Back to Journals » Journal of Pain Research » Volume 10
Response to duloxetine in chronic low back pain: exploratory post hoc analysis of a Japanese Phase III randomized study
Authors Tsuji T ![]() , Itoh N, Ishida M
, Itoh N, Ishida M ![]() , Ochiai T, Konno S
, Ochiai T, Konno S
Received 28 March 2017
Accepted for publication 9 August 2017
Published 4 September 2017 Volume 2017:10 Pages 2157—2168
DOI https://doi.org/10.2147/JPR.S138172
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Katherine Hanlon
Toshinaga Tsuji,1 Naohiro Itoh,1 Mitsuhiro Ishida,2 Toshimitsu Ochiai,3 Shinichi Konno4
1Medical Affairs Department, 2Clinical Research Development, 3Biostatistics Department, Shionogi & Co. Ltd, Osaka, 4Department of Orthopedic Surgery, Fukushima Medical University, Fukushima, Japan
Purpose: Duloxetine is efficacious for chronic low back pain (CLBP). This post hoc analysis of a Japanese randomized, placebo-controlled trial (ClinicalTrials.gov, NCT01855919) assessed whether patients with CLBP with early pain reduction or treatment-related adverse events of special interest (TR-AESIs; nausea, somnolence, constipation) have enhanced responses to duloxetine.
Patients and methods: Patients (N = 456) with CLBP for ≥6 months and Brief Pain Inventory (BPI) average pain severity score of ≥4 were randomized (1:1) to duloxetine 60 mg/day or placebo for 14 weeks. Primary outcome was change from baseline in BPI average pain severity score (pain reduction). Subgroup analyses included early pain reduction (≥30%, 10%–30%, or <10% at Week 4) and early TR-AESIs (with or without TR-AESIs by Week 2). Measures included changes from baseline in BPI average pain severity score and BPI Interference scores (quality of life; QOL), and response rate (≥30% or ≥50% pain reduction at Week 14).
Results: Patients with ≥30% early pain reduction (n = 108) or early TR-AESIs (n = 50) had significantly greater improvements in pain and QOL than placebo-treated patients (n = 226), whereas patients with 10%–30% (n = 63) or <10% (n = 48) pain reduction did not; patients without early TR-AESIs (n = 180) had significant improvements in pain at Week 14. Response rates (≥30%/≥50% pain reduction) were 94.4%/82.4%, 66.7%/49.2%, and 25.0%/18.8% for patients with ≥30%, 10%–30%, and <10% early pain reduction, respectively, 74.0%/64.0% for patients with early TR-AESIs, 67.2%/54.4% for patients without early TR-AESIs, and 52.2%/39.4% for placebo.
Conclusion: Early pain reduction or TR-AESIs may predict which CLBP patients are most likely to respond to duloxetine with improvements in pain and QOL.
Keywords: Brief Pain Inventory, responder, adverse events, duloxetine, low back pain, quality of life
Introduction
Pain in the low back, the most common site of pain in the body,1,2 that persists for >3 months is considered to be chronic low back pain (CLBP).3 CLBP causes substantial physical, mental, and economic burden,4,5 and patients with CLBP experience high levels of disability and low quality of life (QOL).5,6 The pathophysiology of CLBP is complex and may involve dysfunction of central pain pathways.3 Although a range of treatment options for CLBP exists, no single treatment works in all patients.3
Duloxetine is a selective inhibitor of serotonin and norepinephrine reuptake with demonstrated efficacy in the treatment of several chronic pain conditions, including CLBP.7 The effects of duloxetine appear to be mediated by modulation of descending pain pathways that are dysfunctional in chronic pain.7 The efficacy and safety of duloxetine in the treatment of CLBP were established in double-blind, randomized, placebo-controlled trials in which duloxetine at 60 or 120 mg/day for 12–14 weeks reduced average weekly pain severity (measured by the Brief Pain Inventory [BPI] scale) significantly more than placebo.8–11 Duloxetine was also associated with improvements in QOL measures, indicating that pain reduction translates to better outcomes for patients. However, duloxetine was associated with a higher incidence of adverse events (AEs) compared with placebo, primarily nausea, somnolence, constipation, dry mouth, and fatigue.8–11
Despite its overall efficacy, duloxetine, like many pain treatments,12 is more effective in some patients than in others. A responder analysis of clinical trial data for duloxetine in chronic pain (CLBP, fibromyalgia, osteoarthritis, diabetic peripheral neuropathic pain) demonstrated that most patients either responded very well or very poorly.13 Further, the number needed to treat (NNT) varied depending on the targeted level of pain reduction. For CLBP, the NNT varied from 9.4 at a target level of ≥30% pain reduction to 22 at ≥70% pain reduction.13 The authors suggested that the low NNTs at ≥30% and ≥50% pain reduction, levels considered as moderate and substantial improvements, respectively,14 indicate that these pain reduction levels are most able to identify true responses.13 Identification of patients with CLBP who are most likely to benefit from duloxetine would help in the management of this condition. In particular, early indicators of response could encourage patients to persist with treatment.
The aims of this post hoc analysis of a Japanese randomized, placebo-controlled trial8 were (1) to assess whether patients with CLBP who respond to duloxetine 60 mg (Japanese clinical dose) with ≥30% pain reduction at Week 4 (early responders) are more likely than patients without early pain reduction to have clinically significant improvements in pain and QOL at Week 14 compared with placebo, and (2) to assess whether patients who experience treatment-related adverse events of special interest (TR-AESIs; nausea, somnolence, and constipation), especially TR-AESIs with incidence >5% within the first 2 weeks8 (early TR-AESIs), are more likely than patients without early TR-AESIs to have clinically significant improvements in pain and QOL compared with placebo.
Patients and methods
Study design
This was a post hoc analysis of a randomized, double-blind, placebo-controlled Phase III trial of duloxetine monotherapy in Japanese patients with CLBP, conducted in 58 medical institutions in Japan from May 2013 to July 2014, which has been described previously.8 Eligible patients were randomly assigned (1:1) to treatment with oral duloxetine 60 mg once daily or placebo after completion of a pretreatment washout period, using a stochastic minimization procedure and the BPI average pain severity score at baseline (<6, ≥6) as the allocation factor. The BPI average pain severity score measures average pain during the past 24 hours on a scale from 0 (no pain) to 10 (pain as bad as you can imagine); a validated Japanese version of the BPI was used.15 Patients provided informed consent before the start of the trial. The trial was approved by the Institutional Review Board of each study site (Table S1) and was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. The trial is registered at ClinicalTrials.gov (NCT01855919).
Study population
Patients with low back pain persisting for at least 6 months were eligible for inclusion. The main inclusion criteria included: (1) outpatients (male or female) aged ≥20 to <80 years; (2) use of nonsteroidal anti-inflammatory drugs (any dose and administration route) for ≥14 days per month for an average of 3 months and for ≥14 days during the month before the study start; (3) no radiculopathy symptoms or other specific low back diseases; and (4) a BPI average pain severity score of ≥4 at both Visit 1 (Week −1 to −2) and Visit 2 (Week 0). The main exclusion criteria included a history of low back surgery, invasive treatment for the relief of CLBP within 1 month before Visit 1, requiring crutches or a walker, and a diagnosis of major depressive disorder. Exclusion of patients with major depressive disorder minimizes the effect of the antidepressant activity of duloxetine on pain and QOL measures.
Treatment protocol
Patients in the duloxetine group received duloxetine 20 mg/day for 1 week, followed by 40 mg/day for 1 week, and then followed by 60 mg/day for 12 weeks. Patients in the placebo group received placebo for 14 weeks. Patients underwent tapering after the completion of the treatment period or following discontinuation after 2 weeks of treatment.
Outcome measures
In the primary study, the primary efficacy measure was the change in BPI average pain severity score from baseline to Week 14 compared with placebo. Two patients in the duloxetine group who did not have post-baseline data were excluded from both the primary analysis8 and this post hoc analysis.
This post hoc analysis consisted of 2 subgroup analyses for patients receiving duloxetine 60 mg: (1) the extent of early pain response (patients with ≥30% pain reduction, 10%–30% pain reduction, or <10% pain reduction at Week 4), and (2) the presence or absence of early TR-AESIs (patients with or without nausea, somnolence, or constipation within the first 2 weeks). Nausea, somnolence, and constipation were chosen as TR-AESIs because they occurred in >5% of duloxetine-treated patients and were significantly more common in the duloxetine group than in the placebo group (Table S2). Outcome measures included the following: the least squares (LS) mean change in BPI average pain severity score from baseline to Weeks 2, 4, 6, 10, and 14 (end of treatment) in each subgroup and in the placebo group; the proportion of patients with ≥30% or ≥50% reduction in BPI average pain severity score at Week 14 (response rate) in each subgroup and in the placebo group; the LS mean change in BPI Interference scores (rated from 0 [does not interfere] to 10 [completely interferes]15) for 7 daily activities (general activity, mood, walking ability, normal work, relationships with other people, sleep, and enjoyment of life) and the average of the 7 activities from baseline to Weeks 2, 4, 6, 10, and 14 in each subgroup and in the placebo group; and the time of occurrence and resolution of TR-AESIs in the duloxetine group.
Statistical analysis
The full analysis set was used for all analyses and consisted of all randomized patients who received at least 1 dose of study drug and for whom post-baseline BPI average pain severity scores were available. Baseline demographic characteristics are described as mean (standard deviation) and/or median (minimum, maximum) for continuous variables (compared using 1-way analysis of variance) and n (%) for categorical variables (compared using a Fisher exact test). The LS mean changes from baseline in BPI average pain severity score and BPI Interference QOL score in each duloxetine subgroup were compared with changes in the placebo group at each time point using a mixed-effects model repeated measures approach. The model included subgroup, time point, and subgroup-by-time point interaction as fixed effects, as well as baseline value as a covariate. A P value <0.05 was considered statistically significant. The proportions of patients achieving ≥30% pain reduction and ≥50% pain reduction at Week 14 in each duloxetine subgroup and in the placebo group were calculated and compared using a Cochran–Mantel–Haenszel test adjusted by the allocation factor (baseline BPI average pain severity score <6 or ≥6); missing end-of-treatment data were imputed using a last-observation-carried-forward method. All statistical analyses were conducted using SAS version 9.2 (SAS Institute, Cary, NC, USA).
Results
Early improvement in pain
Baseline characteristics of subgroups
At Week 4, 49% (108 of 219) of duloxetine-treated patients who did not withdraw during the first 4 weeks of the study had ≥30% pain reduction, 29% (63 of 219) had between 10% and 30% pain reduction, and 22% (48 of 219) had <10% pain reduction (Table 1). Baseline characteristics were not significantly different between the early improvement subgroups, except that a larger proportion of those with ≥30% pain reduction at Week 4 were women compared with the proportions in the other early improvement subgroups (P = 0.0122; Table 1).
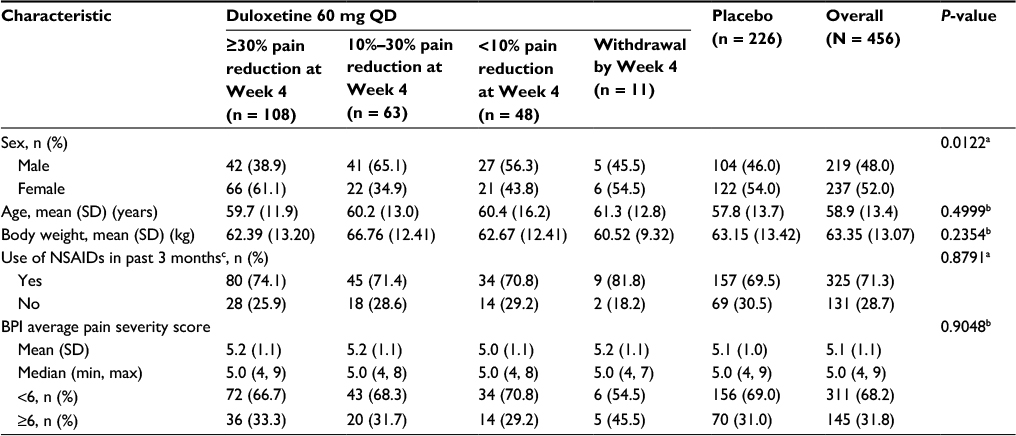 | Table 1 Demographics and baseline disease characteristics of patients with ≥30%, 10%–30%, and <10% reduction in Brief Pain Inventory average pain severity score at Week 4 Notes: aFisher exact test (global comparison across subgroups). bOne-way analysis of variance (global comparison across subgroups). cUse of NSAIDs for at least 14 days per month within the past 3 months before the study start. Abbreviations: BPI, Brief Pain Inventory; max, maximum; min, minimum; NSAIDs, nonsteroidal anti-inflammatory drugs; QD, once daily; SD, standard deviation. |
BPI average pain severity score
Compared with the placebo group, patients in the ≥30% early improvement subgroup had significantly greater decreases (ie, improvements) in BPI average pain severity score at all time points (Figure 1). In contrast, compared with the placebo group, patients in the <10% early improvement subgroup had significantly smaller decreases in BPI average pain severity score at all time points (Figure 1). There were no significant differences in the change from baseline in BPI average pain severity score between the 10%–30% early improvement subgroup and the placebo group (Figure 1).
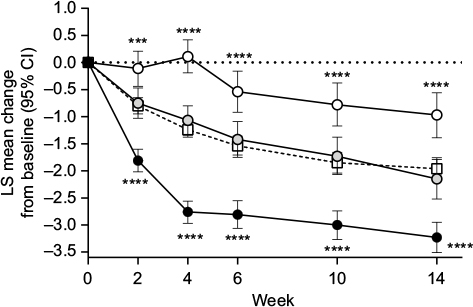 | Figure 1 Least squares mean (95% confidence interval) change from baseline in Brief Pain Inventory average pain severity score through Week 14 in patients with chronic low back pain treated with duloxetine 60 mg/day or placebo (white squares; n = 226). Notes: Duloxetine subgroups were patients who had ≥30% (black circles; n = 108), 10%–30% (gray circles; n = 63), or <10% pain reduction (white circles; n = 48) at Week 4. Mean (standard deviation) Brief Pain Inventory average pain severity scores at baseline were 5.2 (1.1), 5.2 (1.1), 5.0 (1.1), and 5.1 (1.0) for the ≥30% pain reduction, 10%–30% pain reduction, <10% pain reduction, and placebo groups, respectively. ***P ≤ 0.001 and ****P ≤ 0.0001 compared with placebo. Abbreviations: CI, confidence interval; LS, least squares. |
Response rates
The pain response at Week 4 was indicative of the pain response at Week 14 (Figure 2). Patients in the ≥30% early improvement subgroup had the highest response rates for both ≥30% and ≥50% pain reduction at Week 14 (P < 0.0001 versus placebo for both response rates), whereas patients in the <10% early improvement subgroup had the lowest response rates (P = 0.0007 versus placebo for ≥30% pain reduction, P = 0.0070 versus placebo for ≥50% pain reduction). Patients in the 10%–30% early improvement subgroup had a significantly higher ≥30% pain reduction response rate than the placebo group (P = 0.0422); the ≥50% pain reduction response rates did not differ between these 2 groups. In patients with ≥30% early improvement, 94.4% maintained ≥30% pain reduction and 82.4% achieved ≥50% pain reduction at Week 14 (Figure 2).
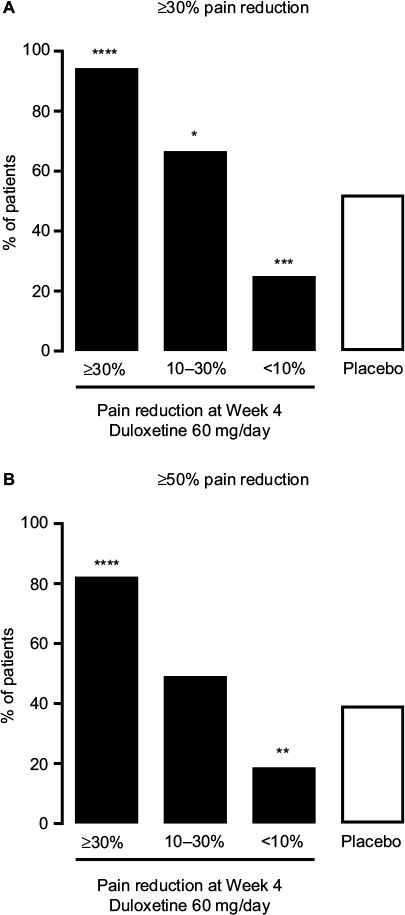 | Figure 2 Proportion of patients with chronic low back pain who achieved (A) ≥30% or (B) ≥50% pain reduction after 14 weeks of duloxetine 60 mg/day treatment (black bars) among those with ≥30% (n = 108), 10%–30% (n = 63), or <10% pain reduction (n = 48) at Week 4, and among those receiving placebo (white bar; n = 226). Note: *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 compared with placebo. |
BPI QOL measures
Compared with the placebo group, patients in the ≥30% early improvement subgroup had significantly greater decreases (improvements) in all BPI Interference (QOL) measures at most time points (Figure 3). For general activity and walking ability, significant differences were seen as early as Week 2. Compared with the placebo group, patients in the <10% early improvement subgroup had significantly smaller decreases in all BPI Interference measures at most time points (Figure 3). Patients in the <10% early improvement subgroup had small, transient increases in relationships with others and sleep scores at Weeks 2 and 4. There were no significant differences in the change from baseline in BPI Interference measures between the 10%–30% early improvement subgroup and the placebo group, except for sleep at Week 4 and enjoyment of life at Week 2 (Figure 3).
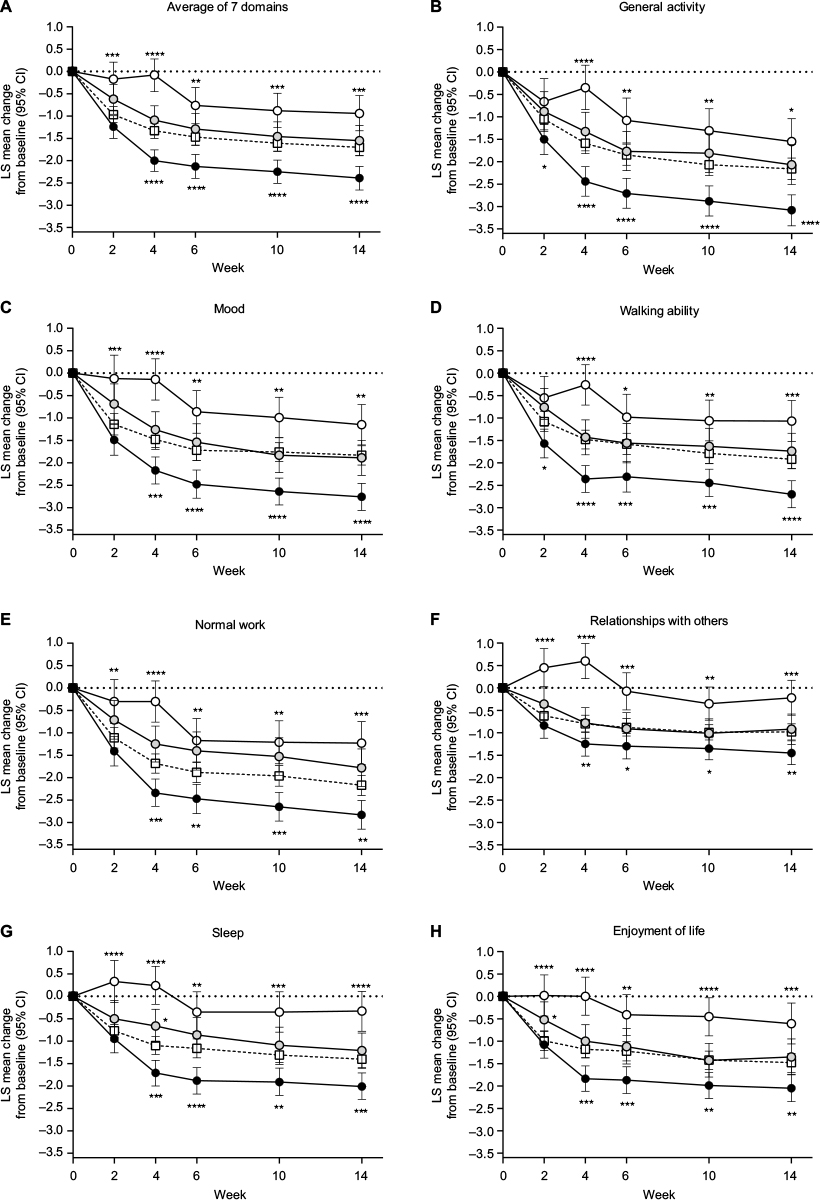 | Figure 3 Least squares mean (95% confidence interval) change from baseline in Brief Pain Inventory Interference quality-of-life domains through Week 14 in patients with chronic low back pain treated with duloxetine 60 mg/day or placebo (white squares; n = 226). Notes: Duloxetine subgroups were patients who had ≥30% (black circles; n = 108), 10%–30% (gray circles; n = 63), or <10% pain reduction (white circles; n = 48) at Week 4. Brief Pain Inventory Interference domains included (B) general activity, (C) mood, (D) walking ability, (E) normal work, (F) relationships with others, (G) sleep, and (H) enjoyment of life, as well as (A) the average of the 7 domains. *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 compared with placebo. Abbreviations: CI, confidence interval; LS, least squares. |
Early TR-AESIs
Baseline characteristics of subgroups
Approximately one-fifth (22%; 50 of 230) of duloxetine-treated patients had at least 1 TR-AESI in the first 2 weeks of treatment (Table 2). Baseline characteristics were generally similar between patients who did and did not have early TR-AESIs (Table 2).
 | Table 2 Demographics and baseline disease characteristics of patients with and without adverse events of special interest (nausea, somnolence, and constipation) in the first 2 weeks Notes: aFisher exact test (global comparison across subgroups). bOne-way analysis of variance (global comparison across subgroups). cUse of NSAIDs for at least 14 days per month within the past 3 months before the study start. Abbreviations: BPI, Brief Pain Inventory; max., maximum; min., minimum; NSAIDs, nonsteroidal anti-inflammatory drugs; QD, once daily; SD, standard deviation; TR-AESIs, treatment-related adverse events of special interest. |
Pattern of TR-AESIs
Most instances of the TR-AESIs (nausea, somnolence, and constipation) occurred within the first 2 weeks of duloxetine treatment (Figure 4). Most cases of nausea resolved within 2 weeks; however, most cases of somnolence and constipation continued for at least 4 weeks.
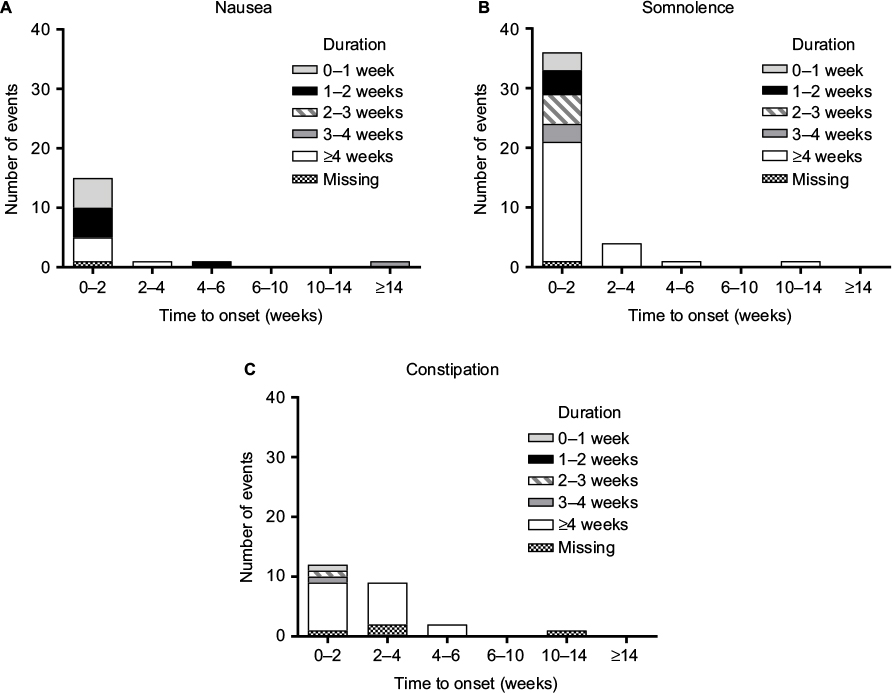 | Figure 4 Adverse events of special interest ([A] nausea, [B] somnolence, [C] constipation) in patients with chronic low back pain treated with duloxetine 60 mg/day for 14 weeks (n = 230). Note: Adverse events are shown according to the time to onset and the duration. |
BPI average pain severity score
Compared with the placebo group, patients without early TR-AESIs had significantly greater decreases (ie, improvements) in BPI average pain severity score at Weeks 4 and 14 (Figure 5). In addition, patients with early TR-AESIs had significantly greater decreases in BPI average pain severity score (compared with placebo) at all time points (Figure 5).
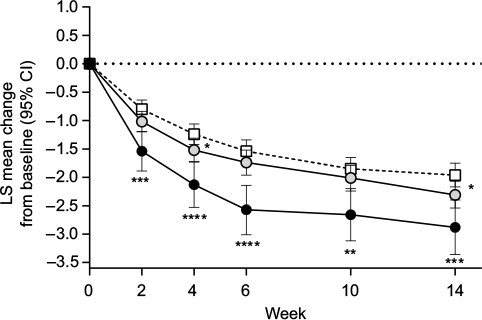 | Figure 5 Least squares mean (95% confidence interval) change from baseline in Brief Pain Inventory average pain severity score through Week 14 in patients with chronic low back pain treated with duloxetine 60 mg/day or placebo (white squares; n = 226). Notes: Duloxetine subgroups were patients who did (black circles; n = 50) or did not (gray circles; n = 180) have an adverse event of special interest (nausea, somnolence, constipation) between baseline and Week 2. Mean (standard deviation) Brief Pain Inventory average pain severity scores at baseline were 5.2 (1.1), 5.1 (1.1), and 5.1 (1.0) for the early adverse event, no early adverse event, and placebo groups, respectively. *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 compared with placebo. Abbreviations: CI, confidence interval; LS, least squares. |
Response rates
Both the ≥30% and ≥50% pain reduction response rates at Week 14 were significantly higher in duloxetine-treated patients than in placebo-treated patients, in both patients with early TR-AESIs (P = 0.0055 versus placebo for ≥30% pain reduction, P = 0.0016 versus placebo for ≥50% pain reduction) and in patients without early TR-AESIs (P = 0.0024 versus placebo for ≥30% pain reduction, P = 0.0026 versus placebo for ≥50% pain reduction) (Figure 6). Patients with early TR-AESIs had slightly higher response rates than patients without early TR-AESIs (Figure 6).
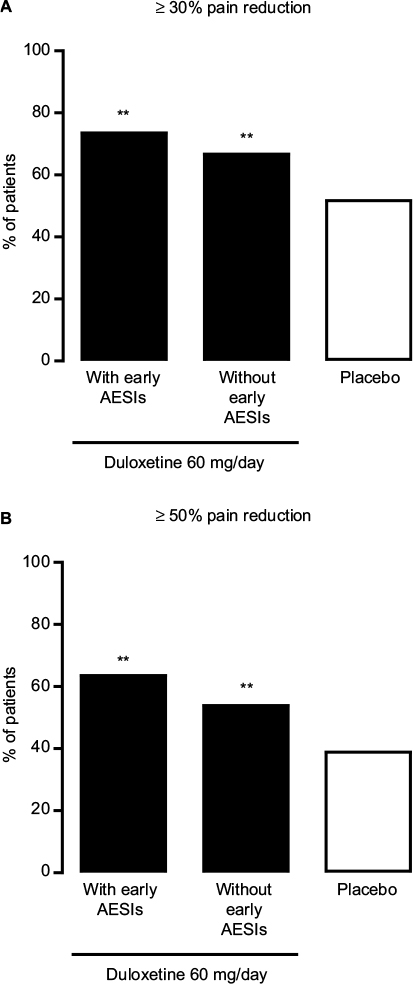 | Figure 6 Proportion of patients with chronic low back pain who achieved (A) ≥30% or (B) ≥50% pain reduction after 14 weeks of duloxetine 60 mg/day treatment (black bars) among those with (n = 50) or without (n = 180) adverse events of special interest between baseline and Week 2, and among those receiving placebo (white bar; n = 226). Note: **P ≤ 0.01 compared with placebo. Abbreviation: AESIs, adverse events of special interest. |
BPI QOL measures
Compared with the placebo group, patients with early TR-AESIs had significantly greater decreases (improvements) in BPI general activity (Week 6), mood (Weeks 6, 10, and 14), walking ability (Weeks 2, 4, 6, and 14), and enjoyment of life (Week 6) (Figure 7). There were no significant differences in the change from baseline in any BPI QOL measure between patients with no early TR-AESIs and the placebo group (Figure 7).
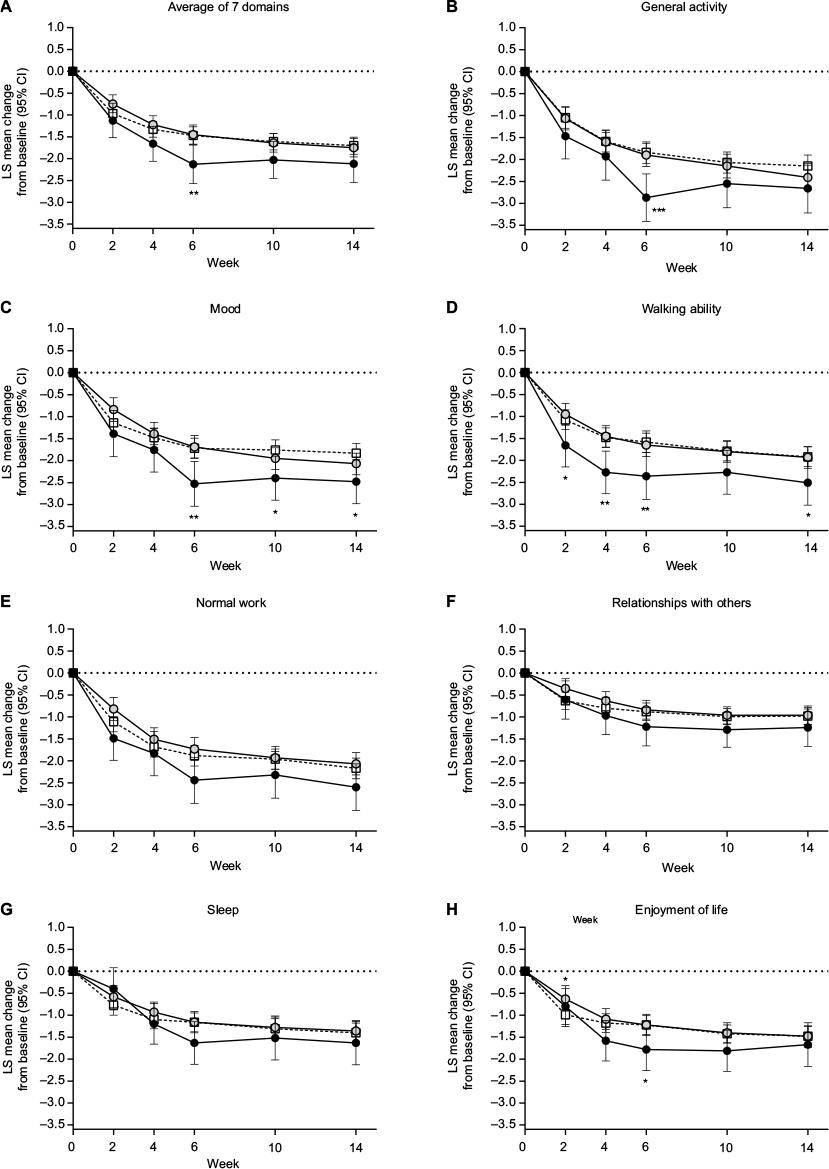 | Figure 7 Least squares mean (95% confidence interval) change from baseline in Brief Pain Inventory Interference quality-of-life domains through Week 14 in patients with chronic low back pain treated with duloxetine 60 mg/day or placebo (white squares; n = 226). Notes: Duloxetine subgroups were patients who did (black circles; n = 50) or did not (gray circles; n = 180) have an adverse event of special interest between baseline and Week 2. Brief Pain Inventory Interference domains included (B) general activity, (C) mood, (D) walking ability, (E) normal work, (F) relationships with others, (G) sleep, and (H) enjoyment of life, as well as (A) the average of the 7 domains. *P < 0.05, **P ≤ 0.01, and ***P ≤ 0.001 compared with placebo. Abbreviations: CI, confidence interval; LS, least squares. |
Discussion
In this post hoc analysis of a randomized, placebo-controlled trial, duloxetine-treated Japanese patients with CLBP who had early responses (at least 30% pain reduction at Week 4) or who had early TR-AESIs achieved greater pain reduction and improvements in QOL by the end of treatment than patients without these early signs of response. In addition, the proportion of patients achieving clinically significant levels of pain reduction (ie, ≥30% or ≥50%) was greatest in early responders. These results are consistent with previous responder analyses that evaluated the relationship between early and end-of-treatment pain reduction responses.13,16,17 However, this is the first analysis to evaluate the relationship between early AEs and pain reduction, and the first to examine the temporal relationship between pain reduction and QOL improvements. Monitoring the level of pain reduction and the occurrence of specific AEs within the first month of treatment may help physicians decide whether individual patients with CLBP are likely to derive clinical benefit from continuing duloxetine treatment.
Consistent with previous studies, patients with CLBP who responded well (≥30% pain reduction) during the first month of duloxetine treatment (early responders) had greater improvements in pain by the end of treatment than patients who responded poorly. Both the extent of pain reduction and the proportion of patients achieving clinically significant levels (≥30% and ≥50%) of pain reduction at the end of treatment were significantly greater in early responders than in the placebo group, suggesting that early responders may be “true responders” to duloxetine. Approximately half of duloxetine-treated patients were early responders. Notably, because of the dose titration during the first 2 weeks of the study, these patients achieved ≥30% pain reduction after only 2 weeks at the full treatment dose of 60 mg/day.
In a previous duloxetine responder analysis, the NNT to achieve 50% pain reduction in patients with CLBP was constant (at approximately 10) after the first 4 weeks of treatment.13 This result suggests that the relative proportion of patients benefitting from duloxetine treatment remains stable after the first month of treatment. In a post hoc analysis of duloxetine for CLBP, only 23% of patients with <10% pain reduction at Week 4 (early nonresponders) achieved ≥30% reduction, and only 9% achieved ≥50% reduction, at Week 12.17 These results are similar to those in our study, in which 25.0% of patients with <10% pain reduction at Week 4 achieved ≥30% reduction, and 18.8% achieved ≥50% reduction by the end of treatment. Early identification of nonresponders to duloxetine has also been reported in patients with fibromyalgia.16 Interestingly, early nonresponders in our study had significantly less pain reduction than the placebo group, most likely because the placebo group included some patients who responded despite not receiving active treatment. This placebo response is well documented in trials of chronic pain and may relate to the increased medical care associated with participation in a clinical trial, regression to the mean, or a genuine neurological response resulting from anticipation of pain reduction.18,19 Further, patients who have an early placebo response often go on to experience sustained pain reduction.20 In contrast, patients in the duloxetine group who had <10% pain reduction by Week 4 clearly had neither a placebo response nor a response to active treatment. Although the overall duloxetine group is likely to have also included patients with a placebo response, it is important to note that the primary trial demonstrated superiority of duloxetine over placebo.8 Our results, together with those of other studies, indicate that both true responders and nonresponders may be identified early, based on the level of pain reduction after 1 month of duloxetine treatment.
Compared with placebo, early responders also had significantly greater improvements in all BPI Interference QOL domains, whereas early nonresponders did not. The greatest changes were seen in the domains of general activity, walking ability, and normal work, with significant changes evident as early as 2 weeks. This result suggests that the rapid reduction in pain among early responders quickly translates to improved physical function, followed by improvements in other aspects of QOL (eg, mood and sleep). According to the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group, patients with CLBP rate all aspects of QOL and physical functioning as very important in evaluating the effectiveness of treatment.21 Early and sustained improvements in QOL evoked by an early reduction in pain severity may help patients maintain lower pain levels.
In the primary analysis of this trial, the most common duloxetine-related AEs were nausea, somnolence, and constipation, all of which occurred primarily in the first 2 weeks.8 We therefore focused on these early AEs when analyzing whether their occurrence was related to pain reduction response. Nausea has been attributed to increased serotonin in the central nervous system,22–24 somnolence is likely to involve complex effects on both serotonin and norepinephrine central pathways,25 and constipation is caused by increased norepinephrine in the gastrointestinal tract.26 Therefore, the occurrence of these TR-AESIs indicates that duloxetine is active at the target serotonin and norepinephrine reuptake transporters in these patients. Patients with longer-lasting TR-AESIs may be less likely to continue duloxetine treatment, unless these AEs are early signs that they will achieve greater pain reduction by the end of treatment.
Duloxetine-treated patients who did not have early TR-AESIs achieved significantly greater pain reduction than placebo-treated patients at Weeks 4 and 14. In contrast, patients with early TR-AESIs (approximately one-fifth of duloxetine-treated patients) achieved significantly greater pain reductions compared with placebo-treated patients at all time points. These differences in pain reduction between subgroups and the placebo group are also reflected in significant improvements in certain QOL measures, such as general activity, mood, walking ability, and enjoyment of life. Because patients who experienced early TR-AESIs also had greater and earlier pain reduction and improvements in QOL than those without TR-AESIs, the early appearance of nausea, somnolence, or constipation may be a sign that the patient is a “true responder” to duloxetine. This is supported by the somewhat higher response rates at Week 14 in patients with early TR-AESIs compared with patients without early TR-AESIs, although the Week 14 response rates in both these duloxetine subgroups were significantly higher than those in the placebo group. In clinical practice, patients who experience early AEs may be encouraged to persist with treatment, especially if there are also early signs of pain reduction. However, it is important to note that the absence of early AEs does not necessarily indicate that duloxetine will be ineffective at reducing pain. Indeed, although 50 patients in our study had early TR-AESIs, 108 patients were early responders who went on to have substantial pain reduction by the end of the trial. Therefore, if physicians base treatment decisions only on the presence or absence of early AEs, a large proportion of patients who could eventually benefit from duloxetine might discontinue the drug prematurely. Thus, both safety and efficacy responses during the early treatment period should be assessed when deciding whether to continue or discontinue duloxetine treatment.
Although post hoc in nature, the strengths of this analysis include the prospective, randomized, placebo-controlled, multicenter design of the original study, the assessment of pain and QOL measures at multiple time points, and the examination of the relationship between the timing of common AEs and subsequent pain reduction. We acknowledge that the limited study period of 14 weeks is insufficient to determine whether early pain reduction or early TR-AESIs can predict long-term benefits of duloxetine treatment. In addition, because this study enrolled only Japanese patients, it is unknown whether these early response or TR-AESIs are associated with overall response in other patient groups.
Conclusion
This post hoc analysis of a randomized placebo-controlled trial suggests that early pain reduction or early occurrence of AEs may be an important indicator for identification of patients who are likely to derive the greatest benefit from duloxetine treatment for CLBP. Both efficacy and safety responses in the early period may predict which patients with CLBP are most likely to respond to duloxetine with a clinically significant reduction in pain and improvements in QOL.
Acknowledgments
This study was sponsored by Shionogi & Co. Ltd, manufacturer of duloxetine. Shionogi & Co. Ltd was involved in the study design, data collection, data analysis, and preparation of the manuscript. Medical writing assistance was provided by Rebecca Lew, PhD, CMPP, and Hiroko Ebina of ProScribe – Envision Pharma Group, and was funded by Shionogi & Co. Ltd. ProScribe’s services complied with international guidelines for Good Publication Practice (GPP3). The authors would like to thank Mr S. Hayashi, Mr A. Noguchi, and Mr M. Furukawa for helpful advice and discussion.
Author contributions
All authors were study investigators, contributed toward data analysis, drafting, and critically revising the manuscript, approved the final version for publication, and agree to be accountable for all aspects of the work. TT, NI, MI, and TO were involved in the study design, MI and TO were involved in data collection, and TO conducted the statistical analysis.
Disclosure
TT, NI, MI, and TO are employees and minor stock holders of Shionogi & Co. Ltd. SK has received consulting fees and honoraria from Eli Lilly Japan K.K. and Shionogi & Co. Ltd, and has received research grants from Shionogi & Co. Ltd. The authors report no other conflicts of interest in this work.
References
Hoy D, Brooks P, Blyth F, Buchbinder R. The epidemiology of low back pain. Best Pract Res Clin Rheumatol. 2010;24(6):769–781. | ||
Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington (DC): National Academies Press; 2011. | ||
Webster LR, Markman J. Medical management of chronic low back pain: efficacy and outcomes. Neuromodulation. 2014;17 (Suppl 2):18–23. | ||
Dagenais S, Caro J, Haldeman S. A systematic review of low back pain cost of illness studies in the United States and internationally. Spine J. 2008;8(1):8–20. | ||
Nakamura M, Nishiwaki Y, Ushida T, Toyama Y. Prevalence and characteristics of chronic musculoskeletal pain in Japan. J Orthop Sci. 2011;16(4):424–432. | ||
Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386(9995):743–800. | ||
Smith HS, Smith EJ, Smith BR. Duloxetine in the management of chronic musculoskeletal pain. Ther Clin Risk Manag. 2012;8:267–277. | ||
Konno S, Oda N, Ochiai T, Alev L. A randomized, double-blind, placebo-controlled phase III trial of duloxetine monotherapy in Japanese patients with chronic low back pain. Spine (Phila Pa 1976). 2016;41(22):1709–1717. | ||
Skljarevski V, Desaiah D, Liu-Seifert H, et al. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine (Phila Pa 1976). 2010;35(13):E578–E585. | ||
Skljarevski V, Ossanna M, Liu-Seifert H, et al. A double-blind, randomized trial of duloxetine versus placebo in the management of chronic low back pain. Eur J Neurol. 2009;16(9):1041–1048. | ||
Skljarevski V, Zhang S, Desaiah D, et al. Duloxetine versus placebo in patients with chronic low back pain: a 12-week, fixed-dose, randomized, double-blind trial. J Pain. 2010;11(12):1282–1290. | ||
Dworkin RH, Turk DC, McDermott MP, et al. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. Pain. 2009;146(3):238–244. | ||
Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions--individual patient data responder analysis. Eur J Pain. 2014;18(1):67–75. | ||
Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9(2):105–121. | ||
Uki J, Mendoza T, Cleeland CS, Nakamura Y, Takeda F. A brief cancer pain assessment tool in Japanese: the utility of the Japanese Brief Pain Inventory--BPI-J. J Pain Symptom Manage. 1998;16(6):364–373. | ||
Wang F, Ruberg SJ, Gaynor PJ, Heinloth AN, Arnold LM. Early improvement in pain predicts pain response at endpoint in patients with fibromyalgia. J Pain. 2011;12(10):1088–1094. | ||
Williamson OD, Schroer M, Ruff DD, et al. Onset of response with duloxetine treatment in patients with osteoarthritis knee pain and chronic low back pain: a post hoc analysis of placebo-controlled trials. Clin Ther. 2014;36(4):544–551. | ||
Häuser W, Sarzi-Puttini P, Tölle TR, Wolfe F. Placebo and nocebo responses in randomised controlled trials of drugs applying for approval for fibromyalgia syndrome treatment: systematic review and meta-analysis. Clin Exp Rheumatol. 2012;30(6 Suppl 74):78–87. | ||
Häuser W, Bartram-Wunn E, Bartram C, Reinecke H, Tölle T. Systematic review: placebo response in drug trials of fibromyalgia syndrome and painful peripheral diabetic neuropathy-magnitude and patient-related predictors. Pain. 2011;152(8):1709–1717. | ||
Tuttle AH, Tohyama S, Ramsay T, et al. Increasing placebo responses over time in U.S. clinical trials of neuropathic pain. Pain. 2015;156(12):2616–2626. | ||
Turk DC, Dworkin RH, Revicki D, et al. Identifying important outcome domains for chronic pain clinical trials: an IMMPACT survey of people with pain. Pain. 2008;137(2):276–285. | ||
Johnston KD, Lu Z, Rudd JA. Looking beyond 5-HT(3) receptors: a review of the wider role of serotonin in the pharmacology of nausea and vomiting. Eur J Pharmacol. 2014;722:13–25. | ||
Shelton RC. Serotonin norepinephrine reuptake inhibitors: similarities and differences. Prim Psychiatry. 2009;16(5):25–35. | ||
Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord. 1998;51(3):215–235. | ||
DeMartinis NA, Winokur A. Effects of psychiatric medications on sleep and sleep disorders. CNS Neurol Disord Drug Targets. 2007;6(1):17–29. | ||
Stahl SM, Grady MM, Moret C, Briley M. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10(9):732–747. |
Supplementary materials
 | Table S1 The list of Institutional Review Boards |
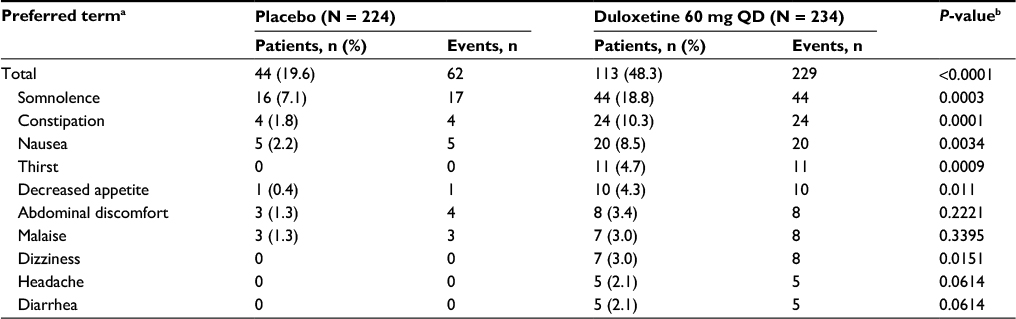 | Table S2 Adverse events related to study treatment (≥2% in either group) Notes: aMedical Dictionary for Regulatory Activities (MedDRA), Version 16.1. bFisher exact test. Abbreviation: QD, once daily. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.