Back to Journals » Clinical Ophthalmology » Volume 9
Retinitis pigmentosa with concomitant essential iris atrophy and glaucoma – case report
Authors Meirelles S, Barreto A, Buscacio ES, Shinzato E, Patrão L, de Oliveira Silva MS
Received 4 October 2014
Accepted for publication 9 January 2015
Published 26 November 2015 Volume 2015:9 Pages 2139—2145
DOI https://doi.org/10.2147/OPTH.S75384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Sérgio Henrique Sampaio Meirelles,1–3 Aline Sá Barreto,1 Eduardo Scaldini Buscacio,1,2 Elke Shinzato,1,2,4 Lia Florim Patrão,1 Mauro Sérgio de Oliveira Silva1
1Piedade Municipal Hospital, Rio de Janeiro, 2Federal University of Rio de Janeiro, 3Estacio de Sá University, Rio de Janeiro, Brazil; 4Ocular Ultrasound Department, Clinical Hospital of the University of São Paulo Medical School, São Paulo, Brazil
Purpose: To report a case of a young patient with retinitis pigmentosa (RP), essential iris atrophy, and glaucoma.
Case report: This report presents a case of a 22-year-old female patient with unilateral glaucoma, increased intraocular pressure, increased cup–disc ratio, iris atrophy, peripheral anterior synechiae, and bilateral RP.
Discussion: The patient presented glaucoma due to the iridocorneal endothelial syndrome, despite low age. RP is a bilateral disorder that may be associated with angle-closure glaucoma.
Keywords: ocular hypertension, ICE syndrome, secondary glaucoma, retinal degeneration
Introduction
Essential iris atrophy is one of the subtypes of iridocorneal endothelial (ICE) syndrome, which is a spectrum of disorders characterized by corneal proliferative endotheliopathy, associated with corneal edema, anterior chamber and iris stroma abnormalities, and glaucoma.1 Other variants of ICE syndrome are Chandler’s syndrome and Cogan-Reese syndrome. The essential iris atrophy usually affects females, generally over 30 years old. It is unilateral, and it is associated with iris holes, peripheral anterior synechiae, and corneal alterations.1 Approximately 50% of the cases present glaucoma, and it is probably caused by peripheral anterior synechiae and by a cellular membrane that covers the trabecular surface. The treatment for glaucoma is clinical; however, there is frequent surgical indication of trabeculectomy.
Retinitis pigmentosa (RP) is characterized by progressive decrease in night vision and progressive visual field loss due to cellular retinal dystrophy. The fundoscopy shows characteristic bone-spicules pigment.2 RP does not present a defined inheritance pattern, and it might occur in an autosomal dominant, recessive, or X-linked fashion.3 It can also be related to other systemic and ocular manifestations.
Case report
A 22-year-old white female patient reported the diagnosis of RP that she had 7 years before. Having gone through many examinations along this period, 1 year ago, it was diagnosed increased intraocular pressure (IOP) in the right eye (OD). The patient had misused hypotensive eye drops: prostaglandin analogs, carbonic anhydrase inhibitors, and B-adrenergic antagonists. She had interrupted the use of eye drops for 3 months. There was no family history of either RP or glaucoma. No ethical approval was required for this procedure. Patient consent was obtained before undergoing treatment.
In the ophthalmic examination, the best-corrected visual acuity was OD: 20/50 (-1,00 cyl ×115) and left eye (OS): 20/25 (+2,00 sph -1,00 cyl ×10). The slit lamp examination showed multiple iris holes and corectopia in OD (Figures 1 and 2), clear cornea in both eyes (OU), and no alterations in OS. IOP by Goldmann applanation tonometry was OD: 34 mmHg and OS: 16 mmHg at 3 pm. The gonioscopy revealed 360° isolated peripheral anterior synechiae in OD (Figure 3) and a visible open-angle up to ciliary body in OS. The fundoscopy (Figures 4 and 5) presented cup–disc ratio 0.9 vertical (V) ×0.9 horizontal (H), visible lamina cribrosa pores, preserved macula, and peripheral pigment mobilization in OD. In OS, the fundoscopy revealed cup–disc ratio 0.3 V ×0.3 H, preserved macula, and peripheral pigment mobilization. The automated perimetry (Figures 6 and 7) and manual perimetry (Figures 8 and 9) showed central island vision in OD and ring scotoma in OS. The ultrasound pachymetry was 524 μm and 530 μm in OD and OS, respectively. The specular microscopy revealed pleomorphism and polymegathism in OU (Figures 10 and 11). Fluorescein angiography featured blocked fluorescence in the peripheral pigment mobilization areas in OU.
 | Figure 1 Iris holes and corectopia in OD. |
 | Figure 2 Iris holes and corectopia in OD. |
 | Figure 3 Peripheral anterior synechiae in OD. |
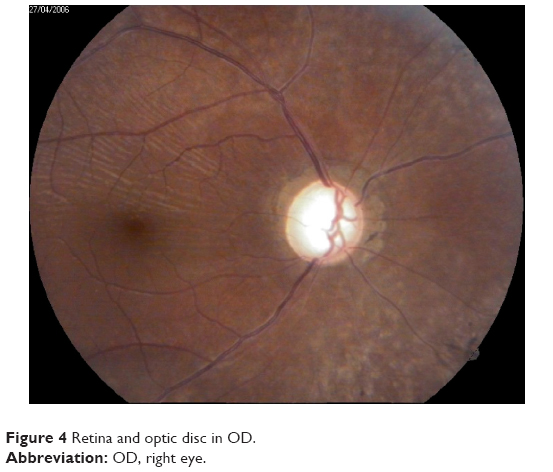 | Figure 4 Retina and optic disc in OD. |
 | Figure 5 Retina and optic disc in OS. |
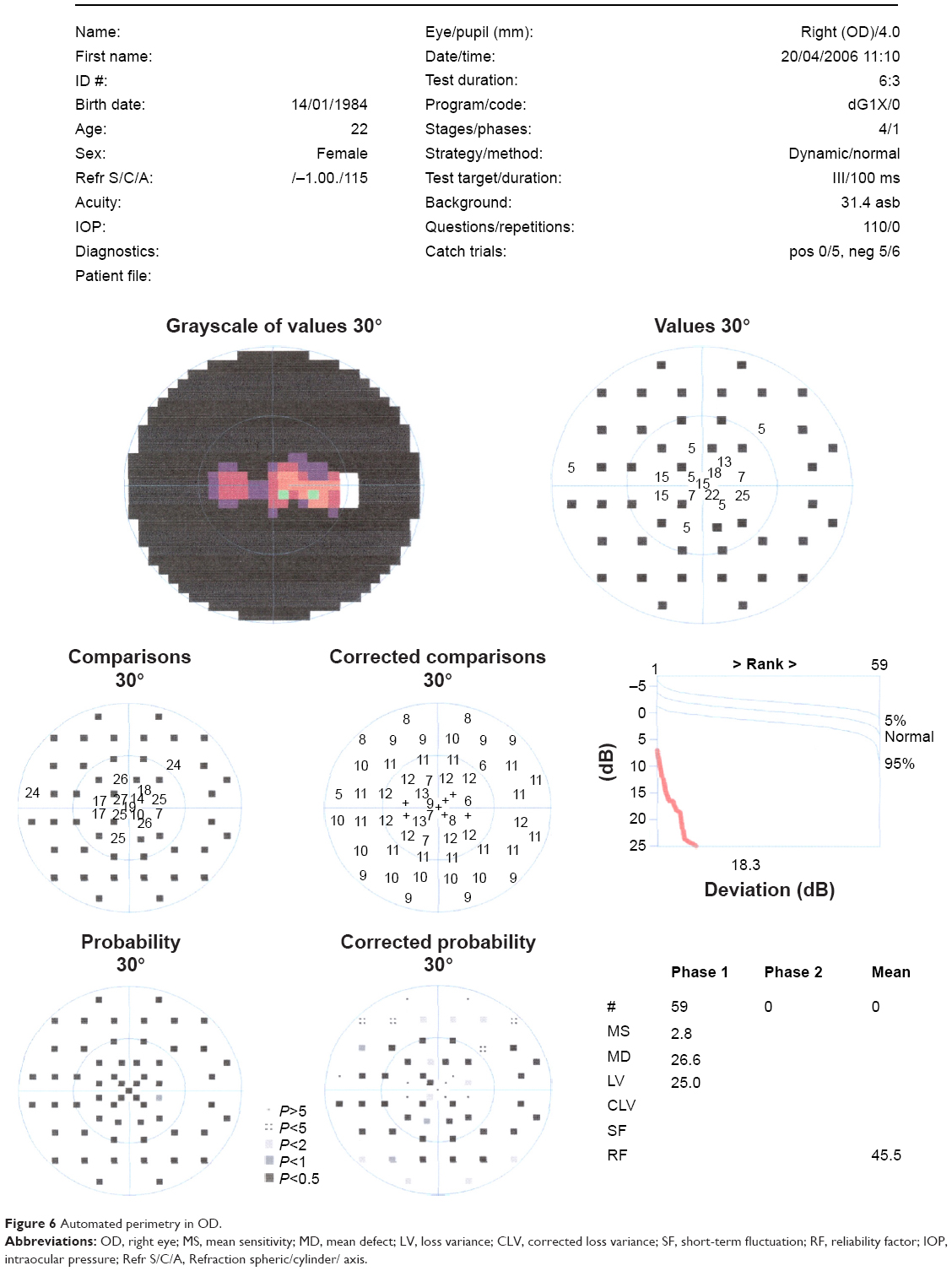 | Figure 6 Automated perimetry in OD. |
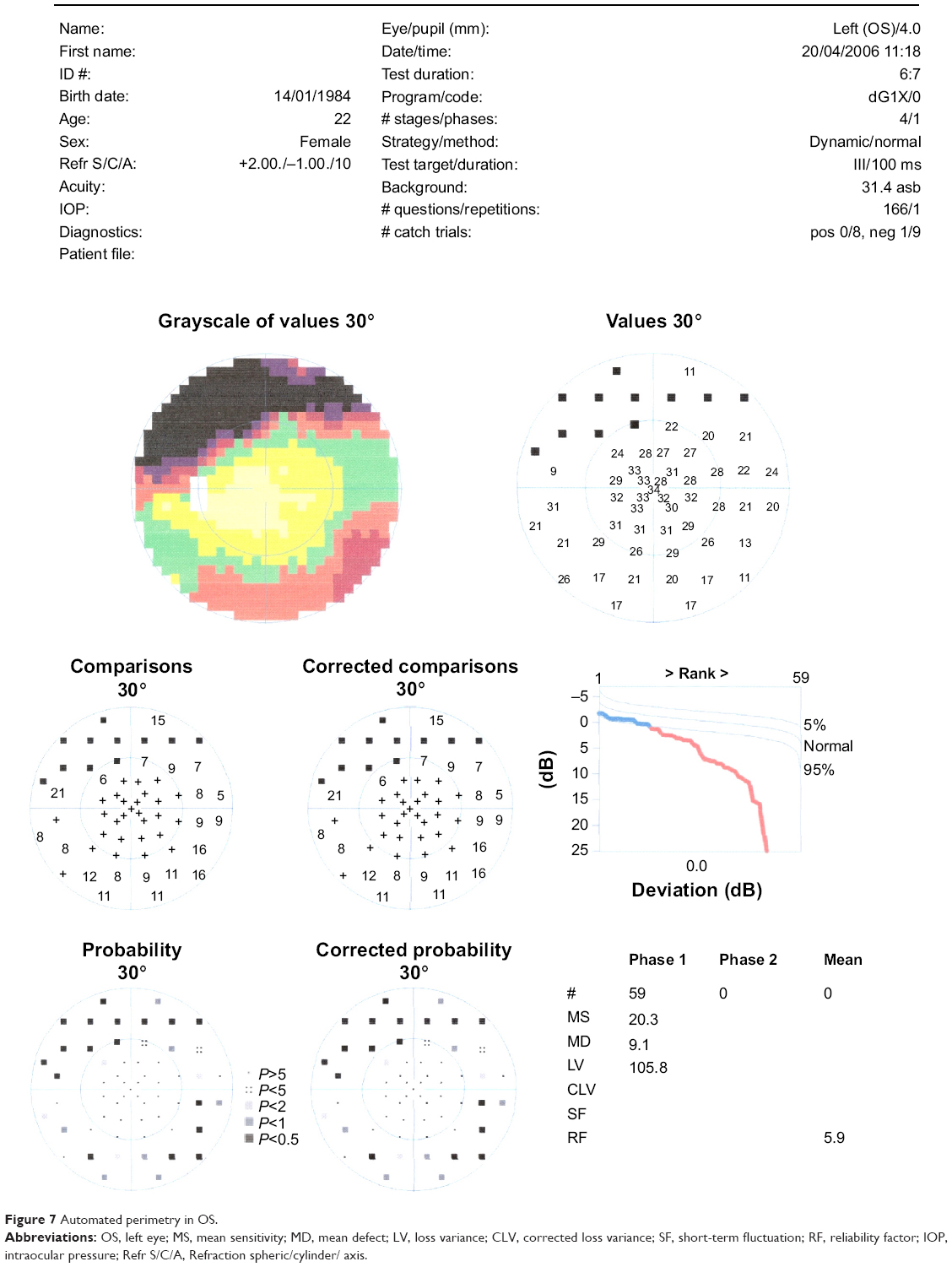 | Figure 7 Automated perimetry in OS. |
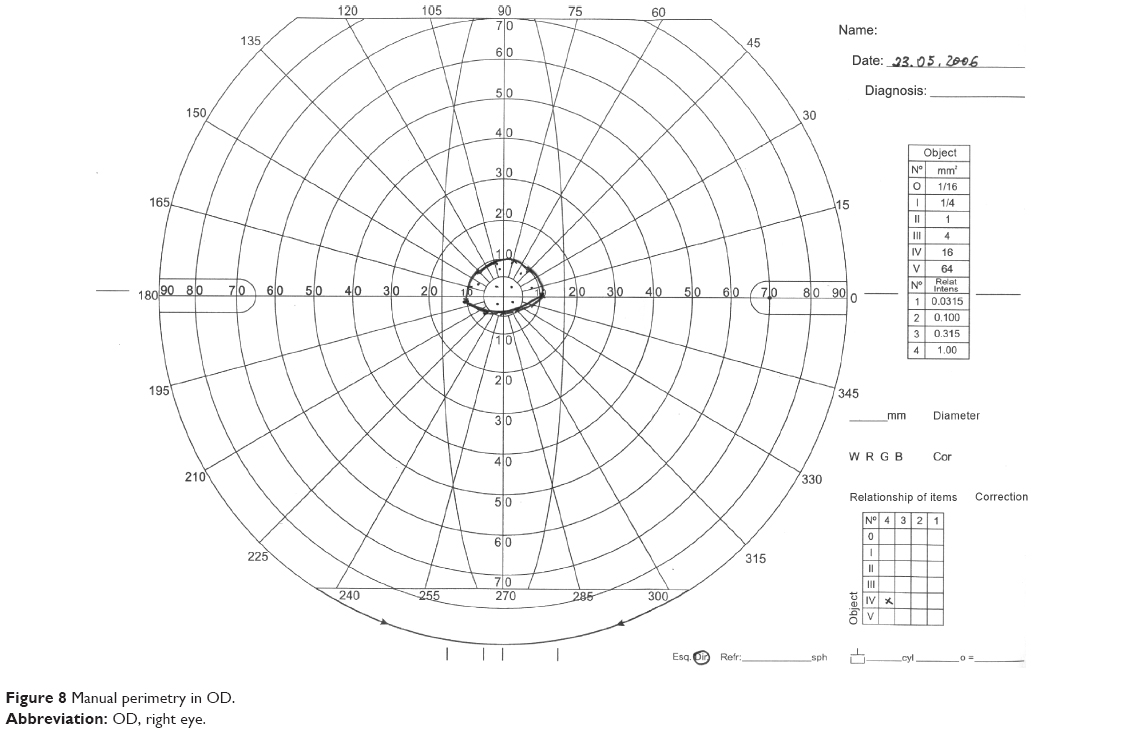 | Figure 8 Manual perimetry in OD. |
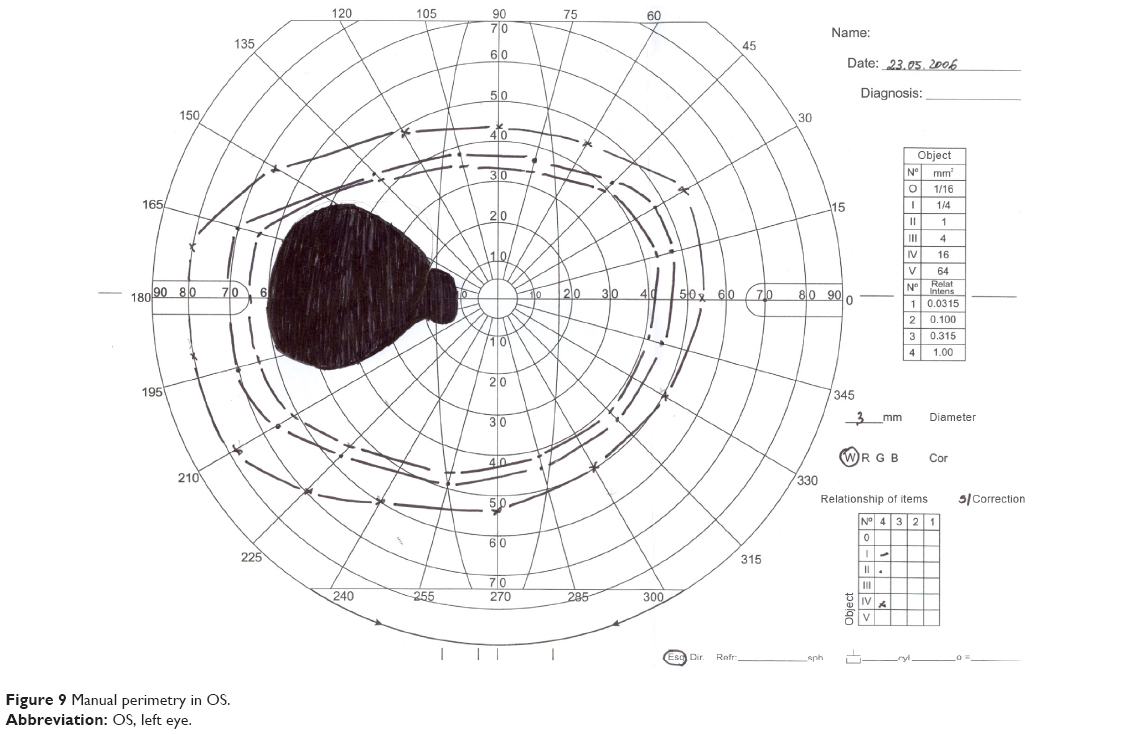 | Figure 9 Manual perimetry in OS. |
 | Figure 10 Specular microscopy in OD. |
 | Figure 11 Specular microscopy in OS. |
After failure of the clinical treatment to diminish IOP in OD, trabeculectomy was performed in this eye and IOP was controlled.
Discussion
We have reported the case of a young patient presenting with RP and ICE syndrome with glaucoma. Although there are some reports of association of RP and glaucoma in the literature, we have not found any report of association between RP and ICE syndrome at PubMed.
The most frequent glaucoma associated with RP is angle-closure glaucoma.2,4–6 The prevalence of glaucoma in patients with RP can be up to 2.3%.4 However, a study of 40 patients with RP found a prevalence of 12.5% of primary glaucoma.7 In the literature, there are reports of glaucoma associated with RP in inherited isolated or secondary cases, such as Sturge–Weber syndrome,8,9 retinal neovascularization,10 familial nephropathy,11 bilateral ectopia lentis,12 and Fuchs heterochromic cyclitis.13 There are also case reports of patients with RP who mimic glaucomatous visual field defect14 and abnormal nerve fiber layer of the retina, similar to those found in patients with glaucoma.15
In this case, we believe that glaucoma is associated with ICE syndrome because the patient has unilateral glaucoma and iris essential atrophy in the same eye. The patient also presents alterations in specular microscopy and in the anterior chamber in OD, which might be related to essential iris atrophy. The perimetry examinations demonstrate a glaucoma typical defect in OD and a RP typical defect in OS.
There was no response to the clinical treatment with hypotensive eye drops. Therefore, the patient underwent trabeculectomy in OD, and the IOP was controlled.
Acknowledgment
Marucia Patrão assisted with the manuscript preparation.
Disclosure
The authors declare that there are no conflicts of interest.
References
Shields MB, Bourgeois JE. Glaucoma associated with primary disorders of the corneal endothelium. In: Retch R, Shields MB, Krupin T, editors. The Glaucomas. St Louis: Mosby; 1996:957–974. | ||
Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc. 1984;81:693–735. | ||
Carr RE, Heckenlively JR. Chapter 24: Hereditary pigmentary degeneration of the retina. In: Duane TD, Jaeger EA, editors. Clinical Ophthalmology. Vol. 3. Philadelphia: Harper and Row;1987:1–28. | ||
Peng DW. Retinitis pigmentosa associated with glaucoma. Chinese Journal of Ophthalmology (Zhonghua yan ke za zhi). 1992;27(5): 262–264. Chinese. | ||
Malta RFS, Takahashi W, Nicolela MT, Soriano D. Glaucoma agudo primário e retinose pigmentar. Rev Bras Oftalmol. 1994;53(2):47–50. | ||
Omphroy CA. Sector retinitis pigmentosa and chronic angle-closure glaucoma: a new association. Ophthalmologica. 1984;189(1–2):12–20. | ||
Kogbe OI, Follmann P. Investigations into the aqueous humour dynamics in primary pigmentary degeneration of the retina. Ophthalmologica. 1975;171(2):165–175. | ||
Maul ED, Kass MA. Retinitis pigmentosa associated with nevus flammeus and unilateral glaucoma. Ann Ophthalmol. 1977;9(5):603–605. | ||
Filatov V, Guyer DR, Lustbader JM, Berkow JW. Dislocation of the crystalline lens in a patient with Sturge-Weber syndrome. Ann Ophthalmol. 1992;24(7):260–262. | ||
Uliss AE, Gregor ZJ, Bird AC. Retinitis pigmentosa and retinal neovascularization. Ophthalmology. 1987;93(12):1599–1603. | ||
Fiore C, Santoni G, Reggiani FM, Buoncristiani U, Pasticci B. Familial nephropathy with retinitis pigmentosa and closed-angle glaucoma. Ophthalmic Paediatr Genet. 1985;5(1–2):39–49. | ||
Halpern BL, Sugar A. Retinitis pigmentosa associated with bilateral ectopia lentis. Ann Ophthalmol. 1981;13(7):823–824. | ||
Vuorre I, Saari M, Tiilikainen A, Rasanen O. Fuch’s heterochromic cyclitis associated with retinitis pigmentosa: a familial study. Can J Ophthalmol. 1979;14(1):10–16. | ||
Dauber SL, Masselos K, Brown TM, Figuera EC, Pandya VB, Francis IC. Atypical retinitis pigmentosa masquerading as primary open angle glaucoma. J Glaucoma. 2008;17(3):248. Available from: http://dx.doi.org/10.1097/IJG.0b013e31816c4d9f. Author reply 248–249. | ||
Trobe JD, Bergsma DR. Atypical retinitis pigmentosa masquerading as a nerve fiber bundle lesion. Am J Ophthalmol. 1975;79(4):681–686. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.