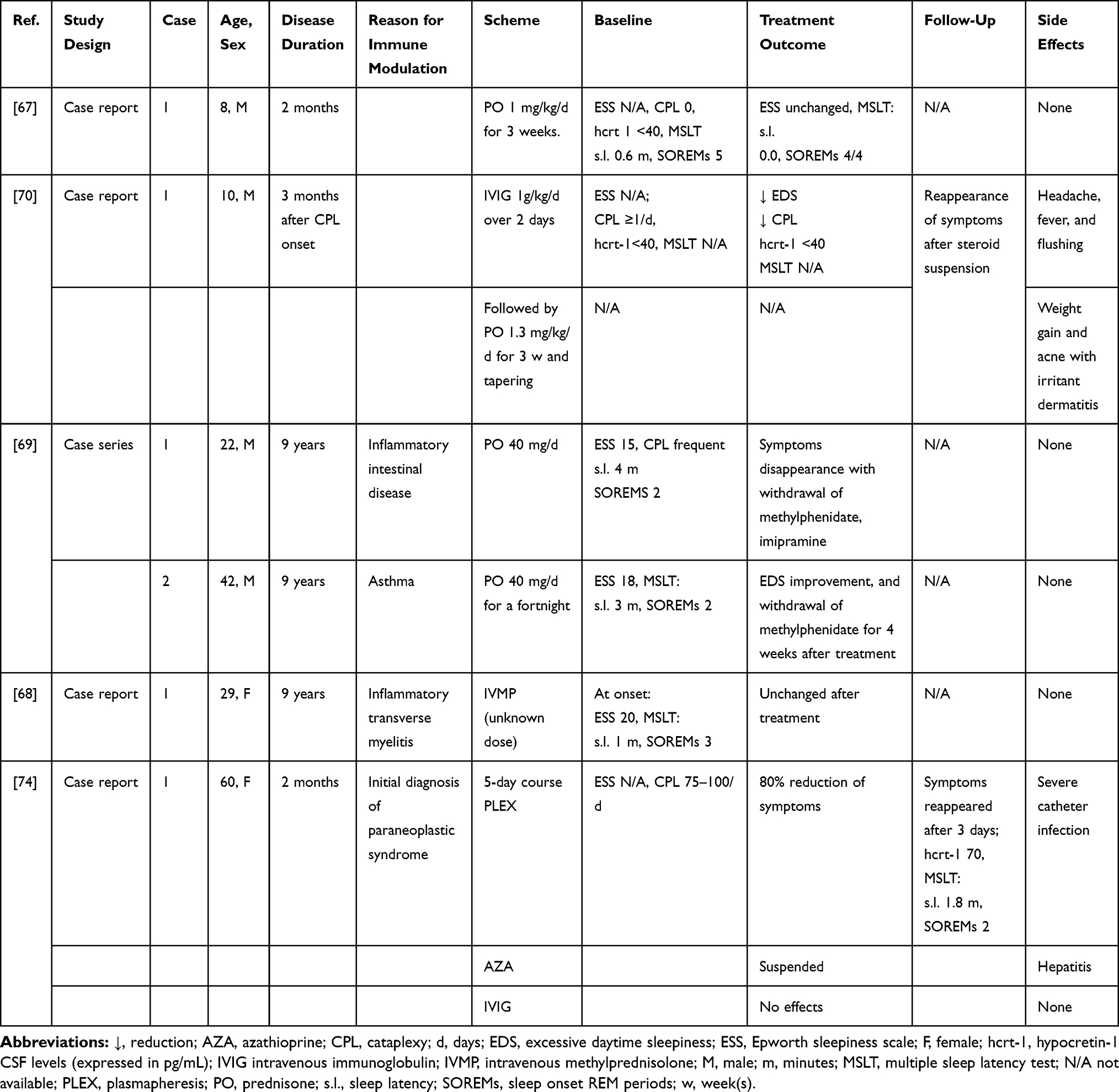 |
Table 1 Summary of NT1 Cases Treated Mainly with Steroids or PLEX |
In other cases, corticosteroids were used as add-on therapy. However, overall results were negative. A 10-year-old child with a 3-month NT1 history was initially treated with IVIG infusion (1g/kg/day for 2 days) but later switched on prednisolone (1.3 mg kg/day) therapy for 3 weeks, due to side effects. Both EDS and cataplexy improved during steroid therapy but reappeared following treatment tapering. Despite the reported symptoms improvement, no changes of CSF hypocretin-1 levels were detected.70 Similarly, two children, one with post-Pandemrix and one with sporadic NT1, respectively, received IVIG (1 g/kg/day for 2 days), followed by IVMP (20 mg/kg/day for 4 days) infusions repeated 3 times at monthly intervals.71 In the first child, a marked improvement of EDS and cataplexy was observed, but symptoms gradually reemerged within 1–2 weeks from treatment, although two follow-up MSLTs showed normal sleep latencies. In the second child, only a transient amelioration of sleepiness and cataplexy was noted. In both cases, hypocretin levels remained low, and indeed dropped despite treatment in patient 1. However, the improvement of cataplexy that occurred during IVMP treatment suggests that steroids could exert an immunomodulating effect, explaining the improvement of cataplexy in these cases71 and in one of the previously reported cases.69 Nevertheless, corticosteroids have several effects on the central nervous system,72 therefore, although unlikely, a direct anticataleptic activity, ie, through their action on noradrenergic and serotoninergic neurotransmission, cannot be excluded.73
PLEX is a very well-established treatment for antibody-mediated disorders. However, only a transitory benefit was observed in the single NT1 case treated so far with this procedure.74 Indeed, despite an early treatment, within 2 months after onset, and an initial amelioration, the symptoms reemerged after few days. The patient was switched on IVIG therapy, but no further improvement was noticed (Table 1). The transient benefit observed with PLEX could be related to the removal of antibodies or other molecules (ie, cytokines) by this procedure, whilst its short duration as well as the lack of response to IVIG, suggest a placebo effect, or a different pathogenic mechanism, as discussed by the same authors. Indeed, the lack of a sustained effect of PLEX is in line with the lack of evidence supporting a central role of antibodies in the pathogenesis of narcolepsy (see above).
IVIG have been more extensively employed in NT1, although mostly in single cases and small case series (without any randomized controlled trial approach to date) possibly because the first few observations seemed to provide positive results. A summary of the results of these studies is given in Table 2 and Figure 1. Dauvilliers et al75 treated with IVIG four typical NT1 patients with low CSF hypocretin-1. The three cases treated close to onset showed a reduction of the frequency and severity of cataplexy, whereas an improvement of the mean sleep latency on the maintenance of wakefulness test (MWT) was observed in the patient with a 9-year disease history. These positive effects continued over time.76 ESS scores improved during IVIG treatment in all cases. However, the concentration of hypocretin-1 in the CSF remained unchanged in two of the three available cases, whereas in one patient a slight increase was detected. This study pinpointed the importance of an early intervention, before the complete loss of hypocretinergic neurons, in ensuring a good outcome. This hypothesis seemed confirmed by a case where IVIG treatment, started 15 days after NT1 onset, led to cataplexy frequency reduction and CSF hypocretin-1 levels normalization. However, treatment discontinuation was followed by a progressive reoccurrence of NT1 symptoms after 4 months.77 In three other cases treated within 1–4 months from symptoms’ onset mixed effects were observed, with some improvement of EDS, and in two cases also of cataplexy, although no significant changes of polysomnographic parameters were documented.78 A 16-year-old girl with NT1 and severe bizarre hallucinations, with an inflammatory CSF (positive oligoclonal bands and pleocytosis) and undetectable hcrt-1 levels at the baseline examination, was treated with IVIG and showed only a transient improvement of the hallucinations. Interestingly, the CSF pleocytosis gradually disappeared after treatment, but CFS hcrt-1 levels remained unchanged.79 Among 4 NT1 children with undetectable CSF hypocretin-1, treated with IVIG for 6 months within 1 year from onset, only one case showed a significant reduction of EDS and cataplexy frequency whereas in the others no persistent clinical changes were noted.80 The short-lived benefit induced by therapy was confirmed by three further pediatric cases, treated close to onset81,82 and in 4 idiopathic adult cases with various disease durations.83 The often-transient nature of the benefit obtained by treatment suggested a possible placebo effect, as indeed demonstrated in a double-blind placebo-controlled single-case trial. A woman with a 7-year history of NT1 received alternatively IVIG or placebo and reported an improvement of her cataplexy whilst under either treatment.84 A larger longitudinal non-randomized, retrospective study including a pediatric NT1 population, evaluated the effects of IVIG in 22 patients compared to 30 controls who received standard therapy.85 The study failed to show an effect of the IVIG treatment, although among patients with more severe symptoms, those receiving IVIG achieved remission earlier than controls. On the other hand, an improvement of symptoms in the treatment group was observed already before IVIG administration. Shorter disease duration did not correlate with response to treatment. The results of this study however cannot be considered as conclusive since the non-randomized study design could have introduced a selection bias. Indeed, baseline symptoms scores were higher in patients treated with IVIG compared to controls.85 This selection bias, together with the significant spontaneous amelioration of cataplexy and EDS documented in non-treated NT1 children underline the probable significant bias in the observations obtained without any randomized study design.86
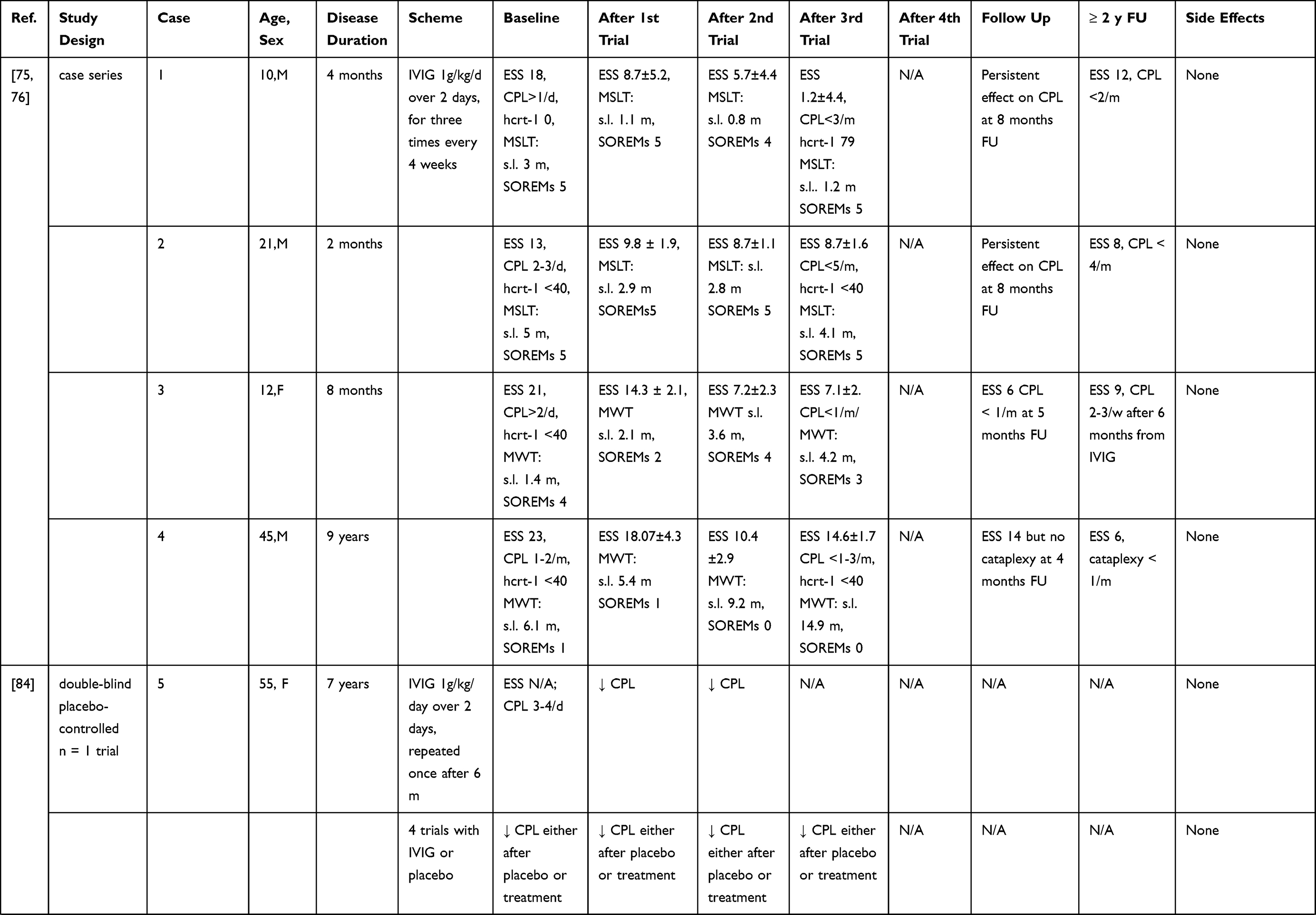 |
 |
 |
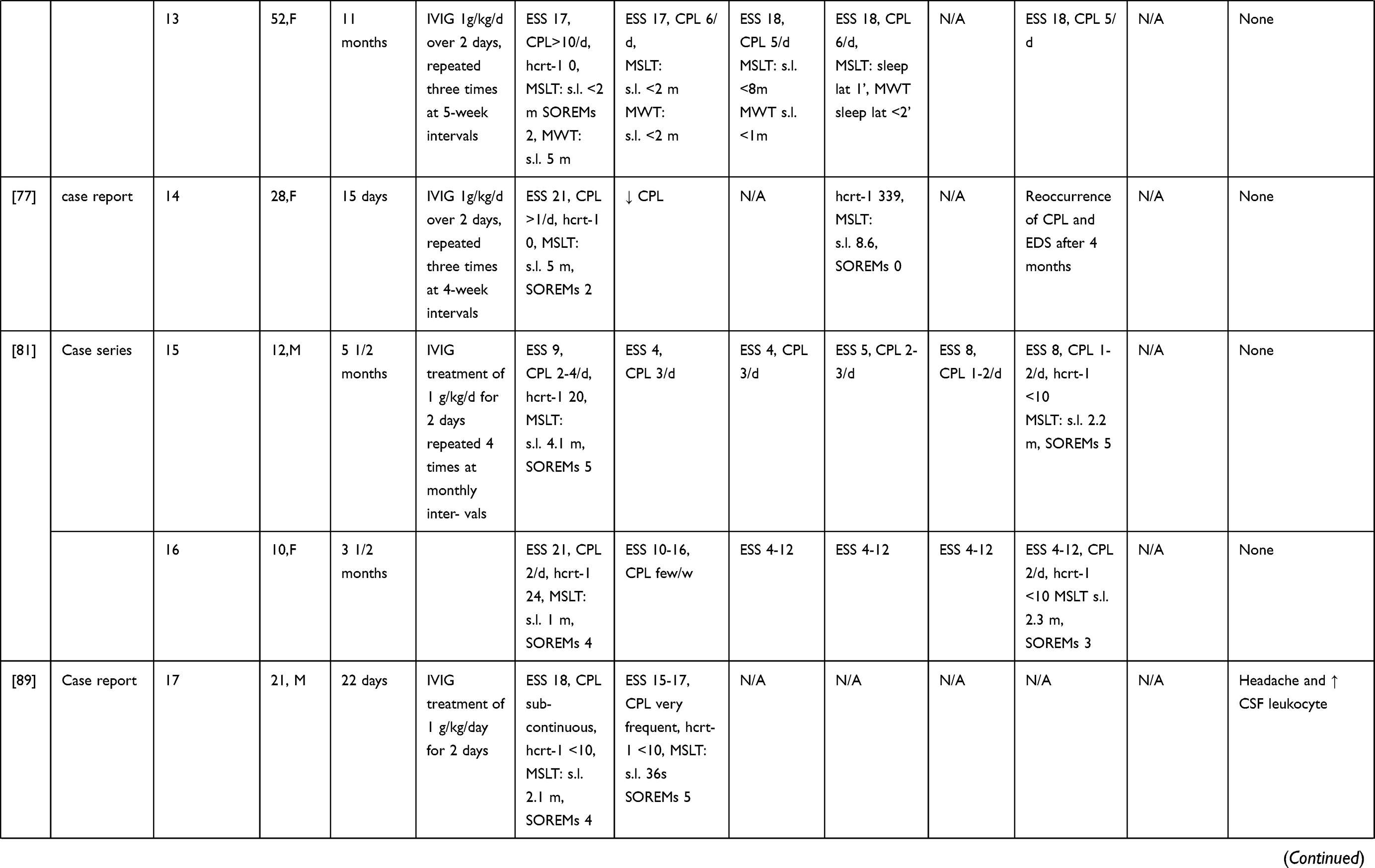 |
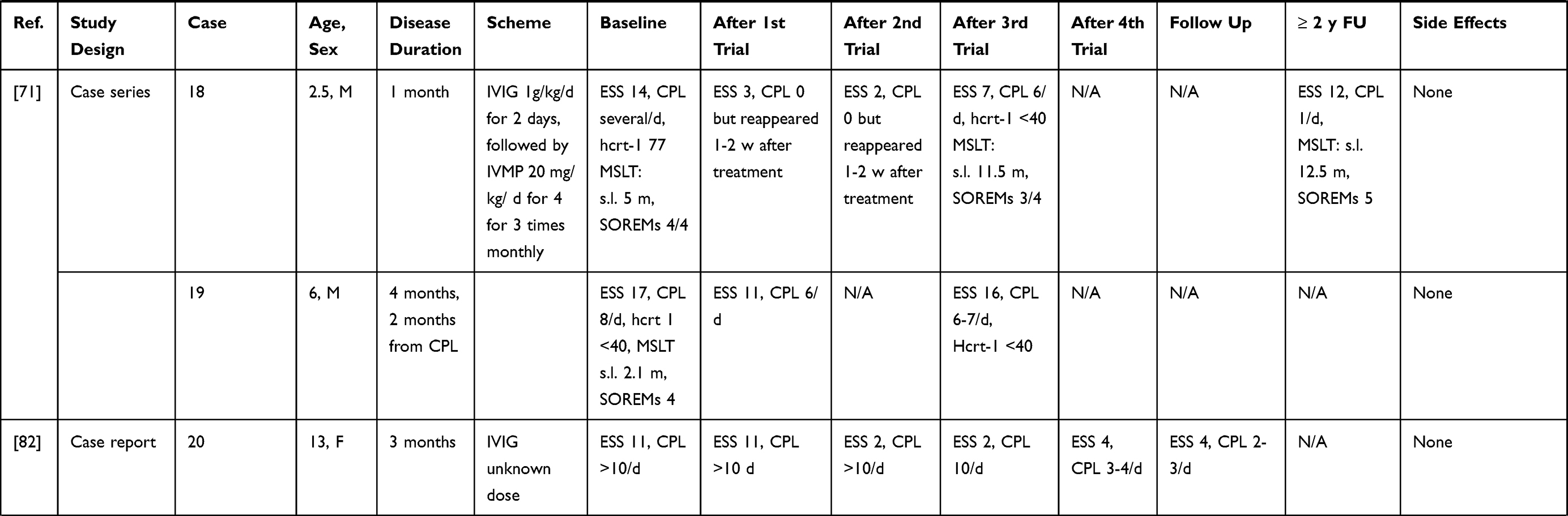 |
 |
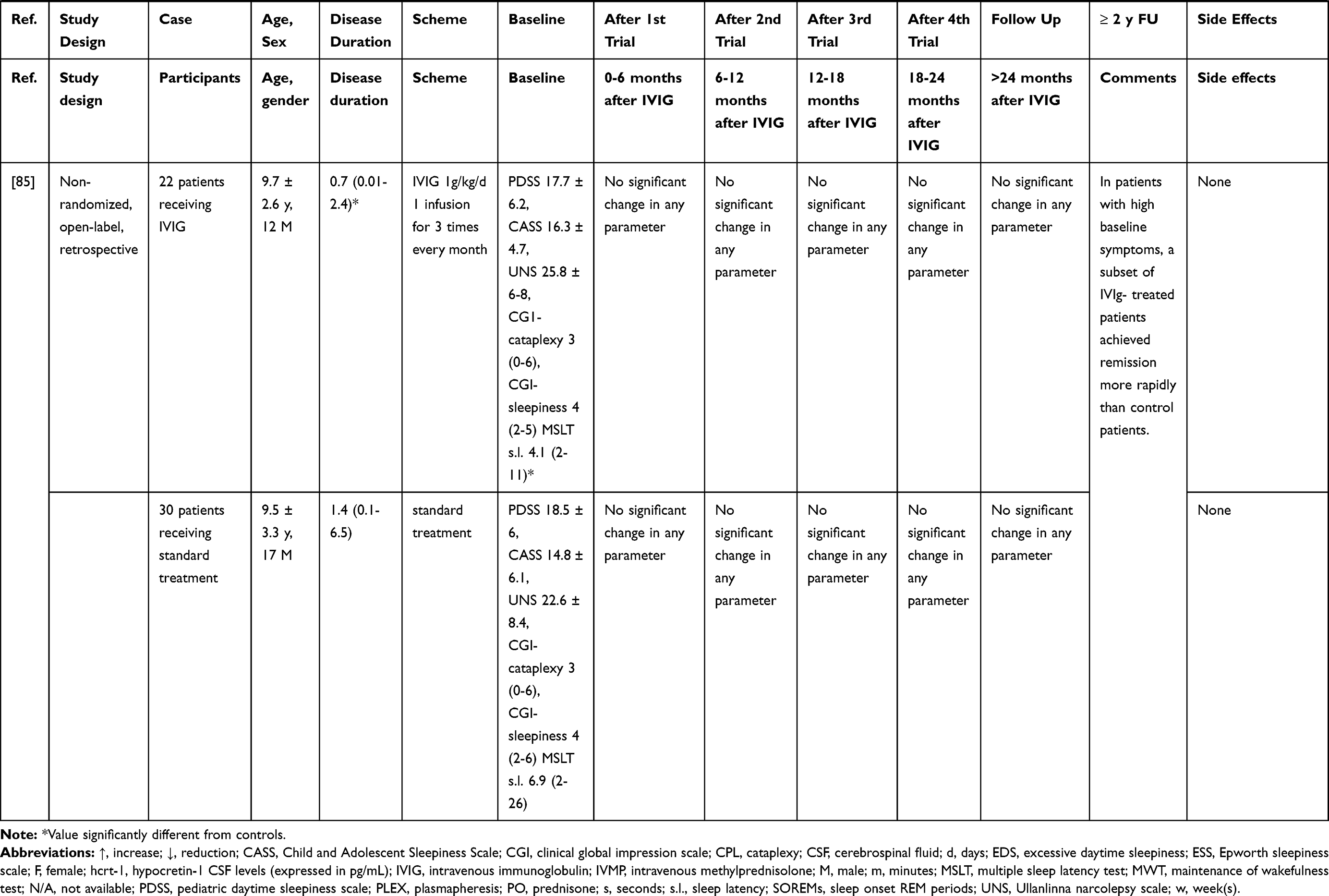 |
Table 2 Summary of NT1 Cases Treated Mainly with IVIG |
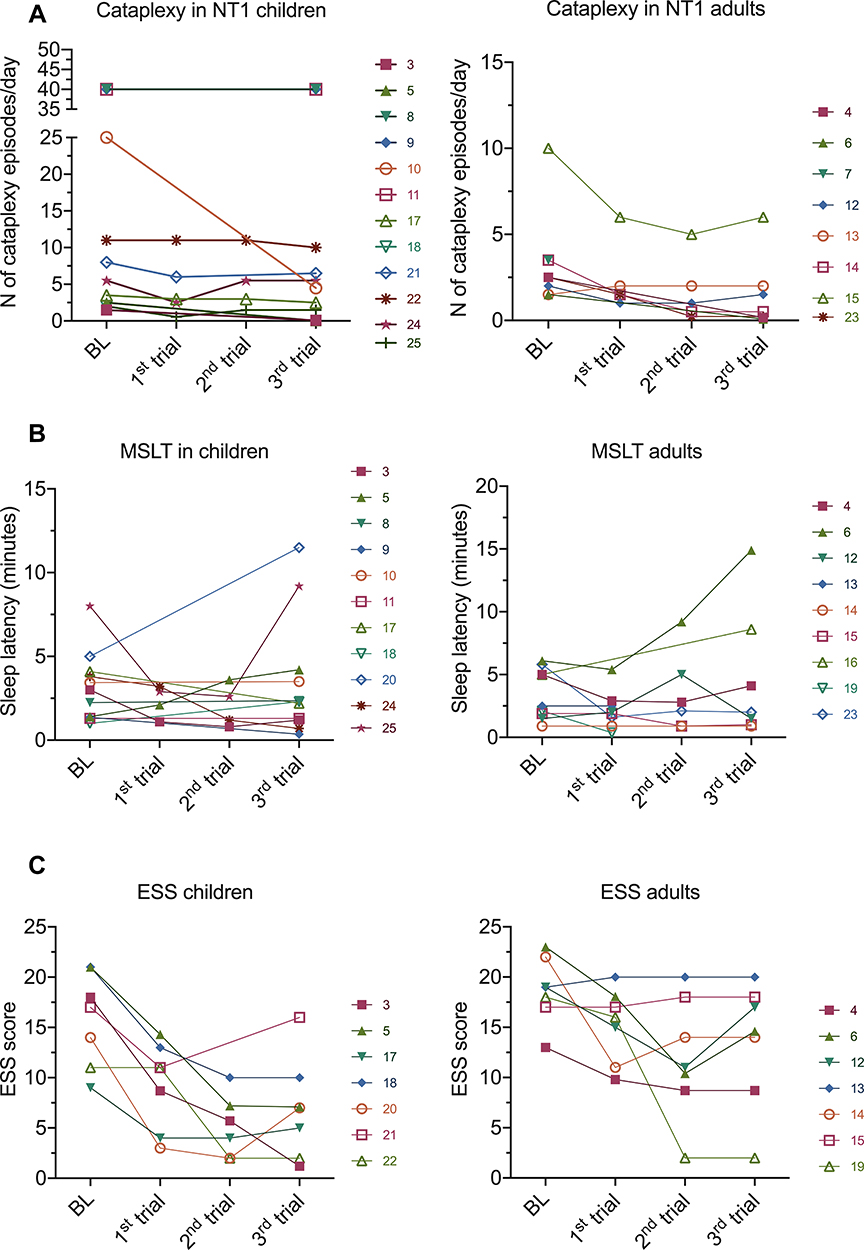 |
Figure 1 Outcomes of IVIG treatment in children and adults NT1 patients. The graphs show the individual patients outcome assessment values at baseline and after each IVIG infusion. Each line represents a single individual, numbered according to the case numbers in Table 2. No significant variations were observed in the majority of cases in respects to cataplexy frequency (A), sleep latency at the multiple sleep latency test (MSLT) (B) or Epworth sleepiness scale (ESS) scores (C) in children and adults. |
Indeed, notwithstanding prospective randomized studies, the utility of IVIG in the treatment of idiopathic NT1 remains to date unclear. In most cases, results have been disappointing or short-lasting. The benefits noted in some anecdotal reports could be related to both placebo effect and a spontaneous improvement of symptoms over time, as observed occasionally.85–88 On the other hand, it cannot be excluded that in rare instances immunotherapy could partially reverse a hypofunction of hypocretin cells preceding their actual destruction, explaining the disappearance of cataplexy in few individual cases.
Limited efficacy of IVIG therapy was also observed in post-vaccine narcolepsy, even when treatment was administered close to onset.89,90
Immunotherapies Targeting B and T Cells
A summary of the results of the studies employing these therapies is given in Tables 3 and 4.
 |
Table 3 Summary of NT1 Cases Treated with Rituximab |
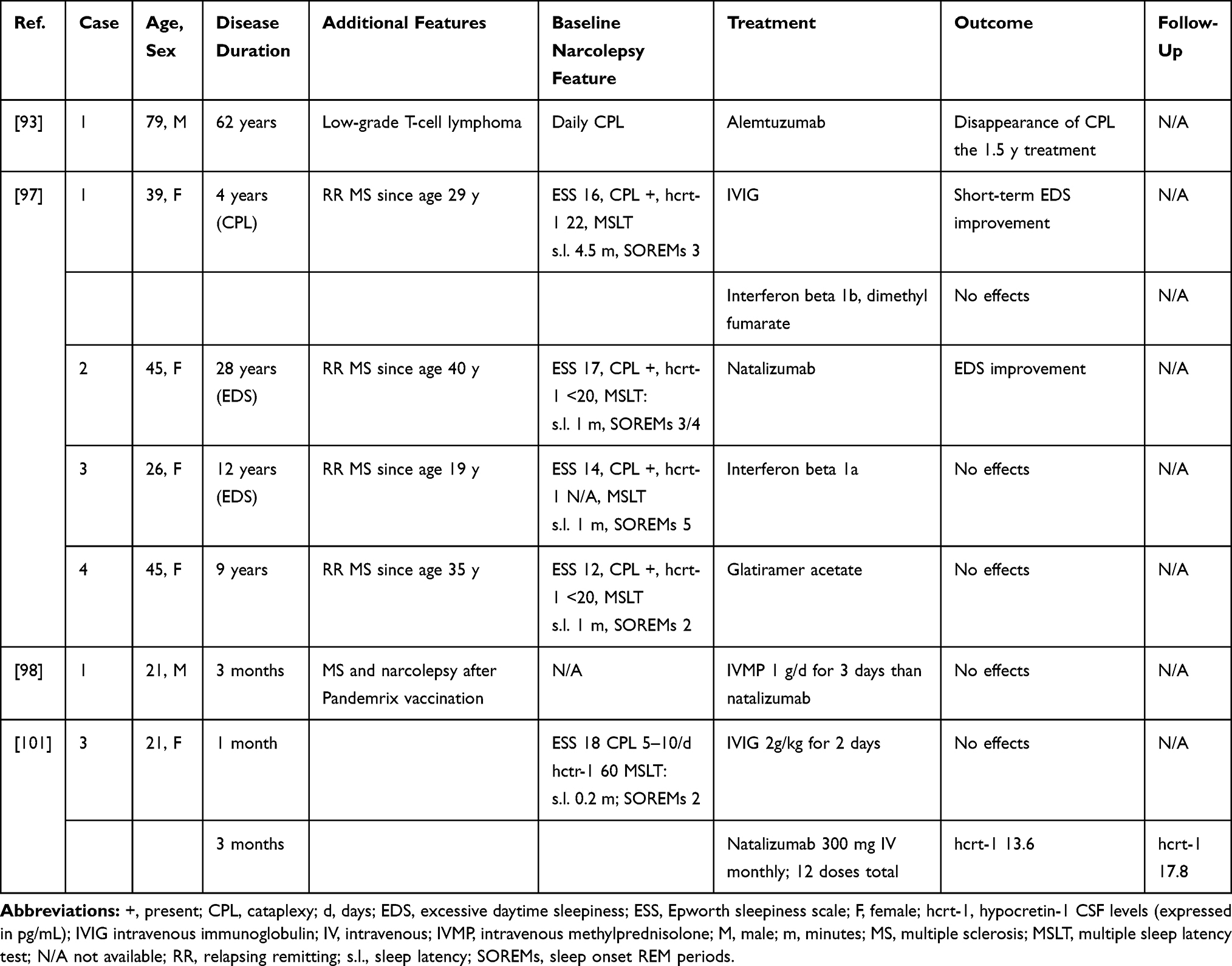 |
Table 4 Summary of NT1 Cases Treated with T Cells Targeting Drugs |
Rituximab is a monoclonal antibody targeting the CD20 antigen, mainly expressed on the surface of B cells. Upon binding to its target, this antibody mediates cells lysis with consequent B cells depletion. This therapy has proven effective in several antibody-mediated neurological disorders, including myasthenia gravis and autoimmune encephalitis. Recently, a 13-year-old boy, with post-Pandemrix NT1, treated with IVIG without benefit, received two rituximab infusions after the onset of a severe psychiatric disorder characterized by daytime hallucinations, behavioral problems and severe aggressivity requiring commitment to a psychiatric department.90 After treatment, an improvement of narcolepsy symptoms and behavioural and psychiatric disorders was observed. However, this beneficial effect lasted only for 2 months and subsequent infusions did not have any effect. Another patient, a 28-year-old man was treated with 5 courses of rituximab (1000 mg every 6 months) 5 months after the onset of NT1. Despite a subjective improvement of EDS 1 month after each infusion, no change of cataplexy frequency was observed. Moreover, longitudinal assessment of hcrt-1 CSF levels showed a progressive decrement from 100 to around 60 pg/mL.91 More recently, a 5-year-old boy, with NT1 and a suspected neuromyelitis optica spectrum disorder (NMOSD) was treated with a combination of IVIG, steroids and rituximab. A transitory improvement was noticed only after high dose IVMP treatment.92 The failure of B cells depletion in reverting the disease course supports the lack of a major role of humoral immunity in driving the pathological process and suggests that treatments targeting the T- cells could be more effective.
A 79-year-old man with a very long NT1 history was treated for a T-cell lymphoma with alemtuzumab,93 a humanized monoclonal antibody directed against the CD52 antigen causing CD4+ T-cells suppression.94 During treatment, the patient reported disappearance of cataplexy, but not of other disease manifestations. Interestingly, methotrexate, another immunosuppressive treatment acting on several immune cells, including T-cells,95 which was administered before alemtuzumab, did not impact narcolepsy symptoms. Why and how alemtuzumab could selectively affect cataplexy, particularly in a patient with a 58-year history of cataplexy, is unclear. It is possible that this drug exerts other effects beside T-cells suppression, such as neuroprotection and repair.96
Rarely, narcolepsy can develop together with multiple sclerosis (MS), either secondarily to hypothalamic demyelinating lesions or as a concomitant disorder.97 In a small series of NT1 cases with concomitant MS, two patients reported EDS improvement upon treatment with IVIG and long-term steroid therapy, respectively.97 Among the five patients receiving disease-modifying MS drugs, no response was observed with glatiramer acetate (n=1) or beta-interferon (n=2) and, of the two patients treated with natalizumab, only one reported reduction of EDS.97,98 It is unclear if this improvement is related to a direct effect of natalizumab on the pathogenic mechanism underlying narcolepsy or to a more complex effect, since natalizumab treatment was shown also to improve fatigue and EDS in MS patients without narcolepsy.99,100 Natalizumab is a recombinant monoclonal antibody directed against the cell adhesion molecule alpha4-integrin expressed on the surface of human leukocytes. This treatment is expected to prevent cellular infiltration of the CNS and could be a promising treatment for NT1. Recently, a 21-year-old woman was treated with IVIG followed by natalizumab 3 months after NT1 onset. Despite treatment, no symptoms improvement was noted and, on the contrary, CSF hypocretin-1 levels dropped from 70 pg/mL to 17 pg/mL.101 Although disappointing, this result could be explained by the relatively delayed treatment, administered when most of the hypocretin secreting cells had already been lost.
Cyclophosphamide (CYC) is an alkylating agent with antineoplastic and immunosuppressive and immunomodulating effects. Its cytotoxic activity is mainly due to DNA cross-linkage leading to cell apoptosis.102 CYC induces B and T cells depletion therefore inhibiting both, humoral and cell-mediated immune response. However, it can also exert beneficial immunomodulatory effect by reducing the number of regulatory T-cells and inducing T-cells grow factors.103,104 CYC is highly effective in the treatment of several autoimmune conditions although is burden by several toxicities.105 To our knowledge, to date, no patients with NT1 treated with CYC have been reported. However, disappearance of hypersomnia was reported in a 36-year-old woman who developed NT2 in the context of a neurolupus and was treated with 4 monthly CYC and IVIM infusions.106 On the other hand, no benefit was observed in four patients with paraneoplastic NT1.107–110
Side Effects of Immunotherapy
Corticosteroids and PLEX have been largely used in clinical practice to treat autoimmune disorders and adverse events are well known. In the few NT1 patients who underwent these treatments, side effects included acne and dermatitis70 and a severe catheter infection.74 Although IVIG treatment is considered safe, side effects, even severe, can occur and were indeed observed also in NT1 patients. In adult cases, headache, sometimes associated with stiff neck101 or rise in CSF leukocytes,89 was the most common complaint, whereas one patient presented a not better-defined allergic reaction.83 In children, side effects included infectious episodes (a flu-like syndrome and viral gastroenteritis),78 skin reactions with urticaria and petechiae,80 hypotension and nausea,90 headache, fever, and flushing70 requiring therapy withdrawal.
Although until now no serious adverse events occurred in NT1 patients treated with monoclonal antibodies, these drugs have potentially lethal side effects, ie, severe infusion-related reactions, secondary autoimmunity94 and life-threatening infections secondary to immunosuppression, in particular progressive multifocal leukoencephalopathy (PML). The risk of PML is higher in patients treated with drugs reducing T-cells trafficking to the brain such as natalizumab, but rare cases have been also reported in association with rituximab.111 Therefore, clinicians should keep in mind this risk and predispose adequate monitoring. Scammel and colleagues101 proposed a treatment trial with natalizumab of a maximum of 1 or 2 years, to reduce the risk of PML in NT1 patients.
Open Issues and Future Directions
Most of the information on the effects of immunotherapy in narcolepsy derive from uncontrolled case studies, with small sample sizes, different treatment schemes and highly heterogeneous outcome measures. Therefore, current data is not sufficient to support the use of immunotherapy for narcolepsy and randomized controlled clinical trials are needed to provide substantial evidence and avoid bias related to placebo effect and to spontaneous disease improvements. To date, most of the attempts to reverse NT1 symptoms have been disappointing, and this could be related to several open issues that need to be addressed and that are summarized below.
Since the autoimmune basis of narcolepsy remains unproven, and our understanding of the immune process involved is still limited (ie, associated or pathogenetic), it is difficult to provide a definite answer to this question. However, mounting evidence underpins the primary role of T-cells in NT1 disease pathogenesis. Many of the studies on immunotherapy have been focused on treatments acting mainly on the antibody-mediated immune response, possibly explaining the lack of a meaningful effect. T-cells targeting drugs have been only rarely employed; therefore, agents such as natalizumab or alemtuzumab could be promising treatments for future clinical trials.
This is an apparently easy question with a seemingly easy answer, the sooner, the better. Indeed, since immunotherapy might prevent the loss of the hypocretinergic neurons, it should be given before this process reach completion. On this assumption, many of the previous studies focused on treating patients close to disease onset. However, cataplexy manifests when most, about 80%, of the hypocretin-secreting neurons are already lost,112 therefore it could be possibly more effective to treat patients who have not yet developed cataplexy. Although the natural history of the hypocretin cells death is unclear, most NT1 patients refer experiencing EDS months or years before the appearance of cataplexy13 implying a progression over time, as documented in few cases by hcrt-1 CSF assessment.7–10,41 These cases show that at least in few cases, narcolepsy symptoms evolve over time along with changes of CSF markers suggesting that in HLA DQB1*06:02 positive subjects with EDS and other narcolepsy features CSF markers should be monitored over time. This could potentially offer a window of opportunity to intervene with immunotherapies in early stages in order to try preventing or reducing the hypocretinergic neuronal loss. Interestingly, Latorre et al41 observed the presence of reactive CD8+ T-cell clones in the CSF of a recently diagnosed NT2 patient who later evolved into NT1 suggesting that these cells could be a potentially reliable early marker. The identification of early markers of progression could be crucial to identify those patients who could benefit from immunotherapy before the complete destruction of hypocretinergic neurons leading to NT1. On the other hand, CSF hypocretin loss can progress quickly and shortly after symptom onset,113 even before the appearance of sleep-onset rapid eye movement periods, therefore prompt diagnosis and CSF examination are crucial for the identification of cases who can benefit from immunotherapy.
Many of the cases reported so far used heterogeneous measurements, including subjective sleepiness scales, CSF hypocretin-1 levels, multiple sleep latency test (MSLT), MWT, alone or in various combinations, or patients’ self-reports to monitor the response to therapy. In certain cases, patients subjectively experienced an improvement of EDS or cataplexy, but the use of objective sleep parameters, often failed to pair these subjective evaluations, showing the intrinsic limitations of current methodology for the assessment of treatment efficacy, as well as their poor correlation with clinically relevant outcome measures.21 For some narcolepsy manifestations including cataplexy or sleep paralysis, there are not appropriate assessment tools and only recently an overall narcolepsy severity scale has been proposed.114 Most of the previous studies on IVIG efficacy involved children and employed the same tools used to evaluate adults, with only few studies adopting age-appropriate scales.80,85 However, children manifest significantly different NT1 symptoms compared to adults thus calling for specific evaluation approaches. Recently, Wang et al115 proposed a subjective sleepiness scale and a cataplexy diary for pediatric narcolepsy, and, although not yet validated, they have been already applied in a clinical trial.116 There is an urgent need of new standardized assessment tolls of disease severity aimed at better following the disease course and documenting treatments’ effects.
Conclusion
To date only symptomatic treatments are available for narcolepsy, with new drugs recently showing promising results;4,19 however, chronic pharmacological treatments met with frequent side effects and may not sufficiently impact on disease burden.21 No disease-modifying cure is available, calling for future research on treatment strategies as well as on diagnostic approaches able to identify patients who will develop NT1 among those complaining only EDS. NT1 is considered an immune-mediated disease, nevertheless the absence of definitive proofs represents a limit to design targeted clinical trials on immunotherapy. To date, indeed, only case series and case reports are available. However, although not yet conclusive, the evidence gathered so far suggest that is the time for randomized, double-blind, placebo-controlled trials that would contribute to answer the question whether immunotherapy is useful in narcolepsy.
Disclosure
Maria Pia Giannoccaro and Fabio Pizza reports no conflicts of interest in this work. Rocco Liguori reports personal fees from Argenx, Biogen, Sanofi-Genzyme, Argon Healthcare s.r.l., Amicus Therapeutics s.r.l. and Alfasigma for Advisory Board consultancy and Lecture fees from Dynamicom Education, SIMG Service, Adnkronos Salute Unipersonale s.r.l. and DOC Congress s.r.l., outside the submitted work. Giuseppe Plazzi participated in advisory board for UCB Pharma, Idorsia, Jazz pharmaceuticals and Bioprojet.
References
1. Medicine AA of S. The International Classification of Sleep Disorders, Third Edition (ICSD-3); 2014. (Medicine AA of S, editor.)
2. Silber MH, Krahn LE, Olson EJ, Pankratz VS. The epidemiology of narcolepsy in Olmsted County, Minnesota: a Population-Based Study. Sleep. 2002;25(2):197–202. doi:10.1093/sleep/25.2.197
3. Dye TJ, Gurbani N, Simakajornboon N. Epidemiology and pathophysiology of childhood narcolepsy. Paediatr Respir Rev. 2018;25:14–18. doi:10.1016/j.prrv.2016.12.005
4. Szabo ST, Thorpy MJ, Mayer G, Peever JH, Kilduff TS. Neurobiological and immunogenetic aspects of narcolepsy: implications for pharmacotherapy. Sleep Med Rev. 2018;43:23–36. doi:10.1016/j.smrv.2018.09.006
5. Thannickal TC, Nienhuis R, Siegel JM. Localized loss of hypocretin (Orexin) cells in narcolepsy without cataplexy. Sleep. 2009;32(8):993–998. doi:10.1093/sleep/32.8.993
6. Tabuchi S, Tsunematsu T, Black SW, et al. Conditional ablation of orexin/hypocretin neurons: a New Mouse Model for the Study of Narcolepsy and Orexin System Function. J Neurosci. 2014;34(19):6495–6509. doi:10.1523/jneurosci.0073-14.2014
7. Andlauer O, Moore H, Hong S-C, et al. Predictors of hypocretin (Orexin) deficiency in narcolepsy without cataplexy. Sleep. 2012;35(9):1247–1255. doi:10.5665/sleep.2080
8. Pizza F, Vandi S, Liguori R, et al. Primary progressive narcolepsy type 1: the other side of the coin. Neurology. 2014;83(23):2189–2190. doi:10.1212/wnl.0000000000001051
9. Lopez R, Barateau L, Evangelista E, et al. Temporal changes in the cerebrospinal fluid level of hypocretin-1 and histamine in narcolepsy. Sleep. 2017;40(1). doi:10.1093/sleep/zsw010
10. Bonakis A, Howard RS, Ebrahim IO, Merritt S, Williams A. REM sleep behaviour disorder (RBD) and its associations in young patients. Sleep Med. 2009;10(6):641–645. doi:10.1016/j.sleep.2008.07.008
11. Hublin C, Partinen M, Kaprio J, Koskenvuo M, Guilleminault C. Epidemiology of narcolepsy. Sleep. 1994;17(suppl_8):S7–S12. doi:10.1093/sleep/17.suppl_8.s7
12. Ohayon MM, Priest RG, Zulley J, Smirne S, Paiva T. Prevalence of narcolepsy symptomatology and diagnosis in the European general population. Neurology. 2002;58(12):1826–1833. doi:10.1212/wnl.58.12.1826
13. Luca G, Haba-Rubio J, Dauvilliers Y, et al. Clinical, polysomnographic and genome-wide association analyses of narcolepsy with cataplexy: a European Narcolepsy Network study. J Sleep Res. 2013;22(5):482–495. doi:10.1111/jsr.12044
14. Thorpy MJ, Krieger AC. Delayed diagnosis of narcolepsy: characterization and impact. Sleep Med. 2014;15(5):502–507. doi:10.1016/j.sleep.2014.01.015
15. Dodel R, Peter H, Walbert T, et al. The socioeconomic impact of narcolepsy. Sleep. 2004;27(6):1123–1128. doi:10.1093/sleep/27.6.1123
16. Jennum P, Ibsen R, Petersen ER, Knudsen S, Kjellberg J. Health, social, and economic consequences of narcolepsy: a controlled national study evaluating the societal effect on patients and their partners. Sleep Med. 2012;13(8):1086–1093. doi:10.1016/j.sleep.2012.06.006
17. Black J, Reaven NL, Funk SE, et al. The Burden of Narcolepsy Disease (BOND) study: health-care utilization and cost findings. Sleep Med. 2014;15(5):522–529. doi:10.1016/j.sleep.2014.02.001
18. Jennum P, Ibsen R, Knudsen S, Kjellberg J. Comorbidity and mortality of narcolepsy: a Controlled Retro- and Prospective National Study. Sleep. 2013;36(6):835–840. doi:10.5665/sleep.2706
19. Abad VC, Guilleminault C. New developments in the management of narcolepsy. Nat Sci Sleep. 2017;9:39–57. doi:10.2147/nss.s103467
20. Thorpy MJ, Dauvilliers Y. Clinical and practical considerations in the pharmacologic management of narcolepsy. Sleep Med. 2015;16(1):9–18. doi:10.1016/j.sleep.2014.10.002
21. Maski K, Steinhart E, Williams D, et al. Listening to the patient voice in narcolepsy: diagnostic delay, disease burden, and treatment efficacy. J Clin Sleep Med. 2017;13(03):419–425. doi:10.5664/jcsm.6494
22. Kornum BR. Narcolepsy type 1: what have we learned from immunology? Sleep. 2020;43(10). doi:10.1093/sleep/zsaa055
23. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today. 1993;14(9):426–430. doi:10.1016/0167-5699(93)90244-f
24. Han F, Lin L, Schormair B, et al. HLA DQB1*06:02 negative narcolepsy with hypocretin/orexin deficiency. Sleep. 2014;37(10):1601–1608. doi:10.5665/sleep.4066
25. Arango M-T, Kivity S, Shoenfeld Y. Is narcolepsy a classical autoimmune disease? Pharmacol Res. 2015;92:6–12. doi:10.1016/j.phrs.2014.10.005
26. Koepsell TD, Longstreth WT, Ton TGN. Medical exposures in youth and the frequency of narcolepsy with cataplexy: a population-based case–control study in genetically predisposed people. J Sleep Res. 2010;19(1–Part–I):80–86. doi:10.1111/j.1365-2869.2009.00756.x
27. Aran A, Lin L, Nevsimalova S, et al. Elevated anti-streptococcal antibodies in patients with recent narcolepsy onset. Sleep. 2009;32(8):979–983. doi:10.1093/sleep/32.8.979
28. Han F, Lin L, Warby SC, et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in china. Ann Neurol. 2011;70(3):410–417. doi:10.1002/ana.22587
29. Partinen M, Saarenpää-Heikkilä O, Ilveskoski I, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS One. 2012;7(3):e33723. doi:10.1371/journal.pone.0033723
30. Szakács A, Darin N, Hallböök T. Increased childhood incidence of narcolepsy in western Sweden after H1N1 influenza vaccination. Neurology. 2013;80(14):1315–1321. doi:10.1212/wnl.0b013e31828ab26f
31. Wijnans L, Lecomte C, de Vries C, et al. The incidence of narcolepsy in Europe: before, during, and after the influenza A(H1N1)pdm09 pandemic and vaccination campaigns. Vaccine. 2013;31(8):1246–1254. doi:10.1016/j.vaccine.2012.12.015
32. Nohynek H, Jokinen J, Partinen M, et al. AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS One. 2012;7(3):e33536. doi:10.1371/journal.pone.0033536
33. Heier MS, Gautvik KM, Wannag E, et al. Incidence of narcolepsy in Norwegian children and adolescents after vaccination against H1N1 influenza A. Sleep Med. 2013;14(9):867–871. doi:10.1016/j.sleep.2013.03.020
34. Luo G, Ambati A, Lin L, et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc Natl Acad Sci. 2018;115(52):201818150. doi:10.1073/pnas.1818150116
35. Schinkelshoek MS, Fronczek R, Kooy-Winkelaar EMC, et al. H1N1 hemagglutinin-specific HLA-DQ6-restricted CD4+ T cells can be readily detected in narcolepsy type 1 patients and healthy controls. J Neuroimmunol. 2019;332:167–175. doi:10.1016/j.jneuroim.2019.04.009
36. Hallmayer J, Faraco J, Lin L, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet. 2009;41(6):708–711. doi:10.1038/ng.372
37. Kornum BR, Kawashima M, Faraco J, et al. Common variants in P2RY11 are associated with narcolepsy. Nat Genet. 2011;43(1):66–71. doi:10.1038/ng.734
38. Han F, Faraco J, Dong XS, et al. Genome wide analysis of narcolepsy in China implicates novel immune Loci and Reveals Changes in Association prior to versus after the 2009 H1N1 influenza pandemic. Plos Genet. 2013;9(10):e1003880. doi:10.1371/journal.pgen.1003880
39. Toyoda H, Miyagawa T, Koike A, et al. A polymorphism in CCR1/CCR3 is associated with narcolepsy. Brain Behav Immun. 2015;49:148–155. doi:10.1016/j.bbi.2015.05.003
40. Faraco J, Lin L, Kornum BR, et al. ImmunoChip Study implicates antigen presentation to T cells in narcolepsy. Plos Genet. 2013;9(2):e1003270. doi:10.1371/journal.pgen.1003270
41. Latorre D, Kallweit U, Armentani E, et al. T cells in patients with narcolepsy target self-antigens of hypocretin neurons. Nature. 2018;562(7725):63–68. doi:10.1038/s41586-018-0540-1
42. Cogswell AC, Maski K, Scammell TE, Tucker D, Orban ZS, Koralnik IJ. Children with Narcolepsy type 1 have increased T-cell responses to orexins. Ann Clin Transl Neurol. 2019;6(12):2566–2572. doi:10.1002/acn3.50908
43. Liblau RS. Put to sleep by immune cells. Nature. 2018;562(7725):46–48. doi:10.1038/d41586-018-06666-w
44. Tafti M, Lammers GJ, Dauvilliers Y, et al. Narcolepsy-Associated HLA class I alleles implicate cell-mediated cytotoxicity. Sleep. 2016;39(3):581–587. doi:10.5665/sleep.5532
45. Ollila HM, Ravel J-M, Han F, et al. HLA-DPB1 and HLA class I confer risk of and protection from narcolepsy. Am J Hum Genetics. 2015;96(1):136–146. doi:10.1016/j.ajhg.2014.12.010
46. Degn M, Dauvilliers Y, Dreisig K, et al. Rare missense mutations in P2RY11 in narcolepsy with cataplexy. Brain. 2017;140(6):1657–1668. doi:10.1093/brain/awx093
47. Dauvilliers Y, Bauer J, Rigau V, et al. Hypothalamic immunopathology in anti-Ma–associated diencephalitis with narcolepsy-cataplexy. JAMA Neurol. 2013;70(10):1305–1310. doi:10.1001/jamaneurol.2013.2831
48. Bernard-Valnet R, Yshii L, Quériault C, et al. CD8 T cell-mediated killing of orexinergic neurons induces a narcolepsy-like phenotype in mice. Proc Natl Acad Sci. 2016;113(39):10956–10961. doi:10.1073/pnas.1603325113
49. Pedersen NW, Holm A, Kristensen NP, et al. CD8+ T cells from patients with narcolepsy and healthy controls recognize hypocretin neuron-specific antigens. Nat Commun. 2019;10(1):837. doi:10.1038/s41467-019-08774-1
50. Jiang W, Birtley JR, Hung S-C, et al. In vivo clonal expansion and phenotypes of hypocretin-specific CD4+ T cells in narcolepsy patients and controls. Nat Commun. 2019;10(1):5247. doi:10.1038/s41467-019-13234-x
51. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol. 2010;10(4):236–247. doi:10.1038/nri2729
52. Black JL, Silber MH, Krahn LE, et al. Analysis of Hypocretin (Orexin) antibodies in patients with narcolepsy. Sleep. 2005;28(4):427–431. doi:10.1093/sleep/28.4.427
53. Tanaka S, Honda Y, Inoue Y, Honda M. Detection of autoantibodies against hypocretin, hcrtr1, and hcrtr2 in narcolepsy: anti-Hcrt system antibody in narcolepsy. Sleep. 2006;29(5):633–638. doi:10.1093/sleep/29.5.633
54. Knudsen S, Mikkelsen JD, Jennum P. Antibodies in narcolepsy–cataplexy patient serum bind to rat hypocretin neurons. Neuroreport. 2007;18(1):77–79. doi:10.1097/wnr.0b013e328010baad
55. Martínez-Rodríguez JE, Sabater L, Graus F, Iranzo A, Santamaria J. Evaluation of hypothalamic-specific autoimmunity in patients with narcolepsy. Sleep. 2007;30(1):27–28. doi:10.1093/sleep/30.1.27
56. Overeem S, Verschuuren JJ, Fronczek R, et al. Immunohistochemical screening for autoantibodies against lateral hypothalamic neurons in human narcolepsy. J Neuroimmunol. 2006;174(1–2):187–191. doi:10.1016/j.jneuroim.2006.02.010
57. Black JL, Krahn LE, Pankratz VS, Silber M. Search for neuron-specific and nonneuron-specific antibodies in narcoleptic patients with and without HLA DQB1*0602. Sleep. 2002;25(7):719–723. doi:10.1093/sleep/25.7.719
58. Cvetkovic-Lopes V, Bayer L, Dorsaz S, et al. Elevated tribbles homolog 2–specific antibody levels in narcolepsy patients. J Clin Invest. 2010;120(3):713–719. doi:10.1172/jci41366
59. Kawashima M, Lin L, Tanaka S, et al. Anti-tribbles Homolog 2 (TRIB2) autoantibodies in narcolepsy are associated with recent onset of cataplexy. Sleep. 2010;33(7):869–874. doi:10.1093/sleep/33.7.869
60. Toyoda H, Tanaka S, Miyagawa T, Honda Y, Tokunaga K, Honda M. Anti-tribbles Homolog 2 autoantibodies in Japanese patients with narcolepsy. Sleep. 2010;33(7):875–878. doi:10.1093/sleep/33.7.875
61. Lind A, Ramelius A, Olsson T, et al. A/H1N1 antibodies and TRIB2 autoantibodies in narcolepsy patients diagnosed in conjunction with the Pandemrix vaccination campaign in Sweden 2009–2010. J Autoimmun. 2014;50:99–106. doi:10.1016/j.jaut.2014.01.031
62. Ahmed SS, Volkmuth W, Duca J, et al. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci Transl Med. 2015;7(294):294ra105. doi:10.1126/scitranslmed.aab2354
63. Giannoccaro MP, Waters P, Pizza F, Liguori R, Plazzi G, Vincent A. Antibodies against Hypocretin receptor 2 are rare in narcolepsy. Sleep. 2016;40(2). doi:10.1093/sleep/zsw056
64. Luo G, Lin L, Jacob L, et al. Absence of anti-hypocretin receptor 2 autoantibodies in post pandemrix narcolepsy cases. PLoS One. 2017;12(12):e0187305. doi:10.1371/journal.pone.0187305
65. Giannoccaro MP, Pizza F, Jacobson L, Liguori R, Plazzi G, Vincent A. Neuronal surface antibodies are common in children with narcolepsy and active movement disorders. J Neurol Neurosurg Psychiatry. 2020:
66. Chatman WW. Glucocorticoid effects on the immune system. In: Orange JS, Marr KA, Feldweg AM, Bond S, editors. Uptodate; 2020. Uptodate (Accessed February 11, 2021).
67. Hecht M, Lin L, Kushida CA, et al. Report of a case of immunosuppression with prednisone in an 8-year-old boy with an acute onset of hypocretin-deficiency narcolepsy. Sleep. 2003;26(7):809–810. doi:10.1093/sleep/26.7.809
68. Peraita-Adrados R, Romero-Martínez J, Guzmán-de Villoria JA, Lillo-Triguero L, Martínez-Ginés ML. Comorbidity of narcolepsy with cataplexy and transverse myelitis: a common autoimmune background? A case report. Sleep Med. 2017;33:167–170. doi:10.1016/j.sleep.2017.02.014
69. Coelho FMS, Pradella-Hallinan M, Alves GR, Bittencourt LRA, Tufik S. Report of two narcoleptic patients with remission of hypersomnolence following use of prednisone. Arq Neuropsiquiatr. 2007;65(2A):336–337. doi:10.1590/s0004-282x2007000200028
70. Lecendreux M, Maret S, Bassetti C, Mouren M, Tafti M. Clinical efficacy of high-dose intravenous immunoglobulins near the onset of narcolepsy in a 10-year-old boy. J Sleep Res. 2003;12(4):347–348. doi:10.1046/j.1365-2869.2003.00380.x
71. Viste R, Soosai J, Vikin T, Thorsby PM, Nilsen KB, Knudsen S. Long-term improvement after combined immunomodulation in early post-H1N1 vaccination narcolepsy. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e389. doi:10.1212/nxi.0000000000000389
72. Fietta P, Fietta P, Delsante G. Central nervous system effects of natural and synthetic glucocorticoids. Psychiatry Clin Neurosci. 2009;63(5):613–622. doi:10.1111/j.1440-1819.2009.02005.x
73. Krugers HJ, Karst H, Joels M. Interactions between noradrenaline and corticosteroids in the brain: from electrical activity to cognitive performance. Front Cell Neurosci. 2012;6:15. doi:10.3389/fncel.2012.00015
74. Chen W, Black J, Call P, Mignot E. Late-onset narcolepsy presenting as rapidly progressing muscle weakness: response to plasmapheresis. Ann Neurol. 2005;58(3):489–490. doi:10.1002/ana.20603
75. Dauvilliers Y, Carlander B, Touchon J, Tafti M. Successful management of cataplexy with intravenous immunoglobulins at narcolepsy onset. Ann Neurol. 2004;56(6):905–908. doi:10.1002/ana.20339
76. Dauvilliers Y. Follow-up of four narcolepsy patients treated with intravenous immunoglobulins. Ann Neurol. 2006;60(1):153. doi:10.1002/ana.20892
77. Dauvilliers Y, Abril B, Mas E, Michel F, Tafti M. Normalization of hypocretin-1 in narcolepsy after intravenous immunoglobulin treatment. Neurology. 2009;73(16):1333–1334. doi:10.1212/wnl.0b013e3181bd1344
78. Ruppert E, Zagala H, Chambe J, et al. Intravenous immunoglobulin therapy administered early after narcolepsy type 1 onset in three patients evaluated by clinical and polysomnographic follow-up. Behav Neurol. 2018;2018:1–5. doi:10.1155/2018/1671072
79. Moreno-Estébanez A, Villabeitia IB, Guibert TE, et al. Positive oligoclonal bands and CSF pleocytosis in narcolepsy type 1: a case report supporting the immune-mediated hypothesis. J Neuroimmunol. 2019;339:577111. doi:10.1016/j.jneuroim.2019.577111
80. Plazzi G, Poli F, Franceschini C, et al. Intravenous high-dose immunoglobulin treatment in recent onset childhood narcolepsy with cataplexy. J Neurol. 2008;255(10):1549–1554. doi:10.1007/s00415-008-0983-7
81. Knudsen S, Mikkelsen JD, Bang B, Gammeltoft S, Jennum PJ. Intravenous immunoglobulin treatment and screening for hypocretin neuron-specific autoantibodies in recent onset childhood narcolepsy with cataplexy. Neuropediatrics. 2010;41(05):217–222. doi:10.1055/s-0030-1267993
82. Seong MJ, Hong SB. Autoimmunity and immunotherapy in narcolepsy. Sleep Med Res. 2017;8(1):1–7. doi:10.17241/smr.2017.00052
83. Valko PO, Khatami R, Baumann CR, Bassetti CL. No persistent effect of intravenous immunoglobulins in patients with narcolepsy with cataplexy. J Neurol. 2008;255(12):1900–1903. doi:10.1007/s00415-008-0996-2
84. Fronczek R, Verschuuren J, Lammers GJ. Response to intravenous immunoglobulins and placebo in a patient with narcolepsy with cataplexy. J Neurol. 2007;254(11):1607–1608. doi:10.1007/s00415-007-0594-8
85. Lecendreux M, Berthier J, Corny J, Bourdon O, Dossier C, Delclaux C. Intravenous immunoglobulin therapy in pediatric narcolepsy: a Nonrandomized, Open-Label, Controlled, Longitudinal Observational Study. J Clin Sleep Med. 2017;13(03):441–453. doi:10.5664/jcsm.6500
86. Pizza F, Franceschini C, Peltola H, et al. Clinical and polysomnographic course of childhood narcolepsy with cataplexy. Brain. 2013;136(12):3787–3795. doi:10.1093/brain/awt277
87. Passouant P. Billiard M. The evolution of narcolepsy with age. In: Guilleminault C, Dement WC, Passouant, P, editors. Narcolepsy. New York: Spectrum; 1976:179–197.
88. Büchele F, Baumann CR, Poryazova R, Werth E, Valko PO. Remitting narcolepsy? Longitudinal observations in a hypocretin-deficient cohort. Sleep. 2018;41(9):zsy118. doi:10.1093/sleep/zsy118
89. Knudsen S, Biering-Sørensen B, Kornum BR, et al. Early IVIg treatment has no effect on post-H1N1 narcolepsy phenotype or hypocretin deficiency. Neurology. 2012;79(1):102–103. doi:10.1212/wnl.0b013e31825dce03
90. Sarkanen T, Alén R, Partinen M. Transient impact of rituximab in H1N1 vaccination–associated narcolepsy with severe psychiatric symptoms. Neurologist. 2016;21(5):85–86. doi:10.1097/nrl.0000000000000099
91. Wasling P, Malmeström C, Blennow K. CSF orexin-A levels after rituximab treatment in recent onset narcolepsy type 1. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e613. doi:10.1212/nxi.0000000000000613
92. Ding Q, Xiao F, Dong X, Zhang J, Han F. Treatment with immune modulators in a child with recent-onset type 1 narcolepsy. Sleep Breath. 2020;1–3. doi:10.1007/s11325-020-02034-z
93. Donjacour CEHM, Lammers GJ. A remarkable effect of alemtuzumab in a patient suffering from narcolepsy with cataplexy. J Sleep Res. 2012;21(4):479–480. doi:10.1111/j.1365-2869.2011.00985.x
94. Guarnera C, Bramanti P, Mazzon E. Alemtuzumab: a review of efficacy and risks in the treatment of relapsing remitting multiple sclerosis. Ther Clin Risk Manag. 2017;13:871–879. doi:10.2147/tcrm.s134398
95. Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol. 2020;16(3):145–154. doi:10.1038/s41584-020-0373-9
96. Jones JL, Anderson JM, Phuah CL, et al. Improvement in disability after alemtuzumab treatment of multiple sclerosis is associated with neuroprotective autoimmunity. Brain. 2010;133(Pt 8):2232–2247. doi:10.1093/brain/awq176
97. Kallweit U, Bassetti CLA, Oberholzer M, et al. Coexisting narcolepsy (with and without cataplexy) and multiple sclerosis. J Neurol. 2018;265(9):2071–2078. doi:10.1007/s00415-018-8949-x
98. Vrethem M, Malmgren K, Lindh J. A patient with both narcolepsy and multiple sclerosis in association with pandemrix vaccination. J Neurol Sci. 2012;321(1–2):89–91. doi:10.1016/j.jns.2012.07.025
99. Penner I-K, Sivertsdotter EC, Celius EG, et al. Improvement in fatigue during natalizumab treatment is linked to improvement in depression and day-time sleepiness. Front Neurol. 2015;6:18. doi:10.3389/fneur.2015.00018
100. Sater RA, Gudesblatt M, Kresa-Reahl K, Brandes DW, Sater P. NAPS-MS: natalizumab effects on parameters of sleep in multiple sclerosis patients. Int J Ms Care. 2015;18(4):177–182. doi:10.7224/1537-2073.2015-033
101. Scammell TE, Luo G, Borker P, Sullivan L, Biddle K, Mignot E. Treatment of narcolepsy with natalizumab. Sleep. 2020;43(7). doi:10.1093/sleep/zsaa050
102. Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol. 2009;6(11):638–647. doi:10.1038/nrclinonc.2009.146
103. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33(4):369–383. doi:10.1007/s00281-011-0245-0
104. Ahlmann M, Hempel G. The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy. Cancer Chemother Pharmacol. 2016;78(4):661–671. doi:10.1007/s00280-016-3152-1
105. McCune WJ, Clowse MB. General principles of the use of cyclophosphamide in rheumatic diseases. In: Furst DE, Curtis MR, editors. Uptodate; 2020. Uptodate (Accessed February 11, 2021).
106. Haddad F, Anouti S, Maalouly G, Koussa S. Narcolepsierévélant un lupus érythémateuxsystémique [Narcolepsy associated with systemic lupus erythematosus]. Rev Med Interne. 2011;32(11):e114–5. French. doi:10.1016/j.revmed.2010.10.352
107. Adams C, McKeon A, Silber MH, Kumar R. Narcolepsy, REM sleep behavior disorder, and supranuclear gaze palsy associated with Ma1 and Ma2 antibodies and tonsillar carcinoma. Arch Neurol. 2011;68(4):521–524. doi:10.1001/archneurol.2011.56
108. Sinsioco C, Silver K, Forrest KM, et al. Narcolepsy with cataplexy as presenting symptom of occult neuroblastoma. Pediatr Neurol. 2013;49(1):64–67. doi:10.1016/j.pediatrneurol.2012.12.038
109. Peters J, Vijiaratnam N, Lo KY, Evans AH. Anti-Ma2-associated paraneoplastic encephalitis eat, sleep and repeat. Intern Med J. 2019;49(7):931–932. doi:10.1111/imj.14349
110. Landolfi JC, Nadkarni M. Paraneoplastic limbic encephalitis and possible narcolepsy in a patient with testicular cancer: case study. Neuro Oncol. 2003;5(3):214–216. doi:10.1215/S1152851702000467
111. Berger JR, Malik V, Lacey S, Brunetta P, Lehane PB. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol. 2018;24(3):323–331. doi:10.1007/s13365-018-0615-7
112. Gerashchenko D, Murillo-Rodriguez E, Lin L, et al. Relationship between CSF hypocretin levels and hypocretin neuronal loss. Exp Neurol. 2003;184(2):1010–1016. doi:10.1016/s0014-4886(03)00388-1
113. Kubota H, Kanbayashi T, Tanabe Y, et al. Decreased cerebrospinal fluid hypocretin-1 levels near the onset of narcolepsy in 2 prepubertal children. Sleep. 2003;26(5):555–557. doi:10.1093/sleep/26.5.555
114. Dauvilliers Y, Beziat S, Pesenti C, et al. Measurement of narcolepsy symptoms. Neurology. 2017;88(14):1358–1365. doi:10.1212/wnl.0000000000003787
115. Wang YG, Benmedjahed K, Lambert J, et al. Assessing narcolepsy with cataplexy in children and adolescents: development of a cataplexy diary and the ESS-CHAD. Nat Sci Sleep. 2017;9:201–211. doi:10.2147/nss.s140143
116. Plazzi G, Ruoff C, Lecendreux M, et al. Treatment of paediatric narcolepsy with sodium oxybate: a double-blind, placebo-controlled, randomised-withdrawal multicentre study and open-label investigation. Lancet Child Adolesc Heal. 2018;2(7):483–494. doi:10.1016/s2352-4642(18)30133-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.