Back to Journals » International Medical Case Reports Journal » Volume 14
Retinitis Pigmentosa and Polydactyly in a Patient with a Heterozygous Mutation on the BBS1 Gene
Authors Guardiola G ![]() , Ramos F
, Ramos F ![]() , Izquierdo N
, Izquierdo N
Received 1 June 2021
Accepted for publication 28 June 2021
Published 6 July 2021 Volume 2021:14 Pages 459—463
DOI https://doi.org/10.2147/IMCRJ.S321961
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Gabriel Guardiola,1 Fabiola Ramos,2 Natalio Izquierdo3
1Universidad Central del Caribe School of Medicine, Bayamon, PR, US; 2Department of Ophthalmology, University of Puerto Rico School of Medicine, University of Puerto Rico, San Juan, PR, US; 3Department of Surgery, University of Puerto Rico School of Medicine, University of Puerto Rico, San Juan, PR, US
Correspondence: Natalio Izquierdo
University of Puerto Rico, 369 De Diego, Torre San Francisco Suite 310, San Juan, PR, 00923, US
Tel +1 (787) 402-0201
Email [email protected]
Purpose: To report retinitis pigmentosa and a history of polydactyly in a Bardet–Biedl syndrome mutation carrier.
Observations: A 25-year-old male presented to the clinic complaining of poor visual acuity since childhood, night-blindness, and progressive peripheral vision loss. The patient also had a history of polydactyly in both feet. Ophthalmic evaluation was remarkable for a best-corrected visual acuity of 20/400 in both eyes. Imaging revealed a “salt-and-pepper” appearance surrounding the macula, bone-spicule retinal pigment epithelium hyperplasia, paravenous retinal pigment epithelium hyperplasia, and arteriolar attenuation. In addition, bilateral macular autofluorescence with a surrounding granular hypoautofluorescence and an additional hyperautofluorescent zone was present. Full-field ERG results showed non-recordable scotopic ERG responses and diminished photopic ERG responses OU, consistent with progressive rod-cone dystrophy. Genetic testing was positive for a pathogenic heterozygous mutation in the BBS1 gene of the variant c.1169T>G (p.Met390Arg) and several variants of uncertain significance in other genes.
Conclusions and Importance: Ascertainment of the inheritance patterns in BBS is an evolving discussion. Our case, a BBS carrier with retinitis pigmentosa and a history of polydactyly, could support previous research suggesting non-Mendelian genetics in this ciliopathy. Furthermore, genetic testing and analyses of additional mutations and variants of uncertain significance could potentially explain the reason for BBS-like phenotype in presumed BBS carriers.
Keywords: Bardet–Biedl syndrome, heterozygous carrier, retinitis pigmentosa, polydactyly
Introduction
Patients with the Bardet–Biedl syndrome (BBS) have a constellation of clinical features associated to several inherited genetic mutations that cause a widespread ciliopathy and thus multisystem complications.1 The most common manifestation and one of the primary features of BBS is a retinal rod-cone dystrophy described as an atypical retinitis pigmentosa, which starts developing at a young age.2 By the third decade of life, patients have markedly impaired vision due to early macular deterioration.3
Even though patients with the BBS have a heterogeneous phenotype, several physical characteristics have been identified and need to be present to establish the diagnosis.4 The BBS diagnosis is clinical, and patients have to display either four primary features or three primary and two secondary features.4 Primary characteristics include retinal degeneration, truncal obesity, polydactyly, renal dysfunction, genital anomalies, and learning difficulties.5 Secondary features include diabetes mellitus, congenital heart disease, hypertension, hepatic disease, among others.5,6
The Bardet–Biedl syndrome is an autosomal recessive disease caused by mutations in at least twenty-four different genes.7 BBS patients are classified according to the gene in which two allelic mutations are present.1,8,9 In addition to homozygotes, compound heterozygotes, which have different mutant alleles within the same gene, also have the syndrome.8 Triallelic inheritance has been suggested, given that some BBS homozygotes with an additional heterozygous mutation exhibit more severe phenotypes than their primary mutation would have predicted.10–13 Confirmation of the syndrome can be made through genetic sequencing 80% of the time.4,14,15 We report on a patient with retinitis pigmentosa and a history of polydactyly with a single pathogenic heterozygous mutation in the BBS1 gene.
Case Report
A 25-year-old male presented to the clinic complaining of poor visual acuity since childhood, night-blindness, and progressive peripheral vision loss in both eyes (OU). The patient also had a history of post-axial polydactyly in both feet (Figure 1). Ophthalmic evaluation was remarkable for a best-corrected visual acuity of 20/400 OU.
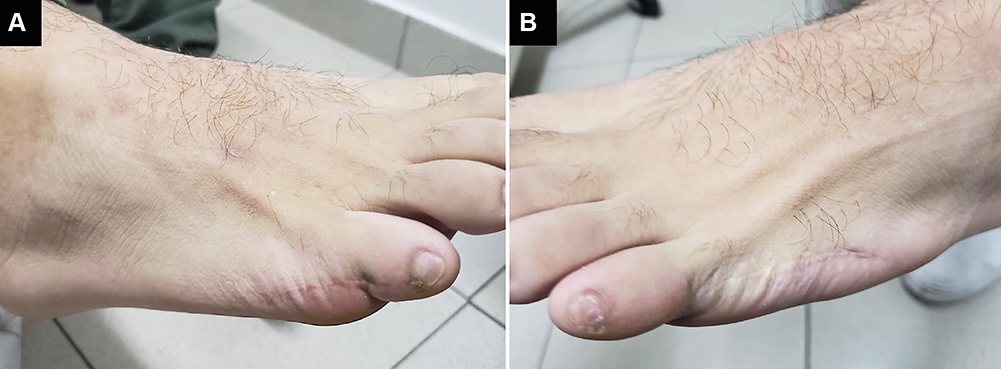 |
Figure 1 Evidence of post-axial polydactyly surgery. (A) Right foot, surgical scar where additional digit was removed. (B) Left foot, surgical scar where additional digit was removed. |
Infrared fundus photography (Optos, Inc.) showed a “salt-and-pepper” appearance surrounding the macula, bone-spicule retinal pigment epithelium (RPE) hyperplasia, paravenous RPE hyperplasia, and arteriolar attenuation OU (Figure 2A and B). Fundus autofluorescence showed macular autofluorescence with a surrounding granular hypoautofluorescence and an additional hyperautofluorescent zone OU (Figure 2C and D). Macular optical coherence tomography revealed decreased macular thickness of 212 microns and 199 microns in the right (OD) and left eye (OS), respectively. Visual field testing (30–2) revealed a mean deviation of −31.52 dB (p<0.5%) and −33.04 dB (p<0.5%) in OD and OS, respectively. Full-field ERG results showed non-recordable scotopic ERG responses and diminished photopic ERG responses OU, consistent with progressive rod-cone dystrophy.
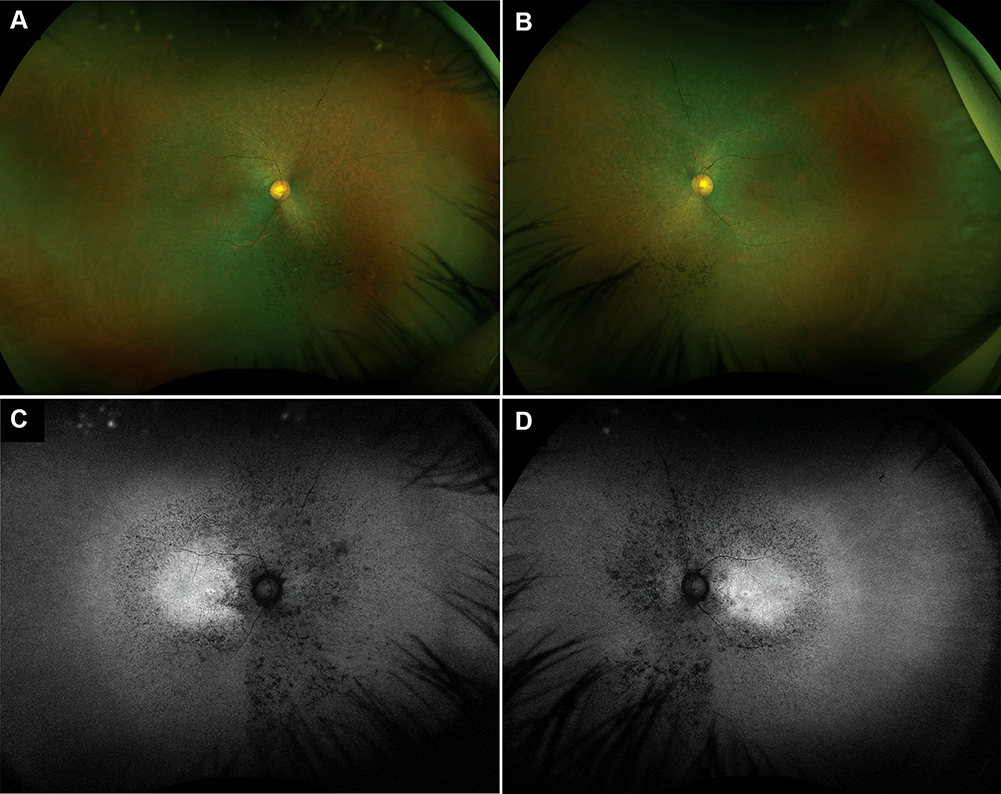 |
Figure 2 Color fundus photography and autofluorescence showing Retinitis Pigmentosa changes. (A) Right eye, there is a “salt-and-pepper” appearance surrounding the macula, which extends towards the nasal mid-periphery. There is bone-spicule RPE hyperplasia, predominantly present in the nasal mid-periphery, and paravenous RPE hyperplasia. Arteriolar attenuation is also evident in all quadrants. (B) Left eye, same findings as the right eye. (C) Right eye, increased macular autofluorescence surrounded by a ring of granular hypoautofluorescence extending towards the mid-periphery surrounded by an additional zone of hyperautofluorescence. (D) Left eye, same findings as the right eye. |
Saliva sample was sent for genetic testing. Full-gene sequencing and deletion/duplication analysis using next-generation sequencing, covering select non-coding variants, coding exons and 10–20 base pairs of adjacent intronic sequence (Invitae Corporation, San Francisco, California), was positive for a single pathogenic heterozygous mutation in the BBS1 gene of the variant c.1169T>G (p.Met390Arg). He had five additional variants of unknown uncertain (VUS) at the ADGVR1, CACNA2D4, COL2A1, IMPG1, and MKKS genes (Table 1).
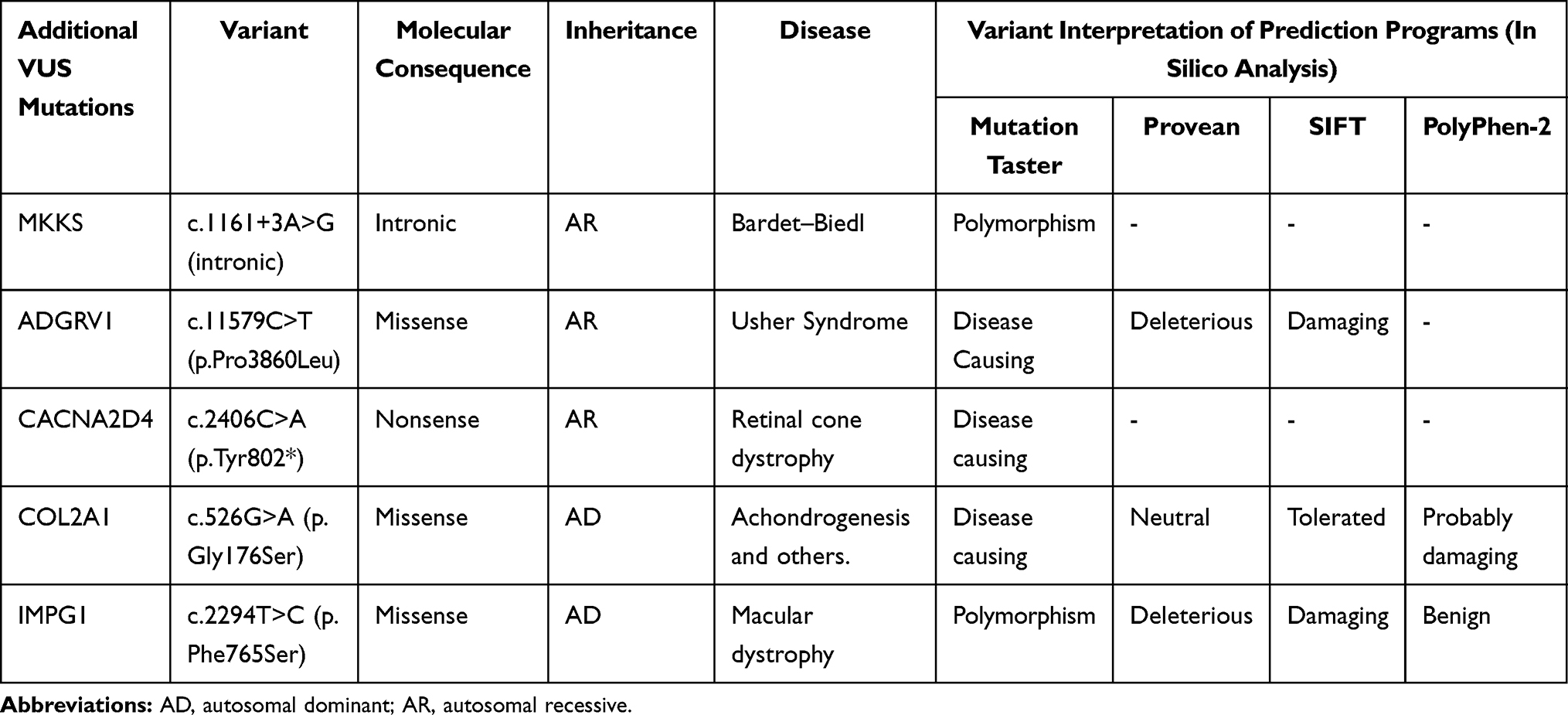 |
Table 1 In silico Analysis of Additional Mutations in Our Patient |
Discussion
The Bardet–Biedl syndrome is an autosomal recessive disease with great genetic heterogeneity.7 Our patient, being heterozygous for a single pathogenic mutation in the BBS1 gene (c.1169T>G (p.Met390Arg)), was therefore classified as a carrier and, according to Mendelian inheritance, not expected to show the phenotype. Yet, heterozygous carriers have been reported to be somewhat affected.16,17
Previous studies have explored the possibility of BBS heterozygotes having an increased risk of certain BBS characteristics.18,19 Beales et al20 correlated an increased risk of renal cancer with BBS heterozygous carriers. However, these findings have not been constant throughout the literature. For example, after analyzing a large cohort of BBS carriers, Hjortshøj et al21 found no association between carriers and renal cancer risk. Croft and Swift19 suggested that some carriers could have mild characteristics of the condition, like high blood pressure, obesity, and renal complications. Still, a study of Newfoundland’s BBS population by Webb et al,18 with a cohort largely composed of patients with mutations in the BBS1 gene, stated that there is no association between being heterozygous for BBS and an increased frequency of obesity, high blood pressure, diabetes and renal impairment.
Electroretinography (ERG) has shown retinal structural and functional abnormalities in visually asymptomatic BBS carriers with a normal appearing fundus.2,22 In contrast, our patient presented with advanced retinitis pigmentosa and severe visual decline.
Genetic testing of our patient showed a heterozygous mutation in the MKKS gene of the variant c.1161+3A>G (Intronic) which was listed in ClinVar as a variant of uncertain significance (VCV000860468.2, rs192968747).23 This MKKS gene sequence change, occuring within intron 4, does not alter the amino acid sequence of the protein.23 In silico analysis using Mutation Tester described the c.1161+3A>G (Intronic) variant as a probably harmless polymorphism.24 However, the mutation disturbs a nucleotide in the intron’s consensus splice site, a known cause of aberrant splicing.24–26 Pathogenic MKKS variants are responsible for approximately 6.3% of BBS cases, specifically BBS6.22
One possibility for our patient’s phenotypic and genetic findings is that the MKKS variant found is indeed pathogenic and that he is not a BBS carrier but an actual BBS patient. The occurrence of digenic heterozygous BBS cases, where one allele in two different affected genes is involved, has been previously supported by some researchers.16,17,27 Another explanation could be that there exists a variation within the BBS1 gene that was undetected by the methodology used, making our patient a compound heterozygote.28
The fact that our patient does not meet the full clinical criteria for a BBS diagnosis at present does not mean that he never will, as some primary and secondary characteristics could develop later in life. There are reports of genetically confirmed BBS patients who do not necessarily meet the clinical diagnosis criteria at the time of evaluation.15,29 Additionally, some BBS patients can present with independent retinal dystrophy without any other BBS feature.4 Furthermore, some BBS patients with fewer clinical features could have a weakened form of the syndrome.15
On the other hand, our patient could truly be a BBS carrier, which would support previous theories regarding BBS inheritance and phenotype penetrance. Some studies suggest that inheritance in patients with the BBS follows a classical Mendelian pattern.22,28 However, it has been proposed that, like with some other ophthalmic diseases (eg, Leber congenital amaurosis with CRB1 mutations), BBS inheritance is not as purely Mendelian as previously thought.10,12,17,27,30 Findings of asymptomatic carriers of biallelic BBS mutations pointed out the possibility of incomplete penetrance in certain BBS genes.10,31 Concurrently, the discovery of an additional BBS heterozygous mutation in many affected homozygous patients implied potential triallelic inheritance.10–13 Seemingly, the findings in our patient, who had a single pathogenic BBS1 mutation but developed BBS-related features, support previous reports showing the existence of complex inheritance in BBS.10–13,17
Conclusion
Ascertainment of the inheritance patterns in BBS is an evolving discussion. Our case, a BBS carrier with retinitis pigmentosa and a history of polydactyly, could support previous research suggesting non-Mendelian genetics in this ciliopathy. Furthermore, genetic testing and analyses of additional mutations and variants of uncertain significance could potentially explain the reason for BBS-like phenotype in presumed BBS carriers.
Ethics Approval and Informed Consent
Institutional review board (IRB) approval for this study was not required. The patient provided informed written consent for the case and images to be published.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tsang SH, Aycinena ARP, Sharma T. Ciliopathy: Bardet-Biedl syndrome. Adv Exp Med Biol. 2018;1085:171–174. doi:10.1007/978-3-319-95046-4_33
2. Kim LS, Fishman GA, Seiple WH, Szlyk JP, Stone EM. Retinal dysfunction in carriers of Bardet-Biedl syndrome. Ophthalmic Genet. 2007;28(3):163–168. doi:10.1080/13816810701537440
3. Schachat AP, Maumenee IH. Bardet-Biedl syndrome and related disorders. Arch Ophthalmol. 1982;100(2):285–288. doi:10.1001/archopht.1982.01030030287011
4. Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet-Biedl syndrome-now and in the future. Front Pediatr. 2018;6:23. doi:10.3389/fped.2018.00023
5. Torrefranca AB, Santiago APD, Lingao MD, Racoma MJC. Novel compound heterozygous pathogenic BBS5 variants in Filipino siblings with Bardet-Biedl syndrome (BBS). Ophthalmic Genet. 2020;41(6):621–624. doi:10.1080/13816810.2020.1810282
6. Forsyth R, Gunay-Aygun M. Bardet-Biedl syndrome overview. GeneReviews® [Internet]. 2020.
7. Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum Mutat. 2019;40(11):2068–2087. doi:10.1002/humu.23862
8. Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Hejtmancik JF, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(7):901–907. doi:10.1001/archophthalmol.2012.89
9. Hirano M, Satake W, Ihara K, et al. The first nationwide survey and genetic analyses of bardet-biedl syndrome in Japan. PLoS One. 2015;10(9):e0136317. doi:10.1371/journal.pone.0136317
10. Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293(5538):2256–2259. doi:10.1126/science.1063525
11. Bandano JL, Kim JC, Hoskins BE, et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Beidl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12(14):1651–1659. doi:10.1093/hmg/ddg188
12. Beales PL, Badano JL, Ross AJ, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72(5):1187–1199. doi:10.1086/375178
13. Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet. 2004;13:R65–R71. doi:10.1093/hmg/ddh092
14. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. doi:10.1038/ejhg.2012.115
15. Suspitsin EN, Imyanitov EN. Bardet-Biedl syndrome. Mol Syndromol. 2016;7(2):62–71. doi:10.1159/000445491
16. Hirano M, Satake W, Moriyama N, et al. Bardet–Biedl syndrome and related disorders in Japan. J Hum Genet. 2020;65(10):847–853. doi:10.1038/s10038-020-0778-y
17. Fauser S, Munz M. Further support for digenic inheritance in Bardet-Biedl syndrome. J Med Genet. 2003;40:104. doi:10.1136/jmg.40.8.e104
18. Webb MP, Dicks EL, Green JS, et al. Autosomal recessive Bardet-Biedl syndrome: first-degree relatives have no predisposition to metabolic and renal disorders. Kidney Int. 2009;76(2):215–223. doi:10.1038/ki.2009.116
19. Croft JB, Swift M. Obesity, hypertension, and renal disease in relatives of Bardet-Biedl syndrome sibs. Am J Med Genet. 1990;36(1):37–42. doi:10.1002/ajmg.1320360109
20. Beales PL, Reid HAS, Griffiths MH, Maher ER, Flinter FA, Woolf AS. Renal cancer and malformations in relatives of patients with Bardet-Biedl syndrome. Nephrol Dial Transplant. 2000;15(12):1977–1985. doi:10.1093/ndt/15.12.1977
21. Hjortshøj TD, Grønskov K, Rosenberg T, Brøndum-Nielsen K, Olsen JH. Risk for cancer in patients with Bardet-Biedl syndrome and their relatives. Am J Med Genet A. 2007;143(15):1699–1702. doi:10.1002/ajmg.a.31805
22. Mykytyn K, Nishimura DY, Searby CC, et al. Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1). Am J Hum Genet. 2003;72(2):429–437. doi:10.1086/346172
23. VCV000860468.2 - ClinVar - NCBI. https://www.ncbi.nlm.nih.gov/clinvar/variation/860468/.
24. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi:10.1038/nmeth.2890
25. Zhang MQ. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7(5):919–932. doi:10.1093/hmg/7.5.919
26. Buratti E, Chivers M, Královičová J, et al. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35(13):4250–4263. doi:10.1093/nar/gkm402
27. Gazzo A, Raimondi D, Daneels D, et al. Understanding mutational effects in digenic diseases. Nucleic Acids Res. 2017;45(15):e140–e140. doi:10.1093/nar/gkx557
28. Shaheen R, Szymanska K, Basu B, et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17(1):242. doi:10.1186/s13059-016-1099-5
29. Castro-Sánchez S, Álvarez-satta M, Cortón M, Guillén E, Ayuso C, Valverde D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J Med Genet. 2015;52(8):503–513. doi:10.1136/jmedgenet-2015-103099
30. Yzer S, Fishman GA, Racine J, et al. CRB1 heterozygotes with regional retinal dysfunction: implications for genetic testing of leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47(9):3736–3744. doi:10.1167/iovs.05-1637
31. Estrada-Cuzcano A, Koenekoop RK, Senechal A, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to bardet-biedl syndrome. Arch Ophthalmol. 2012;130(11):1425–1432. doi:10.1001/archophthalmol.2012.2434
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.