Back to Journals » Eye and Brain » Volume 13
Retinal Damage in Amyotrophic Lateral Sclerosis: Underlying Mechanisms
Authors Soldatov VO, Kukharsky MS, Belykh AE ![]() , Sobolev AM, Deykin AV
, Sobolev AM, Deykin AV
Received 7 January 2021
Accepted for publication 4 March 2021
Published 12 May 2021 Volume 2021:13 Pages 131—146
DOI https://doi.org/10.2147/EB.S299423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Margaret Wong-Riley
Vladislav O Soldatov,1,2 Michail S Kukharsky,3,4 Andrey E Belykh,5 Andrey M Sobolev,4 Alexey V Deykin2,6
1Core Facility Centre, Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russia; 2Department of Pharmacology and Clinical Pharmacology, Belgorod State National Research University, Belgorod, Russia; 3Department of General and Cell Biology, Faculty of Medical Biology, Pirogov Russian National Research Medical University, Moscow, Russia; 4Laboratory of Genetic Modelling of Neurodegenerative Processes, Institute of Physiologically Active Compounds, Russian Academy of Sciences, Chernogolovka, Russia; 5Department of Pathophysiology, Kursk State Medical University, Kursk, Russia; 6Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russia
Correspondence: Vladislav O Soldatov
Belgorod State National Research University, 85 Pobedy St., Belgorod, Belgorod Region, 308015, Russian Federation
Tel +7 91 0325 8496
Fax +7 47 2230-1012
Email [email protected]
Abstract: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease resulting in a gradual loss of motor neuron function. Although ophthalmic complaints are not presently considered a classic symptom of ALS, retinal changes such as thinning, axonal degeneration and inclusion bodies have been found in many patients. Retinal abnormalities observed in postmortem human tissues and animal models are similar to spinal cord changes in ALS. These findings are not dramatically unexpected because retina shares an ontogenetic relationship with the brain, and many genes are associated both with neurodegeneration and retinal diseases. Experimental studies have demonstrated that ALS affects many “vulnerable points” of the retina. Aggregate deposition, impaired nuclear protein import, endoplasmic reticulum stress, glutamate excitotoxicity, vascular regression, and mitochondrial dysfunction are factors suspected as being the main cause of motor neuron damage in ALS. Herein, we show that all of these pathways can affect retinal cells in the same way as motor neurons. Furthermore, we suppose that understanding the patterns of neuro-ophthalmic interaction in ALS can help in the diagnosis and treatment of this disease.
Keywords: ALS, retina, retinal involvement, neuro-ophthalmology, mitochondrial dysfunction, excitotoxicity
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the development of progressive paralysis and muscle atrophy, leading to disability and death on average 3–5 years after the first clinical manifestations. ALS has a hereditary origin with a traceable contribution of familial predisposition in 5–10% of cases.1,2 In the remaining 90–95% of cases, the disease develops sporadically without a strong connection with genetic links.3,4 According to twin-based data, the concordance of ALS is about 65%.5 Currently, 8 genes (SOD1, TARDPB, FUS, OPTN, VCP, UBQLN2, C9ORF72 and PFN1) are known to be most commonly mutated in ALS, and more than 100 genes have been identified as candidates for the association with sporadic ALS.6–9 There also exist evidences, confirming role of environmental factors such as heavy metals exposition, pesticides poisoning, soccer playing and others, in the development of sporadic ALS.10
Paralysis and other ALS symptoms are caused by damage to the upper motor neurons in the primary motor cortex and lower motor neurons in the brainstem and spinal cord. Degeneration of motor neurons results in the decline of the ability to control skeletal muscles and finally in death by respiratory failure. About half of patients, along with impaired motor function, have cognitive and behavioral deficits, which often are signs of the development of frontotemporal dementia (FTD), a condition that is a part of the same disease continuum as ALS.11,12
The pathways of the development and progression of ALS still has many unclarified details and are subject to active study. The formation of pathological aggregates, mitochondrial dysfunction, oxidative stress, glutamate-induced excitotoxicity and impaired protein utilization processes are now considered among the most significant factors in the pathogenesis of ALS. There are still a number of uncleared points in our knowledge about the molecular pathogenesis of ALS. For instance, there is inexplicable contrast between the duration of the asymptomatic and clinical stages of ALS. The pathological process can occur in motor neurons for decades and not manifest itself clinically but, in just 1–2 years, can lead to a dramatic loss of neuromuscular transmission.13 Moreover, it remains unclear why many individuals with ALS-associated mutations do not develop any clinical or pathological signs of the disease throughout their entire life.4
Are Motor Neurons the Only Target of ALS?
The progressive paralysis is the basic and pathognomonic symptom of ALS. However, the conception describing ALS as an only neuromuscular disease is outdated.12 Motor symptoms are often complicated by cognitive deficits14 and olfactory nerve damage.15
Furthermore, in some syndromic forms ALS might be complicated by extrapyramidal functions and hearing impairment16,17 as well as the presence of schizophrenia.18 There are also well documented skin19 and sensory20 abnormalities in ALS patients that may precede/accompany ALS.
Currently, the diagnosis of ALS is based on the El Escorial (or its revised version) criteria, which assess the signs of damage to the upper and lower motor neurons in various areas of the body, defined as bulbar, cervical, thoracic and lumbar areas. Due to the heterogeneity and ambiguity of clinical manifestations in the early stages of ALS, misdiagnosis remains a very common problem.
In this review, we summarize the available information on another non-classical ALS target and present data about possible mechanisms of retinal damage in this disease.
Premises for Retinal Damage in ALS
Although the visual impairment is not a routine complaint in ALS patients, a range of oculomotor disorders have been reported in ALS, including ophthalmoplegia, defective pursuit eye movements, saccadic impairments, nystagmus, and the abnormal Bell’s phenomenon.21,22 Furthermore, a growing body of evidence have revealed that visual dysfunction in ALS can also be caused by impaired retinal function.23 Several studies have indicated the presence of direct morphological and functional signs of retinal degeneration in patients and model animals with ALS.
The phenomenon of retinal damage in ALS may be considered as a consequence of the embryonic commonality of nervous and retinal tissues. Several proteins prone to the formation of aggregates in motor neurons are highly expressed in the retina. One more premise for the retinal involvement is the presence of genetic associations common to glaucoma and ALS, including polymorphic variants of OPTN, TBK1, PFN1, and ATXN2 genes. Finally, sufficient evidence exists that most neurodegenerative processes in the central nervous system (CNS) tend to involve the retina in the pathogenetic process.24
Clinical Evidence for Retinal Damage in ALS
In 2014, Fawzi et al revealed the in vivo and postmortem signs of retinal damage using ophthalmologic examination, contrast and color vision testing, immunohistochemistry and morphometry in two patients with the C9orf72 mutation and the clinical picture of ALS.25 In 2015, the same research group published another study confirming the association between retinopathy and ALS. The authors found axonal degeneration of ganglion cells by autopsy, as well as significant thinning of the retinal nerve fiber layer (RNFL) and reduced total macular thickness by optical coherence tomography (OCT). The researchers also presented the results of the eye fundus study in transgenic UBQLN2P497H mice. They reported large ubiquilin-2-positive aggregates, mainly in the inner plexiform layer (IPL) and less often in the outer plexiform layer.26
Ringelstein et al reported significant thinning of the RNFL and inner nuclear layer (INL) according to the OCT data in ALS patients.27 Similar results, expanding views about the retinal involvement, were then obtained by other research teams. Rohani et al examined 20 ALS patients and 25 healthy subjects. The average thickness of the RNFL was significantly reduced in ALS patients compared with controls. Notably, it was found in this study that retinal thinning positively correlated with the patients’ motor functions, as determined by the revised ALS functional rating scale (ALSFRS-R). An important finding of this study was the revealed asymmetry of the contralateral retinal lesions. The unequal degree of the left and right eye damage indicates that asymmetric CNS damage in ALS is not limited to the motor system.28
Hübers et al, using OCT and ophthalmic tests, performed an intravital morphological and functional study of the eye fundus in 71 ALS patients and 20 control subjects. The research was supplemented by an assessment of the motor neuron degeneration degree using fractional anisotropy, making it possible to analyze the correlation between motor and ophthalmic disorders. The study also revealed a significant thinning of the INL and RNFL in patients compared with controls, as well as a significant correlation between retinal thickness and the values of corticospinal tract fractional anisotropy.29 These results were then confirmed, by another clinical exam, where Simonett et al observed a positive correlation between macular RNFL thickness and some spirometric parameters in ALS patients.30
Boven et al found that 64% of 25 examined ALS patients had color vision deficiency. Additionally, 72% of the ALS patients had color vision below the 50th percentile (of normal), 52% below the 25th percentile, 24% below the 10th percentile, and 8% below the 2nd percentile comparing with distribution in normal subjects. Thus, a diffuse color discrimination deficit was found in ALS subjects at a younger age than in control subjects. The method used in this work may have high diagnostic value because color vision assessment is a simple and quick test that can be easily performed in a neurological clinic, where most ALS patients are treated.31
In another recent study ophthalmologic examination was conducted for 5 ALS patients (10 eyes) and 10 healthy volunteers (20 eyes) revealed several changes comprising the following: 1) a significant increase in the inner ring macular thickness of the temporal and lower regions of the eye fundus; 2) significant macula lutea thinning in the lower regions of the inner and outer macular rings; and 3) significant thinning of the peripapillary RNFL in the upper and lower quadrants. Additionally, some pathological changes in the retina were correlated with motor functions according to the ALSFRS-R exam.32
However, the notion about correlation between ALS and structural changes in the retina remains controversial as some studies did not find retinal changes in ALS patients significantly different compared to healthy volunteers. Roth et al analyzed the retinas of 76 ALS patients (144 eyes) and 54 healthy controls (108 eyes) using OCT. The study did not obtain data confirming a difference in the retinal thickness of patients and healthy subjects, and no correlations were found between retinal and neurological state.33 Moss et al also found no associations between visual acuity and an ALS diagnosis (generalized estimating equation models accounting for age).34
Notably, neurodegenerative changes in ALS were also found in other parts of the visual system in addition to the eye fundus. In 1991, Kushner et al examined the postmortem brain tissue slices of ALS patients and observed generalized reactive gliosis of cortical areas. The glial response in “cerebral infarction-like” cytological manner was particularly expressed in the occipital zone, containing the primary and extrastriate visual cortex regions.35 Later, these findings were confirmed by electrophysiological data. Münte et al recorded the evoked potentials in three different visual experiments. The evoked potentials in all 14 healthy subjects contained the P1 component, which was absent in ALS patients. The P1 component is another indicator of visual cortex function because it emanates from the lower temporal-occipital regions.36
Results of studies discussed above are summarized in Table 1.
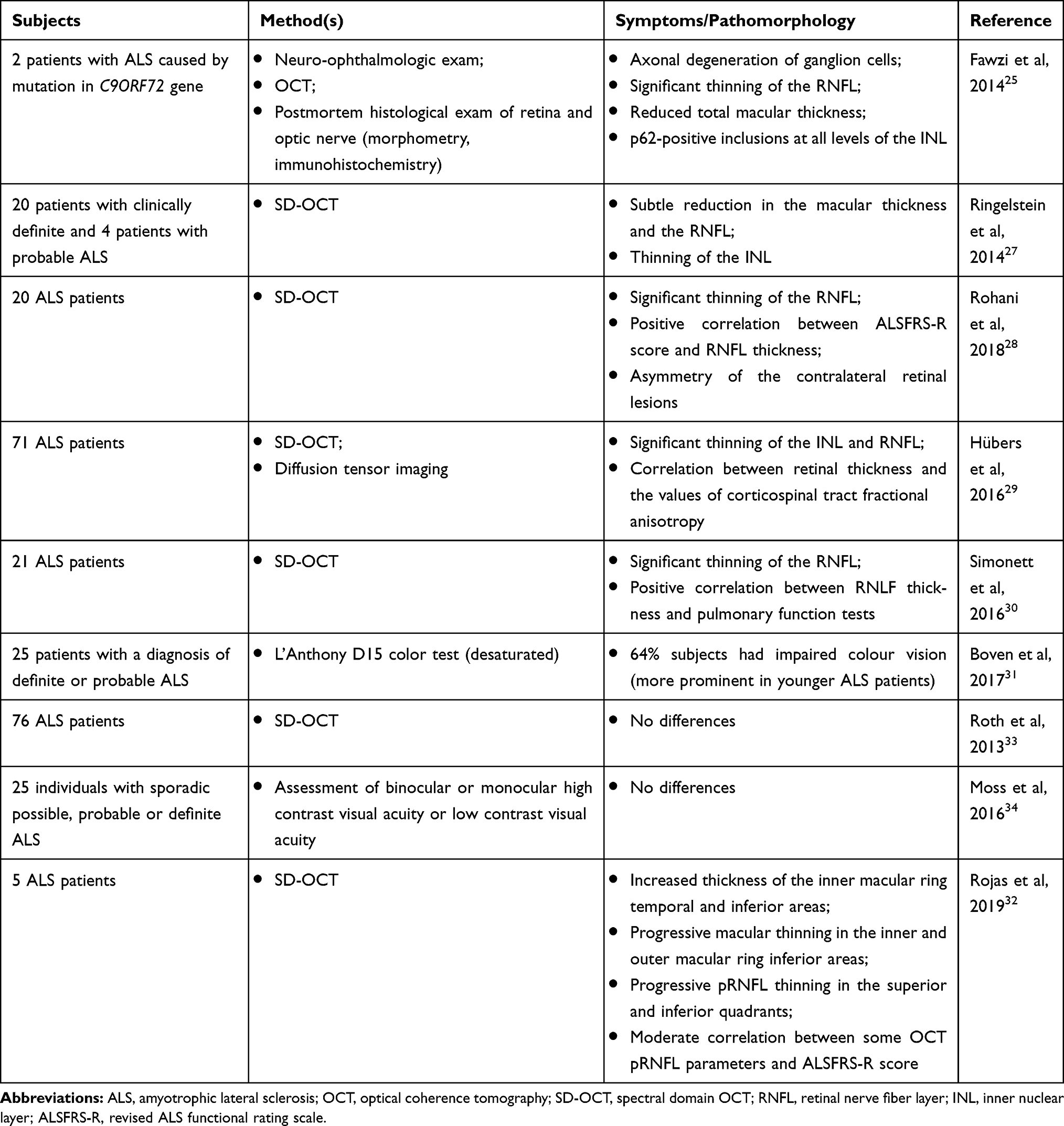 |
Table 1 Retinal Pathology in ALS Patients, Clinical Observations |
Interestingly, retinal abnormalities were regularly observed in patients with Western Pacific ALS and parkinsonism-dementia complex (ALS/PDC) which is clinically indistinguishable from classical ALS elsewhere. ALS/PDC is purely environmental disease, most plausibly caused by exposure to certain plant-derived neurotoxins in local food or medicine, or both.37 These neurotoxins cause ALS-specific neuromuscular phenotype and ophthalmological sign described as linear retinal pigmentary epitheliopathy. Despite there is no understanding of the etiology and nature of Western Pacific ALS, neuro-ophthalmologic correlations in this pathology may indicate retinal involvement in classical ALS. Data, revealing the link of this rare sporadic form of ALS and the retinal pathology detailed in the review.38
Possible Mechanisms of Retinal Damage in ALS
Protein Aggregation and Dysfunction
The presence of proteinaceous inclusions in spinal cord is an obligatory sign of ALS. These inclusions may include diverse components such as neurofilaments, ubiquitin, RNA-binding proteins, and the antioxidant enzyme superoxide dismutase 1 (SOD1) and others. In most cases, such inclusions are immunopositive to ubiquitin/p62, ubiquilin-2 and p-TDP43.39,40
SOD1
SOD1 is a Cu/Zn-dependent enzyme whose function is to remove excess superoxide anion from the cell by converting it to oxygen. The gene encoding this protein was the first found to be associated with ALS. The gene encoding this protein was the first gene showed to be linked to ALS. Mutant SOD1 have instable structure with loss of enzyme function and prone to formation of insoluble aggregates resulting in oxidative stress and neurotoxicity.41 To date, more than 180 different mutations have been identified in the SOD1 gene, causing about 12% of familial and approximately 2% of sporadic cases of ALS.11
Retinal involvement of SOD1 was confirmed in murine model with ALS-causative hSOD1G93A transgene. Studying these mice, Ringer et al found a pronounced vacuolization of neurons both in olfactory bulb and retina on the 60th day after birth. The study of retinal tissue dynamics at different periods revealed an increase in the vacuole size to the disease’s final stage. hSOD1-immunoreactive vacuoles were localized mainly in the dendrites of excitatory neurons in the retina IPL.42
Mutant SOD1 may be involved in retinal pathology not only because of its proteotoxic effect but also because of the antioxidant function loss. A study conducted by Hashizume et al identified several retinal abnormalities in SOD1-deficient mice. For instance, aged Sod1−/- mice exhibited decreased amplitudes of a- and b-waves of the electroretinogram, evoked by stimuli of different intensities. Histological examination showed progressive retinal degeneration and abnormalities in the inner and outer layer of nuclear cells. Additionally, electron microscopy revealed swollen cells and degenerated mitochondria in the inner nuclear layer and outer layer of nuclear cells in transgenic animals, indicating cell death by necrosis.43 Further, the same research group complemented these results by clinical data reporting significant decrease in the serum level of SOD1 in normal tension glaucoma patients.44
TDP-43
The TDP-43 (encoded by the TARDBP gene) protein is the most often detected component of pathological inclusions in various forms of ALS. In healthy neurons, this protein is located mainly in the nucleus,45,46 where its functions are associated with the regulation of RNA metabolism and transcription as well as with the formation of intranuclear structures – paraspeckles.47 However, a certain amount of TDP-43 is also found in the cytoplasm. Generally, TDP-43 is a DNA- and RNA-binding protein involved in biological processes such as DNA repair,48 transcription suppression, alternative splicing regulation, microRNA synthesis and RNA transport.45,49,50 More than 20 different mutations in TARDBP have been identified, together accounting for about 1–5% of all ALS familial cases.51
At the moment, there are no studies revealing the involvement of TDP-43 in ALS-associated retinal injury. However, Ward et al have found the linkage between the depletion of the TDP-43 nuclear pool and retinal thinning in the murine model of FTD on the background of a Grn gene mutation.52 This finding indicates the possible involvement of TDP-43 in retinal damage for genetically and pathologically overlapped ALS and FTD.
Recently, histological evaluation of postmortem enucleation specimens collected from subjects with chronic traumatic encephalopathy (CTE) showed accumulation of phosphorylated TDP-43 (pTDP-43) in retinal inner nuclear layer interneurons on the background of normal morphological state and retinal thickness. TDP-43 staining was commonly found in CTE eyes (7/8 cases), but rarely seen in control eyes (1/8) suggesting that TDP-43 is an important player in the visual impairment reported in patients with CTE.53
FUS
About 5% of familial cases of ALS are associated with mutations in the gene encoding the FUS protein (fused in sarcoma).54–56 FUS is closely similar to TDP-43 in its structural and functional features: both proteins have a glycine-rich, prion-like domain, which determines their tendency to aggregation, contain RNA-recognizing RRM motifs.57–60
TDP-43 and FUS are involved in the net of interconnected nuclear and cytoplasmic processes, including DNA damage repair, mRNA processing and transport, translation, cellular stress response.60–63 That emphasizes the importance of impaired DNA/RNA metabolism and protein homeostasis as a factor determining the sensitivity of motor neurons to damage and, consequently, to the development of neurodegeneration.64
At the moment, there are no direct evidence showing the association between pathogenic FUS and retinal damage. Nevertheless, FUS contributes in N-methyl-D-aspartate receptors (NMDARs) subunits transport in RGN,64–66 suggesting its dysfunction can affect glutamate transmission.
OPTN
Defective optineurin (OPTN gene) is another causal protein for the ALS development. An association between OPTN polymorphism and ALS was first discovered in a small group of patients in the Japanese population.67 Further research confirmed the strong linkage between OPTN mutations and ALS development.68–70 Normally, optineurin is responsible for the inhibition of the nuclear factor kappa-B (NF-kB).71 It acts as an autophagy receptor72 as well as participates in the regulation of vesicular transport and maintenance of the Golgi apparatus functions.73 In pathological conditions, mutant optineurin forms thread-like neuronal TDP-43- and ubiquitin-positive inclusions. Inclusions of a similar composition are also formed during mutations in UBQLN2 gene, coding another ALS-causative protein ubiquilin-2.74
As already mentioned, OPTN is a gene associated with both ALS and glaucoma. In a 2002 study of families with normal tension glaucoma, Rezaie et al found that OPTN mutations are associated with 16.7% of disease cases.75 Glaucoma-related OPTN mutations are mostly missense mutations, while ALS-related mutations include deletions, missense mutations, and nonsense mutations. In general, glaucoma-associated mutations are not associated with ALS, with one exception, a two-base pair insertion in exon 6, which is very rare.76 Nevertheless, optineurin was shown to be involved in retinal damage and it can be important link in our understanding of ophthalmic side of ALS.
C9ORF72
The most common mutation in ALS is hexanucleotide repeat expansion at the C9ORF72 locus, that was found in more than 30% of familial and about 7–9% of sporadic cases of ALS in the European population.11,77 Patients with this mutation have up to hundreds or thousands GGGGCC repeats in C9ORF72 locus whereas no more than 25 repeats are presented normally.78 Patients with this type of mutation can develop both ALS and FTD. The functions of the protein encoded by C9ORF72 have not been finally determined, but its interaction with the family of small Rab-GTPases, in which it acts as a regulator of autophagy79 and endosomal transport80 has been shown. This protein interacts with various components of the nuclear pore complex, participating in nuclear-cytoplasmic transport.81 It was shown that mutations in C9ORF72 lead to increased nuclear permeability.82 Protein product of C9ORF72 is also involved in the regulation of lysosome function83 and formation of stress granules.84 GGGGCCs translated into dipeptides (DPR, dipeptide repeat proteins) through ATG-independent translation (repeat-associated non-ATG (RAN) translation) can form intranuclear and cytoplasmic aggregates.85–88 Another mechanism of C9ORF72 expansion toxicity is forming of GGGGCC-contained RNA foci that can interact with some proteins, important for RNA metabolism, for example ADARB2, thereby disrupting their function.89–91
The connection between hexanucleotide expansion and ophthalmologic disorders demonstrated by Fawzi et al has already been mentioned previously.25 In this study, dipeptide aggregates were identified as a probable cause of decreased visual function in a patient with familial ALS and expansion of the GGGGCC hexanucleotide repeats in the C9ORF72 gene (C9-ALS). After the death of this patient, a morphological and immunohistochemical examination of the retina, optic nerve and CNS was performed, revealing p62-positive, pTDP43-negative perinuclear inclusions in the inner nuclear layer of the retina and brain. In-depth analysis showed that most of the p62-positive retinal inclusions were found in bipolar cells of the cones, as well as in some amacrine and horizontal cells.
Endoplasmic Reticulum Stress
Endoplasmic reticulum (ER) stress represents the state when ER is unable to maintain a balance between the synthesis and utilization of produced proteins. As a result of this imbalance, misfolded proteins accumulate in the cytoplasm that leads to cell damage.92–94 Chronic or severe ER stress triggers apoptotic cell death.95,96 ER stress is considered as a mechanism of vulnerability of motor neurons to pathology in ALS.97–99 Signs of ER stress were found in tissues from patients with familial and sporadic forms of ALS, as well as in cellular and animal models of the disease.100–103 Moreover, ER stress is one of the earliest pathological event in the familial forms of ALS. Additionally, mutations in PDIA1 and PDIA3 genes, which are directly involved in the maintenance of protein homeostasis, were associated with ALS.104,105
Nerve cells, including photoreceptors, have a high basic level of metabolism and protein synthesis, making them more sensitive to ER stress. Inability of retinal cells to cope with ER stress caused by abnormal protein aggregation or dysfunction of key proteins involved in this process can be considered as a mechanism for the development of ophthalmic diseases.106 Malfunction of key ER stress signaling proteins was shown to be associated with retinal pathology. For instance, mutations in ER stress sensor gene ATF6 lead to age-dependent degradation of photoreceptors in both humans107 and mice.108 Dysfunction of another ER stress mediator XBP-1 also leads to retinal degeneration.109 Induction of ER stress by the chemical substance tunicamycin causes the death of photoreceptor cells.110,111 Additionally, activation of ER stress is observed in light-induced retinal degeneration112 and death of retinal ganglion cells (RGC) caused by damage to the optic nerve.113
Disruption of Nuclear Import
As already noted, dysregulation of nuclear-cytoplasmic transfer is one of the important factors in the pathogenesis of ALS. A disruption in the structure of the nuclear localization domain promotes the release of several proteins into the cytoplasm, where they form aggregates.82 Such events occur, for instance, with the mutant FUS or TDP-43. Another reason for the loss of nuclear localization is mutations in proteins that provide nuclear-cytoplasmic transfer.114
Disruption of nuclear import has also been shown in the retinas of mice with FTD symptoms.52 Cho et al demonstrated that retinal RGN and motor neurons share common mechanisms of dysregulation of nuclear-cytoplasmic transport due to the loss of Ranbp2. Moreover, similar to motor neurons, RGN respond by hypertrophy and a decrease in the axonal caliber of axons. Functionally, these effects are accompanied by a delay in visual cortical responses to light stimuli.115
Vascular Regression
The retinal vasculature is one of the most vulnerable tissue to various pathological processes.116 In this regard, the data concerning changes in retinal vessels in ALS have particular interest. Clinical and experimental data accumulated in recent years has indicated that vascular abnormalities can precede neurodegeneration and aggravate it. For example, changes in the microvessels of the skin, muscles, and CNS were found in ALS patients.117,118
Transgenic mice expressing mutant forms of SOD1 and FUS are predisposed to vascular disorder development whereas vasotropic drugs can delay disease onset in them. Thus, Crivello et al found that the systemic administration of recombinant human angiopoietin increased survivability, retarded motor dysfunction, and protected against the loss of motor neurons and vascular regression in SOD1G93A mice.119 Later, the same research group found a significant decrease in the vascular density of the lumbar spinal cord in FUS (1–359) mice.120
An endothelium pathology,121,122 accompanied by decreased Ang1 and Vegf expression,123 develops in mutant SOD1 G93A mice even before disease onset. Some studies show a decrease in CNS vascular perfusion in mice and ALS patients.121,124,125
Moreover, pathological changes in the macro- and microvasculature of the eye fundus have been directly described in ALS patients. In particular, an increase in the thickness of the outer wall of the superior or inferior temporal retinal artery was found compared with that in healthy controls. At the same time, the character of visible changes indicates a possible deposition of b-amyloid in the vascular wall.126
Excitotoxicity
Another obvious pathway for retinal damage during ALS may be an increase in glutamatergic transmission.127,128 The involvement of glutamate, its receptors and transporters in neuronal death has been found in many neurodegenerative processes, including ALS,129 Parkinson’s disease,130,131 Alzheimer’s disease,132–134 dementia with Lewy bodies132 and Huntington’s disease.135 Interestingly, the only drug that prolongs survival of ALS patients is riluzole, the glutamate neurotransmission blocker.136,137 At the same time, the toxic effects of glutamate on the retina are well-known.138,139 Actually, retina is the first tissue in which glutamate-induced injury was discovered.140
Acting as a classic excitatory neurotransmitter,141 glutamate is involved in various processes: synaptogenesis, synaptic plasticity, long-term potentiation, and energy metabolism.142,143 This amino acid realizes its effects through ionotropic and metabotropic receptors. NMDA-, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA-) and kainate belong to ionotropic glutamate receptors.144 Metabotropic glutamate receptors are presented by 8 types (mGluR1-8) and divided into three groups depending on the structure and function. Their stimulation is associated with the activation of G-proteins and has a broad effect on neuronal functioning, including the glutamate release level and ion influx degree.143
An increase in glutamatergic transmission due to several pathological processes leads to the damage and death of neurons – so called “glutamate excitotoxicity”. Excessive glutamatergic stimulation results in the increased intake of calcium, zinc, sodium and potassium. The main important role in glutamate excitotoxicity is shown for the calcium overload. Influx of Ca2+-ions activates the caspase-dependent and caspase-independent pathways of apoptosis145,186-188 as well as stimulates oxidative and nitrosative stress.146,147 The key cellular transductors of glutamate excitotoxicity are ionotropic NMDA receptors (NMDAR’s). The stimulation of glial NMDARs leads to an increase in the production of proinflammatory cytokines, whereas mice with type 1 NMDAR deficiency are significantly protected from glutamate excitotoxicity.148 Similarly, administration of NMDAR’s-blockers such as ketamine or memantine, ameliorates retinal injury in rodents.149,150
Another intracellular factor mediating the retinotoxic effects of glutamate is GTPase RhoA.151,152 Activated RhoA triggers nerve cell damage cascades153 that upregulate inflammatory pathways.154 Pharmacological inhibition of this factor significantly reduces damage to the ganglion cell layer and IPL of the rat retina with NMDA-induced retinopathy.151,152 A more than twofold increase in brain RhoA kinase is found in mice with the SODG93A mutation and an ALS clinical and morphological picture.155 Thus, brain cells become much more sensitive to the toxic effects of glutamate. If the same increase in the expression of this enzyme occurs in the retina (which is very likely), it is logical to assume that the eye fundus becomes much more sensitive to the glutamatergic effects during ALS.
According to its wide range of functions mGluRs may also be involved in retinal damage during ALS. Accumulated data show that stimulation of glial mGluRs may contribute to glial proliferation and astrogliosis in ALS.156 Important role of mGluRs was especially shown for group I. Bonifacino et al showed a significant increase in the expression of group I mGluRs (types 1 and 5) in SOD1G93A mice. The authors suggest that exactly group I mGluRs can cause an increase in glutamate release, triggering glutamate-induced neurodegenerative processes.157 mGluR5 levels were also increased in human ALS brains in vitro. Simultaneously, group I mGluRs play an important role in physiology and pathology of retina. For instance, in Müller cells, mGluR5 receptors are known to modulate the transcription and translation of an inward-rectifying potassium ion channel, Kir4.1.158 Furthermore, mGluR5 is activated in a rat model of chronic ocular hypertension, leading to suppression of Kir currents and reduced expression of Kir4.1.159 Interestingly, suppression of mGluR5 activity can ameliorate both neurodegenerative and retinal pathology. Inhibition of mGluR5 ameliorated retinitis pigmentosa in rats160 whereas pharmacological allosteric mGluR5 inhibitor CTEP reversed the course of neurodegeneration in transgenic murine models of AD.161
Mitochondrial Dysfunction
Mitochondria, known as “energy plants of the cell”, are responsible for the production of the cell’s most “energy currency” in the form of ATP.162 In addition to ATP production, mitochondria are involved in various other cellular processes, including amino acid and nucleotide metabolism, protein synthesis, fatty acid metabolism, ion homeostasis, and apoptosis.163,164 Impaired mitochondrial function or mitochondrial dysfunction (MD) is a key link in cell damage during many hereditary and acquired disorders. MD arises from an inadequate number of mitochondria or an inability to provide necessary substrates to mitochondria, as well as a dysfunction in their electron transport and ATP-synthesis machinery.165
MD has been implicated to play a role in motor neuron death in ALS. The fragmentation of mitochondria as well as changes in mitochondrial morphology and expression of fusion/fission proteins are well described in ALS and have pronounced effects on normal mitochondrial function.166 Defective mitochondrial transport may be responsible for the accumulation of abnormal mitochondria in motor neuron axons seen in the animal models of ALS and also in human patients.167
It has been shown that many genes and proteins associated with both familial and sporadic ALS and FTD, including FUS,168,169 TDP-43,170,171 SOD1172,173 and C9ORF72,174 are involved in mitochondrial function, causing mitochondrial damage associated with neurodegeneration.175,176
Like motor neurons, retinal cells, have a very high mitochondrial density and dependence on aerobic metabolism.177,178 Approaches to treat mitochondria are seen as an effective way of therapy for retinal diseases such as glaucoma.179 Mitochondrial genes are among the most highly expressed in the retina, accounting for about 10% of the transcriptome.180 Mutations in genes associated with mitochondrial function are very often manifested in the form of ophthalmic diseases, such as Leber’s hereditary optic neuropathy,181,182 dominant optic atrophy,183 Wolfram’s syndrome,184 and age-related macular degeneration.185 All of these findings indicate that mitochondrial dysfunction arising in ALS influences the retina state and MD in ALS is one of the likely pathways for retinal degeneration.
Possible mechanisms of retinal damage in ALS that have been discussed above are represented in outline in Figure 1.
 |
Figure 1 Pathological pathways that retina cells can share with motor neurons in ALS. Notes: Excessive release of glutamate leads to the stimulation of NMDARs, AMPARs and mGluRs. Activated NMDARs and AMPARs open up and create Ca2+-influx. Overload by Ca2+ causes mitochondrial permeability transition pore (mPTP) opening. As aresult, the mitochondrion swells, collapses, and releases caspase activators, which lead to caspase-dependent apoptosis.175,176,186 Additionally, activation of NMDARs is closely coupled to the generation of nitric oxide (NO) through neuronal nitric oxide synthase (nNOS),136 which promote reactive nitrogen species (RNS) and subsequent reactive oxygen species (ROS) production.177 Moreover, through mGluRs, glutamate activates RhoA and IP3, which stimulate inflammation and cell damage.187 ALS-associated mutations in SOD-1, Ubiquilin-2 and Optineurin, affecting their functions and causing protein aggregation, enhance mitochondrial dysfunction. Furthermore, mitochondrial dysfunction leads to the rupture of the oxidative phosphorylation (OXPHOS) process, resulting in the production of ROS. The same phenomenon occurs with the FUS proteins and TDP-43, which lose their nuclear localization with increasing nuclear permeability. The hexanucleotide expansion of the C9ORF72 gene and dysfunction of the RANBP2 protein contribute to the decrease in nuclear permeability. Dipeptide repeats (DPRs) that are encoded by disrupted C9ORF72 are also involved in aggregate formation. These processes can also be complicated by vascular regression. |
Perspectives
Significant point in the field of ALS studies is the understanding that the disease can extend far beyond motor neurons. Detected morphological and functional retinal disorders in patients indicate the retina is also one of possible ALS targets. Further study of retinal state in ALS is important because retinal changes can serve as one of the signs, contributing to early diagnostics or disease prediction. Indeed, the late diagnosis is one of the main problems in neurodegenerative diseases. While the process has already started at the molecular level, the patient does not feel any symptoms for a long time. Unfortunately, by the time the disease manifests itself, about 50% of neurons can be damaged.
At the moment, only one drug, riluzole, has been clinically proven to be effective in delaying death in ALS. A recent study has shown that riluzole acts by prolonging stage 4 ALS rather than by slowing the entire disease course or prolonging intermediate stages.137 Edaravone, the second agent received FDA approval for ALS can slow the various stages progression but not influence lifespan.189 Therefore, currently, the early detection of the disease for early initiation of therapy has not been confirmed to be strategically important for extending a patient’s life. However, early detection or prediction of ALS remains important for studying drugs influencing early stages of ALS and for development approaches preventing or delaying the disease onset such as gene or cell therapy.
In addition to the practical aspect, the problem of retinal involvement in ALS is of fundamental importance. The retina is a very delicate tissue sensitive to any damaging influences. For instance, the retina is a prime target of hypertension,116 diabetes190 and neurotoxic poisons191. Furthermore, the retina is predominantly consisted of nerve tissue and has a proteomic composition similar to the brain.192 However, retinal damage is not a classic symptom of any neurodegenerative disease. As previously mentioned, some researchers have found no pathological changes or thinning of the retina in ALS patients. These findings can be explained by a simpler organization of the retina than that in the brain. Despite all the similarities in the organization of the retinal and nervous tissues, it is known that about 80–95% of all human genes are involved in the development and functioning of the brain, but for the retina, this number is about 65%.180 However, there may be other undisclosed reasons for this phenomenon and elucidating the molecular nature of such paradox may shed light on the defense mechanisms that the retina, but not motor neurons, uses. Mimicking these mechanisms can help us in the development of new ways of neuroprotection.
Abbreviations
ALS, amyotrophic lateral sclerosis; ALSFRS-R, revised ALS functional rating scale; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CNS, central nervous system; CTE, chronic traumatic encephalopathy; INL, inner nuclear layer; IPL, inner plexiform layer; FTD, frontotemporal dementia; mGluR, metabotropic glutamate receptor; MD, mitochondrial dysfunction; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NLS, nuclear localization signal; NMDAR, N-methyl-D-aspartate receptor; NR1, NMDAR subunit NR1; OCT, optical coherence tomography; OXPHOS, oxidative phosphorylation; PY-NLS, proline-tyrosine nuclear localization signal; Rab, Ras-related in brain; RGNs, retinal ganglion neurons; RhoA, Ras homolog family member A; RNFL, retinal nerve fiber layer; RNS, reactive nitrogen species; ROS, reactive oxygen species; SD-OCT, spectral domain OCT; SOD1, superoxide dismutase 1; TDP-43, transactive response DNA binding protein 43 kDa.
Acknowledgments
We are grateful to Professor Mikhail Pokrovskiy and Professor Vladimir Buchman for productive discussions and Nikita Sivkov for preparing the graphic material.
Funding
The reported study was funded by Russian Foundation for Basic Research (RFBR), project number 19-315-90114 and by grant 075-15-2019-1661 from the Ministry of Science and Higher Education of the Russian Federation. Institute of Gene Biology of the Russian Academy of Sciences (IGB RAS) facilities are supported by the Ministry of Science and Education of the Russian Federation.
Disclosure
Vladislav O Soldatov reports grants from Russian Foundation for Basic Research. Alexey V Deykin reports grants from Ministry of Science and Higher Education of the Russian Federation. The authors report no other potential conflicts of interest in this work.
References
1. Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015;1607:75–93. doi:10.1016/j.brainres.2014.10.009
2. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi:10.1016/S0140-6736(10)61156-7
3. Guerreiro R, Bras J, Hardy J. SnapShot: genetics of ALS and FTD. Cell. 2015;160(4):798–798 e791. doi:10.1016/j.cell.2015.01.052
4. Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta. 2015;1852(4):679–684. doi:10.1016/j.bbadis.2014.08.010
5. Al-Chalabi A, Fang F, Hanby MF, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. 2010;81(12):1324–1326. doi:10.1136/jnnp.2010.207464
6. van Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48(9):1043–1048. doi:10.1038/ng.3622
7. Pensato V, Tiloca C, Corrado L, et al. TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J Neurol. 2015;262(5):1376–1378. doi:10.1007/s00415-015-7739-y
8. Yanagi KS, Wu Z, Amaya J, et al. Meta-analysis of genetic modifiers reveals candidate dysregulated pathways in amyotrophic lateral sclerosis. Neuroscience. 2019;396:A3–A20. doi:10.1016/j.neuroscience.2018.10.033
9. Pochet R. Genetics and ALS: cause for Optimism. Cerebrum. 2017;2017.
10. Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. 2016;31(1):7–19. doi:10.11138/fneur/2016.31.1.007
11. Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. doi:10.1038/nn.3584
12. Tao QQ, Wu ZY. Amyotrophic lateral sclerosis: precise diagnosis and individualized treatment. Chin Med J (Engl). 2017;130(19):2269–2272. doi:10.4103/0366-6999.215323
13. Al-Chalabi A, Calvo A, Chio A, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13(11):1108–1113. doi:10.1016/S1474-4422(14)70219-4
14. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(11):994–1003. doi:10.1016/S1474-4422(07)70265-X
15. Viguera C, Wang J, Mosmiller E, Cerezo A, Maragakis NJ. Olfactory dysfunction in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2018;5(8):976–981. doi:10.1002/acn3.594
16. Fang T, Jozsa F, Al-Chalabi A. Nonmotor symptoms in amyotrophic lateral sclerosis: a systematic review. Int Rev Neurobiol. 2017;134:1409–1441.
17. Philippou E, Joubert K. The audiological profile and perceptions of hearing loss in individuals with amyotrophic lateral sclerosis. In: Segawa K, Ijichi R, editors. Amyotrophic Lateral Sclerosis: Symptoms, Treatment and Prognosis. New York, NY: Nova Science Publishers, Inc; 2013:1–56.
18. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327–336. doi:10.1016/0010-440x(90)90039-u
19. Ono S, Toyokura Y, Mannen T, Ishibashi Y. Increased dermal collagen density in amyotrophic lateral sclerosis. J Neurol Sci. 1988;83(1):81–92. doi:10.1016/0022-510x(88)90022-6
20. Pugdahl K, Fuglsang-Frederiksen A, de Carvalho M, et al. Generalised sensory system abnormalities in amyotrophic lateral sclerosis: a European multicentre study. J Neurol Neurosurg Psychiatry. 2007;78(7):746–749. doi:10.1136/jnnp.2006.098533
21. Sharma R, Hicks S, Berna CM, Kennard C, Talbot K, Turner MR. Oculomotor dysfunction in amyotrophic lateral sclerosis: a comprehensive review. Arch Neurol. 2011;68(7):857–861. doi:10.1001/archneurol.2011.130
22. Moss HE, McCluskey L, Elman L, et al. Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population. J Neurol Sci. 2012;314(1–2):97–101. doi:10.1016/j.jns.2011.10.016
23. Cerveró A, Casado A, Riancho J. Retinal changes in amyotrophic lateral sclerosis: looking at the disease through a new window [published online ahead of print, 2019 Dec 2]. J Neurol. 2019. doi:10.1007/s00415-019-09654-w
24. Yap TE, Balendra SI, Almonte MT, Cordeiro MF. Retinal correlates of neurological disorders. Ther Adv Chronic Dis. 2019;10:2040622319882205. doi:10.1177/2040622319882205
25. Fawzi AA, Simonett JM, Purta P, et al. Clinicopathologic report of ocular involvement in ALS patients with C9orf72 mutation. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):569–580. doi:10.3109/21678421.2014.951941
26. Volpe NJ, Simonett J, Fawzi AA, Siddique T. Ophthalmic manifestations of amyotrophic lateral sclerosis (An American Ophthalmological Society Thesis). Trans Am Ophthalmol Soc. 2015;113:T12.
27. Ringelstein M, Albrecht P, Sudmeyer M, et al. Subtle retinal pathology in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2014;1(4):290–297. doi:10.1002/acn3.46
28. Rohani M, Meysamie A, Zamani B, Sowlat MM, Akhoundi FH. Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: a window to disease progression. J Neurol. 2018;265(7):1557–1562. doi:10.1007/s00415-018-8863-2
29. Hubers A, Muller HP, Dreyhaupt J, et al. Retinal involvement in amyotrophic lateral sclerosis: a study with optical coherence tomography and diffusion tensor imaging. J Neural Transm (Vienna). 2016;123(3):281–287. doi:10.1007/s00702-015-1483-4
30. Simonett JM, Huang R, Siddique N, et al. Macular sub-layer thinning and association with pulmonary function tests in Amyotrophic Lateral Sclerosis. Sci Rep. 2016;6:29187. doi:10.1038/srep29187
31. Boven L, Jiang QL, Moss HE. Diffuse colour discrimination as marker of afferent visual system dysfunction in amyotrophic lateral sclerosis. Neuroophthalmology. 2017;41(6):310–314. doi:10.1080/01658107.2017.1326153
32. Rojas P, de Hoz R, Ramírez AI, et al. Changes in Retinal OCT and their correlations with neurological disability in early ALS Patients, a Follow-Up Study. Brain Sci. 2019;9(12):337. doi:10.3390/brainsci9120337
33. Roth NM, Saidha S, Zimmermann H, et al. Optical coherence tomography does not support optic nerve involvement in amyotrophic lateral sclerosis. Eur J Neurol. 2013;20(8):1170–1176. doi:10.1111/ene.12146
34. Moss HE, Samelson M, Mohan G, Jiang QL. High and low contrast visual acuity are not affected in amyotrophic lateral sclerosis. PLoS One. 2016;11(12):e0168714. doi:10.1371/journal.pone.0168714
35. Kushner PD, Stephenson DT, Wright S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol. 1991;50(3):263–277. doi:10.1097/00005072-199105000-00008
36. Munte TF, Troger MC, Nusser I, et al. Alteration of early components of the visual evoked potential in amyotrophic lateral sclerosis. J Neurol. 1998;245(4):206–210. doi:10.1007/s004150050206
37. Spencer PS, Fry RC, Palmer VS, Kisby GE. Western Pacific ALS-PDC: a prototypical neurodegenerative disorder linked to DNA damage and aberrant proteogenesis? Front Neurol. 2012;3:180. doi:10.3389/fneur.2012.00180
38. Spencer PS. Etiology of retinal and cerebellar pathology in western pacific amyotrophic lateral sclerosis and parkinsonism-dementia complex. Eye Brain. 2020;12:97–104. doi:10.2147/EB.S260823
39. Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi:10.1016/j.bbrc.2006.10.093
40. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi:10.1126/science.1134108
41. Pansarasa O, Bordoni M, Diamanti L, Sproviero D, Gagliardi S, Cereda C. SOD1 in amyotrophic lateral sclerosis: “ambivalent” behavior connected to the disease. Int J Mol Sci. 2018;19(5):1345. doi:10.3390/ijms19051345
42. Ringer C, Weihe E, Schutz B. SOD1G93A mutant mice develop a neuroinflammation-independent dendropathy in excitatory neuronal subsets of the olfactory bulb and retina. J Neuropathol Exp Neurol. 2017;76(9):769–778. doi:10.1093/jnen/nlx057
43. Hashizume K, Hirasawa M, Imamura Y, et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am J Pathol. 2008;172(5):1325–1331. doi:10.2353/ajpath.2008.070730
44. Yuki K, Ozawa Y, Yoshida T, et al. Retinal ganglion cell loss in superoxide dismutase 1 deficiency. Invest Ophthalmol Vis Sci. 2011;52(7):4143–4150. doi:10.1167/iovs.10-6294 PMID: 21421868.
45. Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(6):777–794. doi:10.1007/s00401-013-1125-6
46. Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659–667. doi:10.1016/j.molmed.2011.06.004
47. Nishimoto Y, Nakagawa S, Hirose T, et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol Brain. 2013;6:31. doi:10.1186/1756-6606-6-31
48. Mitra J, Guerrero EN, Hegde PM, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. 2019;116(10):4696–4705. doi:10.1073/pnas.1818415116
49. Mackenzie IR, Rademakers R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol. 2008;21(6):693–700. doi:10.1097/WCO.0b013e3283168d1d
50. Vanden Broeck L, Callaerts P, Dermaut B. TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol Med. 2014;20(2):66–71. doi:10.1016/j.molmed.2013.11.003
51. van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084–2098. doi:10.1016/S0140-6736(17)31287-4
52. Ward ME, Taubes A, Chen R, et al. Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J Exp Med. 2014;211(10):1937–1945. doi:10.1084/jem.20140214
53. Goodwill V, DiLoreto C, Chea L, et al. Chronic traumatic encephalopathy is associated with TDP-43 retinal pathology. Invest Ophthalmol Vis Sci. 2020;61(7):5175.
54. Muller K, Brenner D, Weydt P, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. 2018;89(8):817–827. doi:10.1136/jnnp-2017-317611
55. Naumann M, Peikert K, Gunther R, et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2019;6(12):2384–2394. doi:10.1002/acn3.50930
56. Ticozzi N, Silani V, LeClerc AL, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73(15):1180–1185. doi:10.1212/WNL.0b013e3181bbff05
57. An H, Skelt L, Notaro A, et al. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol Commun. 2019;7(1):7. doi:10.1186/s40478-019-0658-x
58. Guerrero EN, Wang H, Mitra J, et al. TDP-43/FUS in motor neuron disease: complexity and challenges. Prog Neurobiol. 2016;145–146:78–97.
59. Law WJ, Cann KL, Hicks GG. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic. 2006;5(1):8–14. doi:10.1093/bfgp/ell015
60. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. doi:10.1016/S1474-4422(10)70195-2
61. Kawaguchi T, Rollins MG, Moinpour M, et al. Changes to the TDP-43 and FUS Interactomes Induced by DNA Damage. J Proteome Res. 2020;19(1):360–370. doi:10.1021/acs.jproteome.9b00575
62. Birsa N, Bentham MP, Fratta P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin Cell Dev Biol. 2020;99:193–201. doi:10.1016/j.semcdb.2019.05.023
63. Aksoy YA, Deng W, Stoddart J, et al. “STRESSED OUT”: the role of FUS and TDP-43 in amyotrophic lateral sclerosis. Int J Biochem Cell Biol. 2020;126:105821. doi:10.1016/j.biocel.2020.105821
64. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. doi:10.1016/j.neuron.2013.07.033
65. Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3(7):661–669. doi:10.1038/76615
66. Selamat W, Jamari I, Wang Y, Takumi T, Wong F, Fujii R. TLS interaction with NMDA R1 splice variant in retinal ganglion cell line RGC-5. Neurosci Lett. 2009;450(2):163–166. doi:10.1016/j.neulet.2008.12.014
67. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. doi:10.1038/nature08971
68. Del Bo R, Tiloca C, Pensato V, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1239–1243. doi:10.1136/jnnp.2011.242313
69. Toth RP, Atkin JD. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front Immunol. 2018;9:1017. doi:10.3389/fimmu.2018.01017
70. Tumer Z, Bertelsen B, Gredal O, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging. 2012;33(1):208 e201–205. doi:10.1016/j.neurobiolaging.2011.07.001
71. Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17(16):1438–1443. doi:10.1016/j.cub.2007.07.041
72. Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi:10.1126/science.1205405
73. Sahlender DA, Roberts RC, Arden SD, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005;169(2):285–295. doi:10.1083/jcb.200501162
74. Osaka M, Ito D, Yagi T, Nihei Y, Suzuki N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24(6):1617–1629. doi:10.1093/hmg/ddu575
75. Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. doi:10.1126/science.1066901
76. Swarup G, Sayyad Z. Altered functions and interactions of glaucoma-associated mutants of optineurin. Front Immunol. 2018;9:1287. doi:10.3389/fimmu.2018.01287
77. Mathis S, Goizet C, Soulages A, Vallat JM, Masson GL. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. 2019;399:217–226. doi:10.1016/j.jns.2019.02.030
78. Babic Leko M, Zupunski V, Kirincich J, et al. Molecular mechanisms of neurodegeneration related to C9ORF72 hexanucleotide repeat expansion. Behav Neurol. 2019;2019:2909168. doi:10.1155/2019/2909168
79. Webster CP, Smith EF, Bauer CS, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016;35(15):1656–1676. doi:10.15252/embj.201694401
80. Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–3595. doi:10.1093/hmg/ddu068
81. Xiao S, MacNair L, McGoldrick P, et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568–583. doi:10.1002/ana.24469
82. Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi:10.1038/nature14973
83. Amick J, Ferguson SM. C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 2017;18(5):267–276. doi:10.1111/tra.12477
84. Maharjan N, Kunzli C, Buthey K, Saxena S. C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol Neurobiol. 2017;54(4):3062–3077. doi:10.1007/s12035-016-9850-1
85. Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. doi:10.1016/j.neuron.2013.02.004
86. Freibaum BD, Taylor JP. The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci. 2017;10:35. doi:10.3389/fnmol.2017.00035
87. Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. doi:10.1126/science.1232927
88. Nonaka T, Masuda-Suzukake M, Hosokawa M, et al. C9ORF72 dipeptide repeat poly-GA inclusions promote intracellular aggregation of phosphorylated TDP-43. Hum Mol Genet. 2018;27(15):2658–2670. doi:10.1093/hmg/ddy174
89. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention [published correction appears in Neuron. 2013 Nov 20;80(4):1102.Heusler, Aaron R [corrected to Haeusler, Aaron R]]. Neuron. 2013;80(2):415–428. doi:10.1016/j.neuron.2013.10.015
90. Gendron TF, Belzil VV, Zhang YJ, Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127(3):359–376. doi:10.1007/s00401-013-1237-z
91. Martier R, Liefhebber JM, García-Osta A, et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther Nucleic Acids. 2019;16:26–37. doi:10.1016/j.omtn.2019.02.001
92. Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15(4):233–249. doi:10.1038/nrn3689
93. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi:10.1126/science.1209038
94. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. doi:10.1038/nature17041
95. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi:10.1146/annurev-pathol-012513-104649
96. Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta. 2013;1833(12):3507–3517. doi:10.1016/j.bbamcr.2013.07.024
97. Ng SY, Soh BS, Rodriguez-Muela N, et al. Genome-wide RNA-Seq of human motor neurons implicates selective ER stress activation in spinal muscular atrophy. Cell Stem Cell. 2015;17(5):569–584. doi:10.1016/j.stem.2015.08.003
98. Rozas P, Bargsted L, Martinez F, Hetz C, Medinas DB. The ER proteostasis network in ALS: determining the differential motoneuron vulnerability. Neurosci Lett. 2017;636:9–15. doi:10.1016/j.neulet.2016.04.066
99. Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71(1):35–48. doi:10.1016/j.neuron.2011.06.031
100. Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30(3):400–407. doi:10.1016/j.nbd.2008.02.009
101. Farg MA, Soo KY, Walker AK, et al. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol Aging. 2012;33(12):2855–2868. doi:10.1016/j.neurobiolaging.2012.02.009
102. Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23(19):2294–2306. doi:10.1101/gad.1830709
103. Ilieva EV, Ayala V, Jove M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130(Pt 12):3111–3123. doi:10.1093/brain/awm190
104. Gonzalez-Perez P, Woehlbier U, Chian RJ, et al. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene. 2015;566(2):158–165. doi:10.1016/j.gene.2015.04.035
105. Yang Q, Guo ZB. Polymorphisms in protein disulfide isomerase are associated with sporadic amyotrophic lateral sclerosis in the Chinese Han population. Int J Neurosci. 2016;126(7):607–611. doi:10.3109/00207454.2015.1050098
106. Song JY, Wang XG, Zhang ZY, Che L, Fan B, Li GY. Endoplasmic reticulum stress and the protein degradation system in ophthalmic diseases. PeerJ. 2020;8:e8638. doi:10.7717/peerj.8638
107. Ansar M, Santos-Cortez RL, Saqib MA, et al. Mutation of ATF6 causes autosomal recessive achromatopsia. Hum Genet. 2015;134(9):941–950. doi:10.1007/s00439-015-1571-4
108. Kohl S, Zobor D, Chiang WC, et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet. 2015;47(7):757–765. doi:10.1038/ng.3319
109. McLaughlin T, Falkowski M, Park JW, et al. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol Neurodegener. 2018;13(1):16. doi:10.1186/s13024-018-0250-z
110. Alavi MV, Chiang WC, Kroeger H, et al. In vivo visualization of endoplasmic reticulum stress in the retina using the ERAI reporter mouse. Invest Ophthalmol Vis Sci. 2015;56(11):6961–6970. doi:10.1167/iovs.15-16969
111. Shimazawa M, Inokuchi Y, Ito Y, et al. Involvement of ER stress in retinal cell death. Mol Vis. 2007;13:578–587.
112. Kroeger H, Messah C, Ahern K, et al. Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53(12):7590–7599. doi:10.1167/iovs.12-10221
113. Hu Y, Park KK, Yang L, et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73(3):445–452. doi:10.1016/j.neuron.2011.11.026
114. Ferreira PA. The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell Mol Life Sci. 2019;76(12):2247–2273.
115. Cho KI, Yoon D, Yu M, Peachey NS, Ferreira PA. Microglial activation in an amyotrophic lateral sclerosis-like model caused by Ranbp2 loss and nucleocytoplasmic transport impairment in retinal ganglion neurons. Cell Mol Life Sci. 2019;76(17):3407–3432. doi:10.1007/s00018-019-03078-5
116. Sun Y, Smith LEH. Retinal vasculature in development and diseases. Annu Rev Vis Sci. 2018;4:101–122. doi:10.1146/annurev-vision-091517-034018
117. Kolde G, Bachus R, Ludolph AC. Skin involvement in amyotrophic lateral sclerosis. Lancet. 1996;347(9010):1226–1227. doi:10.1016/S0140-6736(96)90737-0
118. Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, et al. Amyotrophic lateral sclerosis: a neurovascular disease. Brain Res. 2011;1398:113–125. doi:10.1016/j.brainres.2011.04.049
119. Crivello M, O’Riordan SL, Woods I, et al. Pleiotropic activity of systemically delivered angiogenin in the SOD1G93A mouse model. Neuropharmacology. 2018;133:503–511. doi:10.1016/j.neuropharm.2018.02.022
120. Crivello M, Hogg MC, Jirstrom E, et al. Vascular regression precedes motor neuron loss in the FUS (1-359) ALS mouse model. Dis Model Mech. 2019;12(8):dmm040238. doi:10.1242/dmm.040238
121. Zhong Z, Deane R, Ali Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11(4):420–422. doi:10.1038/nn2073
122. Winkler EA, Sengillo JD, Sagare AP, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. 2014;111(11):E1035–1042. doi:10.1073/pnas.1401595111
123. Lu L, Zheng L, Viera L, et al. Mutant Cu/Zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. J Neurosci. 2007;27(30):7929–7938. doi:10.1523/JNEUROSCI.1877-07.2007
124. Rule RR, Schuff N, Miller RG, Weiner MW. Gray matter perfusion correlates with disease severity in ALS. Neurology. 2010;74(10):821–827. doi:10.1212/WNL.0b013e3181d3e2dd
125. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(1):111–120. doi:10.1007/s00401-012-1039-8
126. Abdelhak A, Hubers A, Bohm K, Ludolph AC, Kassubek J, Pinkhardt EH. In vivo assessment of retinal vessel pathology in amyotrophic lateral sclerosis. J Neurol. 2018;265(4):949–953. doi:10.1007/s00415-018-8787-x
127. Blasco H, Mavel S, Corcia P, Gordon PH. The glutamate hypothesis in ALS: pathophysiology and drug development. Curr Med Chem. 2014;21(31):3551–3575. doi:10.2174/0929867321666140916120118
128. Ribeiro FM, Vieira LB, Pires RG, Olmo RP, Ferguson SS. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res. 2017;115:179–191.
129. Paul P, de Belleroche J. The role of D-serine and glycine as co-agonists of NMDA receptors in motor neuron degeneration and amyotrophic lateral sclerosis (ALS). Front Synaptic Neurosci. 2014;6:10. doi:10.3389/fnsyn.2014.00010
130. Mironova YS, Zhukova IA, Zhukova NG, Alifirova VM, Izhboldina OP, Latypova AV. [Parkinson’s disease and glutamate excitotoxicity]. Zh Nevrol Psikhiatr Im S S Korsakova. 2018;118(6.Vyp. 2):50–54. Russian. doi:10.17116/jnevro201811806250
131. Blandini F, Porter RH, Greenamyre JT. Glutamate and Parkinson’s disease. Mol Neurobiol. 1996;12(1):73–94. doi:10.1007/BF02740748
132. Albasanz JL, Dalfo E, Ferrer I, Martin M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol Dis. 2005;20(3):685–693. doi:10.1016/j.nbd.2005.05.001
133. Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004;45(5):583–595.
134. Esposito Z, Belli L, Toniolo S, Sancesario G, Bianconi C, Martorana A. Amyloid beta, glutamate, excitotoxicity in Alzheimer’s disease: are we on the right track? CNS Neurosci Ther. 2013;19(8):549–555. doi:10.1111/cns.12095
135. Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov Today. 2014;19(7):990–996. doi:10.1016/j.drudis.2014.02.006
136. Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47(6 Suppl 4):S233–241. doi:10.1212/WNL.47.6_Suppl_4.233S
137. Fang T, Al Khleifat A, Meurgey JH, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018;17(5):416–422. doi:10.1016/S1474-4422(18)30054-1
138. Shen Y, Liu XL, Yang XL. N-methyl-D-aspartate receptors in the retina. Mol Neurobiol. 2006;34(3):163–179. doi:10.1385/MN:34:3:163
139. Kim JH, Lee NY, Jung SW, Park CK. Expression of N-methyl-d-aspartate receptor 1 in rats with chronic ocular hypertension. Neuroscience. 2007;149(4):908–916. doi:10.1016/j.neuroscience.2007.07.056
140. Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58(2):193–201. doi:10.1001/archopht.1957.00940010205006
141. Hayashi T. Effects of sodium glutamate on the nervous system. Keio J Med. 1954;3:192–193. doi:10.2302/kjm.3.183
142. Watkins JC, Jane DE. The glutamate story. Br J Pharmacol. 2006;147(Suppl 1):S100–108. doi:10.1038/sj.bjp.0706444
143. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460(2):525–542. doi:10.1007/s00424-010-0809-1
144. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61.
145. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407–6418. doi:10.1038/onc.2008.308
146. Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. 2016;2016:1245049. doi:10.1155/2016/1245049
147. Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284(5421):1845–1848. doi:10.1126/science.284.5421.1845
148. Kaindl AM, Degos V, Peineau S, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. 2012;72(4):536–549. doi:10.1002/ana.23626
149. Tsukahara Y, Blair NP, Eappen DC, et al. Ketamine suppresses ischemic injury in the rabbit retina. Invest Ophthalmol Vis Sci. 1992;33(5):1822–1825. PMID: 1532793.
150. Abdel-Hamid AA, Firgany A-D, Ali EM. Effect of memantine: a NMDA receptor blocker, on ethambutol-induced retinal injury. Ann Anat. 2016;204:86–92. doi:10.1016/j.aanat.2015.11.006
151. Kitaoka Y, Kitaoka Y, Kumai T, et al. Involvement of RhoA and possible neuroprotective effect of fasudil, a Rho kinase inhibitor, in NMDA-induced neurotoxicity in the rat retina. Brain Res. 2004;1018(1):111–118. doi:10.1016/j.brainres.2004.05.070
152. Wang Y, Wang Y, Yang Q, et al. Neuroprotective effects of C3 exoenzyme in excitotoxic retinopathy. Exp Eye Res. 2014;125:128–134. doi:10.1016/j.exer.2014.05.018
153. Semenova MM, Maki-Hokkonen AM, Cao J, et al. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10(4):436–443. doi:10.1038/nn1869
154. Wu X, Walker CL, Lu Q, et al. RhoA/Rho kinase mediates neuronal death through regulating cPLA2 Activation. Mol Neurobiol. 2017;54(9):6885–6895. doi:10.1007/s12035-016-0187-6
155. Hu JH, Chernoff K, Pelech S, Krieger C. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem. 2003;85(2):422–431. doi:10.1046/j.1471-4159.2003.01669.x
156. Anneser JM, Chahli C, Ince PG, Borasio GD, Shaw PJ. Glial proliferation and metabotropic glutamate receptor expression in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2004;63(8):831–840. doi:10.1093/jnen/63.8.831
157. Bonifacino T, Rebosio C, Provenzano F, et al. Enhanced function and overexpression of metabotropic glutamate receptors 1 and 5 in the spinal cord of the SOD1G93A mouse model of amyotrophic lateral sclerosis during disease progression. Int J Mol Sci. 2019;20(18):4552. doi:10.3390/ijms20184552
158. Gao F, Li F, Miao Y, et al. Group I metabotropic glutamate receptor agonist DHPG modulates Kir4.1 protein and mRNA in cultured rat retinal Müller cells. Neurosci Lett. 2015;588:12–17. doi:10.1016/j.neulet.2014.12.048
159. Ji M, Miao Y, Dong LD, et al. Group I mGluR-mediated inhibition of Kir channels contributes to retinal Müller cell gliosis in a rat chronic ocular hypertension model. J Neurosci. 2012;32(37):12744–12755. doi:10.1523/JNEUROSCI.1291-12.2012
160. Dai J, Fu Y, Zeng Y, Li S, Qin Yin Z. Improved retinal function in RCS rats after suppressing the over-activation of mGluR5. Sci Rep. 2017;7(1):3546. doi:10.1038/s41598-017-03702-z
161. Hamilton A, Vasefi M, Vander Tuin C, McQuaid RJ, Anisman H, Ferguson SS. Chronic pharmacological mglur5 inhibition prevents cognitive impairment and reduces pathogenesis in an Alzheimer Disease mouse model. Cell Rep. 2016;15(9):1859–1865. doi:10.1016/j.celrep.2016.04.077
162. Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342(3):619–630. doi:10.1124/jpet.112.192138
163. Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. doi:10.1146/annurev-genom-082509-141720
164. Atteia A, Adrait A, Brugiere S, et al. A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the alpha-proteobacterial mitochondrial ancestor. Mol Biol Evol. 2009;26(7):1533–1548. doi:10.1093/molbev/msp068
165. Nicolson GL. Mitochondrial dysfunction and chronic disease: treatment with natural supplements. Integr Med (Encinitas). 2014;13(4):35–43.
166. Sasaki S, Horie Y, Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007;114(6):633–639. doi:10.1007/s00401-007-0299-1
167. Muyderman H, Chen T. Mitochondrial dysfunction in amyotrophic lateral sclerosis - a valid pharmacological target? Br J Pharmacol. 2014;171(8):2191–2205. doi:10.1111/bph.12476
168. Deng J, Yang M, Chen Y, et al. FUS Interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015;11(9):e1005357. doi:10.1371/journal.pgen.1005357
169. Nakaya T, Maragkakis M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci Rep. 2018;8(1):15575. doi:10.1038/s41598-018-33964-0
170. Wang W, Wang L, Lu J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22(8):869–878. doi:10.1038/nm.4130
171. Wang P, Deng J, Dong J, et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019;15(5):e1007947. doi:10.1371/journal.pgen.1007947
172. Li Q, Vande Velde C, Israelson A, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107(49):21146–21151. doi:10.1073/pnas.1014862107
173. Vehvilainen P, Koistinaho J, Gundars G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:126. doi:10.3389/fncel.2014.00126
174. Choi SY, Lopez-Gonzalez R, Krishnan G, et al. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat Neurosci. 2019;22(6):851–862. doi:10.1038/s41593-019-0397-0
175. Lau DHW, Hartopp N, Welsh NJ, et al. Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 2018;9(3):327. doi:10.1038/s41419-017-0022-7
176. Kodavati M, Wang H, Hegde ML. Altered mitochondrial dynamics in motor neuron disease: an emerging perspective. Cells. 2020;9(4):1065. doi:10.3390/cells9041065
177. Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88(2):286–290. doi:10.1136/bjo.2003.027664
178. Kong GY, Van Bergen NJ, Trounce IA, Crowston JG. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009;18(2):93–100. doi:10.1097/IJG.0b013e318181284f
179. Osborne NN, Núñez-álvarez C, Joglar B, Del Olmo-Aguado S. Glaucoma: focus on mitochondria in relation to pathogenesis and neuroprotection. Eur J Pharmacol. 2016;787:127–133. doi:10.1016/j.ejphar.2016.04.032
180. Pinelli M, Carissimo A, Cutillo L, et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016;44(12):5773–5784. doi:10.1093/nar/gkw486
181. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242(4884):1427–1430. doi:10.1126/science.3201231
182. Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111(6):750–762. doi:10.1016/S0002-9394(14)76784-4
183. Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol Genet Metab. 2002;75(2):97–107. doi:10.1006/mgme.2001.3278
184. Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140(3):517–523. doi:10.1016/j.ajo.2005.03.017
185. Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40(7):892–896. doi:10.1038/ng.170
186. Baumgartner HK, Gerasimenko JV, Thorne C, et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284(31):20796–20803. doi:10.1074/jbc.M109.025353
187. Wang Q, Wang D, Shibata S, et al. Group I metabotropic glutamate receptor activation induces TRPC6-dependent calcium influx and RhoA activation in cultured human kidney podocytes. Biochem Biophys Res Commun. 2019;511(2):374–380. doi:10.1016/j.bbrc.2019.02.062
188. Rueda CB, Llorente-Folch I, Traba J, et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim Biophys Acta. 2016;1857(8):1158–1166. doi:10.1016/j.bbabio.2016.04.003
189. Bagnas MA, Reyes NG, Pascual JL. Safety and efficacy of edaravone in delaying functional decline in amyotrophic lateral sclerosis: a meta-analysis (P1.4-025). Neurology. 2019;92(15 Supplement):
190. Nentwich MM, Ulbig MW. Diabetic retinopathy - ocular complications of diabetes mellitus. World J Diabetes. 2015;6(3):489–499. doi:10.4239/wjd.v6.i3.489
191. Garner CD, Lee EW, Terzo TS, Louis-Ferdinand RT. Role of retinal metabolism in methanol-induced retinal toxicity. J Toxicol Environ Health. 1995;44(1):43–56. doi:10.1080/15287399509531942
192. The Human Protein Atlas. The retina proteome. Available from: https://www.proteinatlas.org/humanproteome/tissue/retina.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.