Back to Journals » International Journal of Nanomedicine » Volume 17
Respiratory Exposure to Copper Oxide Particles Causes Multiple Organ Injuries via Oxidative Stress in a Rat Model
Authors Wang K, Ning X ![]() , Qin C, Wang J, Yan W, Zhou X, Wang D, Cao J, Feng Y
, Qin C, Wang J, Yan W, Zhou X, Wang D, Cao J, Feng Y ![]()
Received 15 June 2022
Accepted for publication 14 September 2022
Published 24 September 2022 Volume 2022:17 Pages 4481—4496
DOI https://doi.org/10.2147/IJN.S378727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Farooq A. Shiekh
Kaifang Wang,1,2 Xin Ning,1 Chuanyue Qin,1 Jianlin Wang,1 Wenjie Yan,1 Xin Zhou,1 Deping Wang,1 Jimin Cao,1 Yanlin Feng1
1Key Laboratory of Cellular Physiology at Shanxi Medical University, Ministry of Education, and the Department of Physiology, Shanxi Medical University, Taiyuan, People’s Republic of China; 2Department of Cardiology, First Clinical Medical College, Shanxi Medical University, Taiyuan, People’s Republic of China
Correspondence: Jimin Cao; Yanlin Feng, Email [email protected]; [email protected]
Introduction: The wide application of copper oxide nanoparticles (CuO NPs) in industry, agriculture, environmental remediation, and biomedicine has increased the risk of human exposure to CuO NPs. Recent studies suggested that CuO NPs have genotoxic and cytotoxic effects on various cells. However, little is known about the toxicity of CuO NPs on major peripheral organs after respiratory exposure.
Materials and Methods: We investigated the toxicities of CuO NPs on human bronchial epithelial (BEAS-2B) and human cardiomyocytes (AC16) cells in vitro, and on the lungs, liver, kidneys, and heart of spontaneously hypertensive rats (SHRs) at 24 and 72 h after intrabronchial instillation in vivo.
Results: CuO NPs induced concentration-dependent toxicities in both BEAS-2B and AC16 cells mainly through hierarchical oxidative stress mechanisms, involving generation of reactive oxygen species (ROS), upregulation of heme oxygenase-1 (HO-1), mitochondrial dysfunction, and secretion of proinflammatory and profibrogenic cytokines. Respiratory exposure to CuO NPs induced acute multiple organ injuries in SHRs manifesting through inflammation and fibrosis. However, cardiac injury was relatively less severe than injuries in the lungs, liver, and kidneys. Upregulation of serum C-reaction protein (CRP), tumor necrosis factor α (TNF-α), intercellular adhesion molecule 1 (ICAM-1), endothelin-1 (ET-1), angiotensin converting enzyme (ACE), and von Willebrand factor (vWF) after exposure to CuO NPs indicated systematic inflammation, endothelial injury, and potential prothrombosis.
Conclusion: Respiratory exposure to CuO NPs induced acute injuries in main peripheral organs, including the lungs, liver, kidneys, and heart. Individuals with existing cardiovascular diseases were susceptible to exposure to CuO NPs. This study provides a warning about the extensive toxic effects of CuO NPs, especially in the susceptible population.
Keywords: CuO nanoparticle, nanotoxicology, hierarchical oxidative stress response, intratracheal instillation, spontaneously hypertensive rat
Introduction
Epidemiological and clinical findings have indicated that inhalation of ultrafine particles (UFP, diameter <100 nm) increase the risk of respiratory and cardiovascular diseases in humans1,2 and aggravate their morbidity and mortality in susceptible populations.3–6 The extensive use of nanoparticles (NPs) in various fields during the past decades has increased human exposure, because NPs can enter into the body through respiration, skin contact, and the food chain.7 Among metal oxide NPs, copper oxide nanoparticles (CuO NPs) have been widely used in various fields, including the construction of semiconductors and electronic chips,8,9 wood preservation, face masks, antifouling paints,10–12 production of color ceramic glaze, polishing precise optical instruments,13,14 and as nanofertilizers15 and antibacterial agents,16 because of their special thermophysical, optical, mechanical, electrical, and antibacterial properties. During the production, transportation, and application process, CuO NPs might diffuse in the ambient air as aerosols and be inhaled into the lungs, thus, potentially causing severe hazard to humans.17 Therefore, it is important to evaluate the potential hazardous effects of the exposure of main organs, such as lungs, heart, liver, and kidneys, to CuO NPs and delineate its underlying molecular mechanisms.
Notably, NPs can interact with biological tissues and induce inflammation;18 the proinflammatory effects of NPs have been investigated in various models.19,20 In vitro toxicological studies have demonstrated that CuO NPs induce production of reactive oxygen species (ROS), oxidative stress, cytotoxicity, genotoxicity, and immunotoxicity.21–25 Moreover, the activation of the nucleotide-binding domain and leucine-rich repeat protein 3 (NLRP3) inflammasome and production of interleukin-1β (IL-1β) have been reported to contribute to the inflammatory response to CuO NPs.26 In vivo toxicological studies revealed that the lung is the prime target organ for CuO toxicity after inhalation, especially in occupational settings.27 Lei et al demonstrated that exposure to CuO NPs via intranasal delivery aggravated pulmonary inflammation and induced pulmonary fibrosis by promoting collagen accumulation in C57BL/6 mice.28 To simulate the inhalation of NPs in humans, Yokohira et al established an intratracheal instillation of CuO NPs in F344 male rats and found that CuO NPs induced the formation of pulmonary neoplastic lesions.29 Using the same method, Rani et al found that CuO NPs induced pulmonary fibrosis and granuloma by increasing the levels of alkaline phosphatase and expression of lactate dehydrogenase, whereas simultaneously decreasing the expression of dismutase and catalase in Wistar rats.30 Multiple studies have evaluated immunotoxicity31,32 and alterations in haematological,33 biochemical, urine chemistry, and histological indices of major organs33–35 after exposure to CuO NPs using different administration modes. Unfortunately, research on murine inhalation exposure to CuO NPs is somewhat limited, especially lacking evaluations on potential multiple organ injuries after respiratory exposure to CuO NPs.
Hypertension is a leading cause of cardiovascular incidents worldwide owing to the aging of the population and increased exposure to various lifestyle risk factors such as alcohol consumption, obesity, unhealthy diet, and physical inactivity.36 Approximately 1.39 billion people have hypertension, causing a 4.5% disease burden globally.37–39 Exposure to particles has been shown to trigger acute cardiac responses or promote chronic development of cardiovascular disorders.40 However, limited attention has been paid to the potential cardiovascular impacts of respiratory exposure to CuO NPs. Therefore, there is an urgent need to study the influence of CuO NPs on the respiratory system and the potential cardiovascular outcomes after respiratory exposure.
Considering that inhalation is the primary route of entry for CuO NPs, the adverse effects of CuO NPs on pulmonary and cardiovascular systems were investigated in normal human bronchial epithelial (BEAS-2B) cells41 and human cardiomyocyte (AC16) cells,42 which are representative lung and heart target cells for NPs. We demonstrated here that CuO NPs induced concentration-dependent toxicity in both BEAS-2B and AC16 cells mainly through oxidative-stress mechanisms, including generation of ROS, upregulation of heme oxygenase-1 (HO-1), mitochondrial dysfunction, and secretion of proinflammatory and profibrogenic cytokines. We further investigated the inflammatory and fibrotic responses of the lungs and hearts of mice after inhalation of CuO NPs. Spontaneously hypertensive rats (SHRs) have been widely used as a model of human hypertension to test antihypertensive drugs and analyze responses to a wide range of stimuli.43,44 Because SHRs are more susceptible to fine particles than normal rats,44–46 and fine particles enter the body usually via inhalation, it was rational to use SHRs to study the toxic effects of CuO NPs on major peripheral organs after respiratory exposure. Our findings indicated that respiratory exposure to CuO NPs triggered acute injuries in the lungs, heart, liver, and kidneys likely via hierarchical oxidative-stress signaling.
Materials and Methods
Cell Culture
The BEAS-2B human bronchial epithelial and AC16 human cardiomyocyte cell lines were purchased from ATCC and cultured in BEGM and DMEM media, respectively. Cells were cultured in an incubator at 37 ℃ containing 5% CO2. Cells at 70–80% confluency were passed by trypsin digestion every 1–3 d.
Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) Analysis to Determine Cu Dissolution
ICP-OES analysis was performed to determine Cu ion (Cu2+) dissolution in H2O, BEGM, or DMEM cell culture medium. Briefly, 20 μL of 5 mg/mL CuO NPs was added to 980 μL of H2O or culture medium followed by gentle shaking for 24 h. After centrifugation at 12,000 rpm for 20 min, 500 μL supernatant was used for ICP-OES measurement.
Hemolysis Assay
Fresh heparinized mouse blood was obtained and washed 6 times with PBS. After each wash, the supernatant was discarded. Suspended red blood cells (RBC) were diluted 10-fold with PBS. Triton X-100 at a stock concentration of 1% was used as positive control. A mixture of 300 µL diluted RBC suspension and 1200 µL PBS was used as a negative control. In addition, 300 µL CuO NPs at various concentrations (200, 100, 50, 25, 12.5, 6.2, 3.1, and 1.5 μg/mL) were suspended in 1200 µL PBS. These mixtures were incubated for 2 h with gentle shaking at 37 ℃. The absorbance of the obtained supernatants after centrifugation was measured at 541 nm to calculate the percentage of hemolysis.
Cell Viability Assay
BEAS-2B or AC16 cells were seeded into 96-well plates at a density of 104 cells per well, and grown overnight. The culture medium was exchanged with 100 μL of fresh medium containing 0–200 μg/mL CuO NPs. After 24 h incubation, a CCK-8 assay was performed to evaluate cytotoxicity. Briefly, 10 μL of CCK8 solution was added to each well and incubated for 2 h. Then, absorption at 490 nm was measured to calculate the cell activity. For live/dead cell staining, cells were washed thrice with PBS, stained with calcein AM (2 µmol/L) and propidium iodide (PI, 4 µmol/L) for 2 h, and visualized under a fluorescence microscope.
Measurement of Intracellular Level of Reactive Oxygen Species
Approximately 8×104 BEAS-2B or AC16 cells were seeded into 12-well plates for overnight growth. The culture medium was then replaced with 800 μL of fresh medium containing 0, 12.5, 25, 50, 100 and 200 μg/mL CuO NPs. After 6 h of incubation, H2DCFDA (10 µmol/L) was added to each well and incubated for another 2 h. Cells were then washed twice with PBS and visualized under a fluorescence microscope.
Transmission Electron Microscopy (TEM) Assay
TEM was used to examine the cellular uptake and intracellular localization of CuO NPs after exposure. Approximately 1.6×105 BEAS-2B or AC16 cells were seeded in each well of 6-well plate for 24 h growth. Cells were then treated with 25 μg/mL CuO NPs for 6 h. Cells were washed several times with PBS and fixed with 3% glutaraldehyde. After further fixation in 1% osmium tetroxide and dehydration in a series of acetone solutions, cells were embedded in epoxy resin and cut into ultrathin sections. Finally, sections were stained with foruranyl acetate and lead citrate and examined under TEM (JEM-1400).
Western Blotting
Western blotting was used to analyze the protein expression of HO-1. Approximately 1.6×105 BEAS-2B or AC16 cells were seeded into 6-well plates. After overnight growth, cells were treated with 0, 12.5, 25, 50, 100 and 200 μg/mL CuO NPs for 6 h. Cells were washed with PBS and lysed to measure the protein level in the culture supernatant using the Bradford method. Approximately 20 μg of total protein was electrophoresed in 12% SDS-PAGE and then transferred to a PVDF membrane. After blocking, the membrane was incubated with antihuman/mouse HO-1 monoclonal antibodies (1:1000, Abcam) overnight, followed by incubation with biotinylated secondary antibodies (1:1000, Beyotime) and HRP-conjugated avidin−biotin complex (1:1000, Beyotime), with TBST washes between incubations. After washing with TBST buffer, the membrane was visualized using a Bio-Rad imaging system.
Detection of Mitochondrial Membrane Potential and Superoxide Generation
Mitochondrial membrane potential and superoxide production were detected using JC-1 and Mitosox Red fluorescent dyes. Approximately 8×104 BEAS-2B or AC16 cells were seeded into 12-well plates for overnight growth. The culture medium was then replaced with 800 μL of fresh medium containing 0, 12.5, 50, 100 and 200 μg/mL CuO NPs. After 6 h incubation, cells were washed twice with PBS and then stained with JC-1 (5 μmol/L) or Mitosox Red dye (5 μmol/L) for 15 min. The nuclei were stained with Hoechst (5 μmol/L). Finally, cells were washed twice with PBS and visualized under a fluorescence microscope.
Enzyme Linked Immunosorbent Assay (ELISA) Assay
ELISA was used to quantify the levels of IL-6 and TNF-α released by cells. BEAS-2B or AC16 cells were seeded into 96-well plates at a density of 104 cells per well, and grown overnight. The culture medium was then replaced by 100 μL of fresh medium containing 0–200 μg/mL CuO NPs. After 24 h incubation, cell plates were centrifuged at 2000 rpm for 20 min to acquire the supernatant for measuring the levels of IL-6 or TNF-α using the IL-6 or TNF-α ELISA kits, according to the manufacturer’s instructions.
Animal Experiments
Male spontaneously hypertensive rats (SHRs) (220–250 g, 11–12 weeks old) purchased from Vital River Laboratory Animal Technology (Beijing, China) were selected as model animals to investigate the acute toxicity of CuO NPs in vivo. All procedures involving animals were approved by the Animal Research Ethics Committee of Shanxi Medical University and was in accordance with the guidelines for the Care and Use of Laboratory Animals (NIH, revised 2011) under the approval number SYDL2019012. SHRs were randomly assigned to the following groups (4 rats per group): control (24 h), CuO NPs (24 h), control (72 h), and CuO NPs (72 h). A CuO NPs dose of 2 mg/kg via intratracheal instillation was used to evaluate in vivo toxicity. SHRs were lightly anesthetized by intraperitoneal (i.p.) injection of 30–40 mg/kg sodium pentobarbital, and subsequently intratracheally instilled with 100 μL of CuO NPs (calculated according to 2 mg/kg body weight) or PBS (pH 7.4, negative control) using a ball tipped animal feeding needle. Rats were killed 24 h or 72 h after oropharyngeal aspiration and major peripheral organs (lungs, heart, liver, and kidneys) were harvested for further laboratory tests.
Preparation and Analysis of Bronchoalveolar Lavage Fluid (BALF)
The left bronchus was ligated and the right lung was utilized to obtain BALF by cannulating the trachea and lavaging the lung thrice with sterile PBS (27 mL/kg body weight). The recovered lavage fluid was centrifuged and the pellets were adhered onto microscopic slides for counting the number of neutrophil cells; BALF supernatants were stored at −80 ℃ for cytokine and chemokine analysis using respective kits.
Blood Analysis
The serum and plasma of SHRs were harvested according to the standard procedures of routine blood sampling. Approximately 8 mL of whole blood was collected from the abdominal aorta to acquire serum and plasma, which were stored at −80 ℃ avoiding repeated freeze/thaw cycles. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CRE), and urea nitrogen (BUN) were measured using corresponding assay kits, according to the manufacturer’s instructions. Serum levels of C-reaction protein (CRP), tumor necrosis factor α (TNF-α), intercellular cell adhesion molecule-1 (ICAM-1), endothelin-1 (ET-1), and angiotensin converting enzyme (ACE), and plasma levels of von Willebrand factor (vWF) were measured using commercial ELISA kits, according to the manufacturer’s instructions.
Histopathological Examination of Different Organ Tissues
The lungs, heart, liver, and kidneys of SHRs in each group were dissected and fixed in 4% paraformaldehyde, imbedded in paraffin, cut into 5 μm tissue sections, and mounted on slides. Routine hematoxylin and eosin (H&E) and Masson’s trichrome staining were performed. After staining, sections were photographed using an optical microscope.
Statistical Analysis
All data were expressed as the mean ± standard deviation (SD). All values were obtained from at least 3 independent experiments. Statistical significance was evaluated using one-way analysis of variance (ANOVA). Differences between groups were considered statistically significant when the p value was lower than 0.05.
Results and Discussion
Physicochemical Characterization of CuO NPs
We observed the morphological characteristics of CuO NPs using TEM, and found that most CuO NPs were nearly spherical in shape with a primary size of 50 ± 7.1 nm (Figure 1A). We noticed that CuO NPs were well dispersed in distilled water, BEGM, and DMEM cell culture medium, resulting in hydrodynamic sizes of 58.8 ± 4.1 nm, 91.3 ± 7.4 nm, and 98.5 ± 4.9 nm, respectively (Figure 1B). Zeta potential measurement showed that CuO NPs displayed negative surface charges of –13.3 ± 0.7 in distilled water, which became –26.1 ± 1.9 in BEGM and –25.4 ± 2.3 in DMEM cell culture medium (Figure 1C).
 |
Figure 1 Physicochemical characterization of CuO NPs. (A) TEM images of CuO NPs. (B–D) Hydrodynamic diameters, zeta potential and Cu ion dissolution of CuO NPs in distilled water, BEGM and DMEM cell culture medium, respectively. (E and F) DCF and APF fluorescence spectra of PBS, Cu ion and CuO NPs with different concentrations. |
The toxicity of NPs has been known to be closely associated with their physicochemical properties. As CuO NPs are a dissolvable nanomaterial, we used ICP-OES to compare the dissolution of CuO NPs in water, BEGM, and DMEM. To this end we incubated 100 μg/mL CuO NPs in water or culture medium at 37 ℃ for 24 h. We found that CuO NPs displayed <1% dissolution percentages in distilled water, whereas approximately 10% was dissolved in BEGM and DMEM, mainly because of the addition of fetal bovine serum (FBS) in the culture medium,47 as shown in Figure 1D. Then, we assessed the ability of CuO NPs to induce generation of ROS. We measured the levels of abiotic ROS and hydroxyl radicals (HO•) using 2′,7′-dichlorodihydrofluorescein diacetate (DCF) and 3′-(p-aminophenyl) fluorescein (APF) fluorescent dye. Figure 1E and F demonstrates the progressive dose-dependent fluorescence increase in the levels of DCF and APF; whereas pure Cu ion (200 μg/mL) did not influence the levels of fluorescence, suggesting the ability of CuO NPs for abundant generation of ROS and HO•.
Cytotoxicity of CuO NPs in BEAS-2B Human Bronchial Epithelial Cells
The ability of CuO NPs to engage in Cu ion dissolution and HO• generation under abiotic conditions suggested that CuO NPs could generate oxidative stress in vitro and in vivo. We thus performed cellular studies using BEAS-2B cells, which are representative lung target cells for NPs. We first investigated the uptake and intracellular distribution of CuO NPs in BEAS-2B cells using TEM. We observed that the structure of control (not exposed to CuO NPs) was normal (Figure 2A). However, endocytosis was observed in BEAS-2B cells after coculture with CuO NPs at 25 μg/mL for 6 h (Figure 2B). The internalized CuO NPs were mainly localized inside these endosome-like vesicles and did not accumulate on the cell surface. Then, we examined cell viability using a cell counting kit (CCK-8). We found that the viability of BEAS-2B cells was decreased in a dose-dependent manner after exposure to various concentrations of CuO NPs (0−200 μg/mL) (Figure 2C). To clarify the toxic effect of Cu ions, we used CuCl2 as a control. We detected that CuCl2 yielded only approximately a 20% decrease in cell viability even at the highest concentration (200 μg/mL) without an obvious concentration-dependent trend (Figure 2C). In comparison, CuO NPs induced an 80% decrease in cell viability even at a lower concentration (Figure 2C). These results suggested the Cu ions only mildly contributed to the decline of cell viability; this decline was mainly induced by CuO NPs, rather than dissolute pure Cu ions. Consistent with the CCK-8 assay result, calcein acetoxymethyl (AM)/propidium iodide live/dead cell staining assays further confirmed the dose-dependent decline in cell viability after exposure to CuO NPs (Figure 2D).
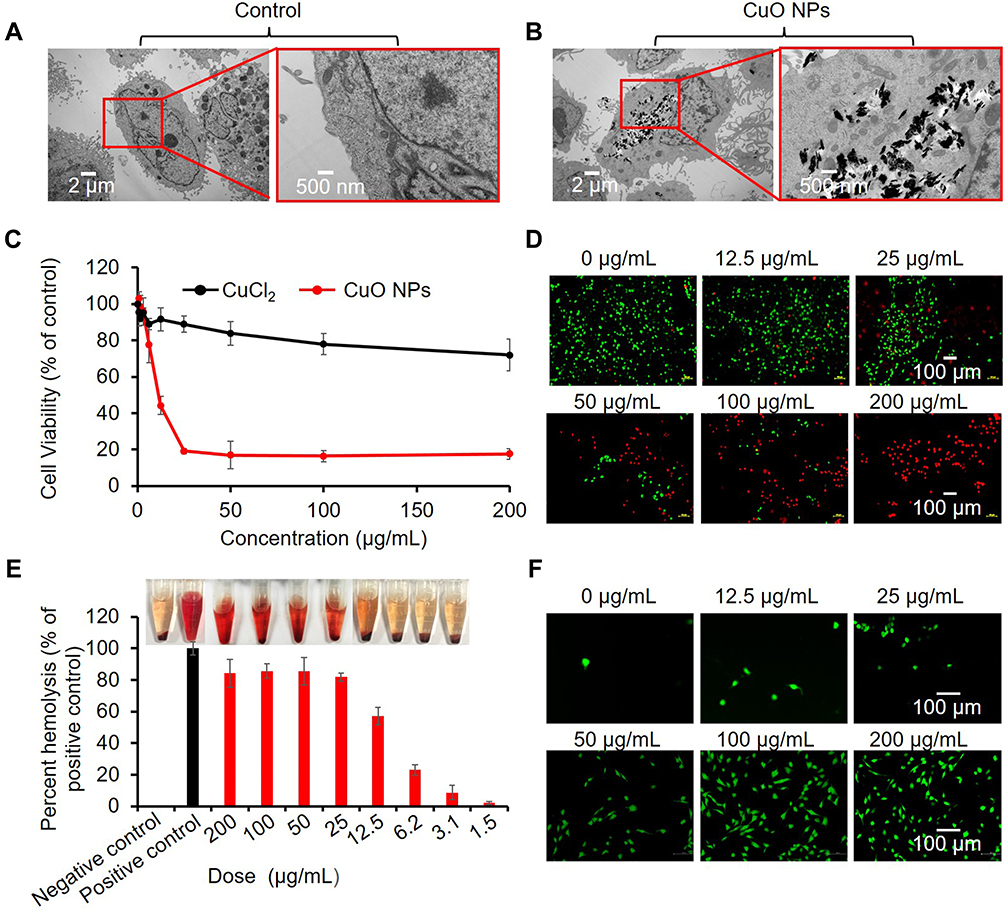 |
Figure 2 Cytotoxic properties of CuO NPs. (A) TEM images of BEAS-2B cells before exposure and (B) after exposure to 25 μg/mL CuO NPs for 6 h. (C) Cell viability assessment of Cu ions (CuCl2) and CuO NPs in BEAS-2B cells. Cells were exposed to serial concentrations (0, 0.4, 0.8, 1.5, 3.1, 6.25, 12.5, 25, 50, 100, and 200 μg/mL) of Cu ions or CuO NPs, and cell viability was assessed by the CCK-8 assay. (D) Live/dead staining of BEAS-2B cells treated with CuO NPs at 200, 100, 50, 25, 12.5 and 0 μg/mL for 24 h. The live and dead cells were stained with calcein AM (green) and PI (red), respectively. (E) Hemolytic effect of CuO NPs. Mouse RBCs were exposed to CuO NPs for 3 h. Inset picture showing hemoglobin appearance (red) in the RBC solution. (F) CLSM images of BEAS-2B cells reflecting intracellular ROS (DCF, green). |
We also performed a hemolysis assay on red blood cells (RBCs) to examine the potential hemolytic effect of CuO NPs. We incubated RBCs with CuO NPs (1.5–200 μg/mL) for 3 h, and determined the release of hemoglobin into the solution due to possible membrane rupture by measuring absorbance at 541 nm. We detected that CuO NPs exhibited a dose-dependent hemolytic effect, as indicated by the red color of the solution in the tubes (inset of Figure 2E). The hemolytic effect of CuO NPs might result from the generation of ROS, because HO• radicals as highly reactive species might interact with the cell membrane, resulting in cell death.48 We assessed the cellular levels of ROS using a dichlorofluorescein (DCFH) assay under confocal laser scanning microscopy (CLSM). CLSM images revealed that CuO NPs induced a dose-dependent enhancement in DCF fluorescence, indicating the abundant generation of ROS in BEAS-2B cells (Figure 2F).
Mechanism of Toxicity of CuO NPs Involving Hierarchical Oxidative Stress in BEAS-2B Cells
The production of intracellular ROS can trigger a series of oxidative stress reactions ranging from antioxidant defense response (Tier 1), initiation of inflammation (Tier 2), to mitochondrial-mediated cytotoxicity (Tier 3). Tier 1 is characterized by phase II enzymes with the most representative being heme oxygenase-1 (HO-1) (Tier 1) that plays a critical role in cytoprotection against ROS. Western blotting showed that CuO NPs increased the abundance of HO-1 in BEAS-2B cells proportionally to the exposure concentrations of CuO NPs (Figure 3A).
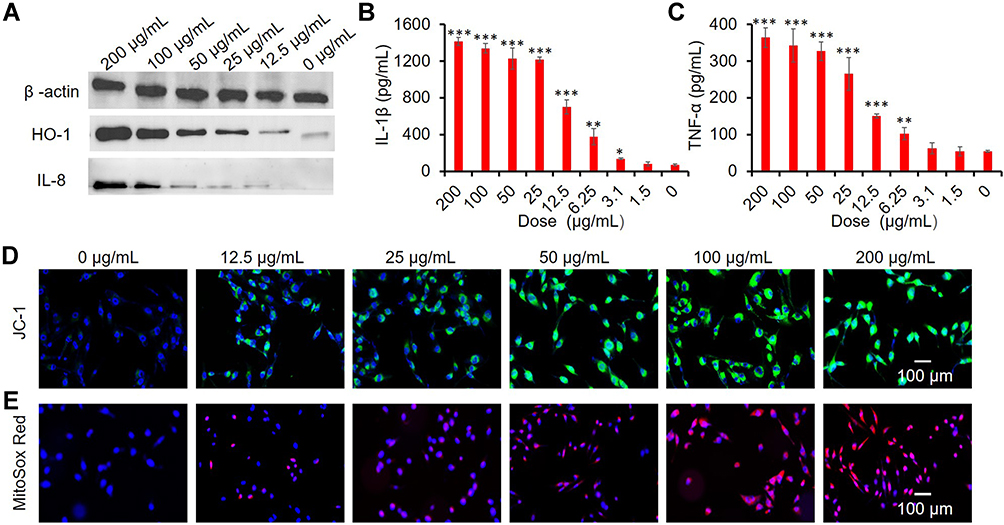 |
Figure 3 Toxicity mechanism of CuO NPs in BEAS-2B cells. (A) Western blots of HO-1, IL-8 expression. (B and C) ELISA results of IL-1β and TNF-α production. *p < 0.05, **p < 0.01, ***p < 0.001 vs control. (D) CLSM images showing mitochondrial membrane depolarization (JC-1, green). (E) Superoxide generation (Mitosox Red, red). Cell nuclei were stained with DAPI (blue). BEAS-2B cells were exposed to CuO NPs (0, 1.5, 3.1, 6.25, 12.5, 25, 50, 100, and 200 μg/mL) for 6 h. |
The Jun kinase (JNK) and NF-κB proinflammatory cascades are activated (Tier 2) after failure to restore redox equilibrium in Tier 1. In Tier 2, multiple proinflammatory cytokines (IL-1β, IL-8, IL-6, TNF-α, MIP-1, and MIP-2) are involved. We investigated the release of IL-8, IL-1β, and TNF-α, which is a typical proinflammatory response, to demonstrate the inflammatory effects of CuO NPs in BEAS-2B cells. Western blotting analysis of IL-8 (Figure 3A) and ELISA analysis of IL-1β and TNF-α (Figure 3B and C) indicated that CuO NPs significantly increased the levels of proinflammatory cytokines in a dose-dependent manner.
Upon failure of proinflammatory responses, oxidative stress responses are activated in Tier 3, which is associated with mitochondrial dysfunctions, such as generation of mitochondrial superoxide and depolarization of mitochondrial membrane, ultimately leading to initiation of programmed cell death. Here, we applied the JC-1 and Mitosox Red fluorescent dyes to detect the depolarization of mitochondrial membrane and production of mitochondrial superoxide, respectively. We found that exposure to CuO NPs significantly increased the intensities of JC-1 green fluorescence and Mitosox Red fluorescence in a concentration-dependent manner (Figure 3D and E), indicating that CuO NPs induced the depolarization of mitochondrial membrane potential and generation of superoxide.
Cytotoxicity of CuO NPs in AC16 Cells and Mechanism of Toxicity
We also examined the effects of CuO NPs on AC16 cells to estimate the potential toxicity of NPs on cardiomyocytes. We observed a substantial endocytosis of CuO NPs in AC16 cells compared with that in control AC16 cells not exposed to CuO NPs, as shown in Figure 4A and B. Both the CCK-8 (Figure 4C) and calcein acetoxymethyl (AM)/propidium iodide live/dead cell staining (Figure 4D) assays demonstrated similar cytotoxic effects of CuO NPs on AC16 cells. We specifically observed a burst generation of ROS in AC16 cells after exposure to CuO NPs (Figure 4E). We further tested the hierarchical oxidative stress response to CuO NPs in AC16 cells. Figure 4F shows that the levels of expression of HO-1 and IL-8 were proportional to the exposure concentrations of CuO NPs, whereas those of β-actin did not change. We observed similar effects of CuO NPs on the release of IL-1β and TNF-α from AC16 cells (Figures 4G and H). We also found that exposure of AC16 cells to CuO NPs resulted in the depolarization of the mitochondrial membrane potential and mitochondrial production of superoxide (Figures 4I and J).
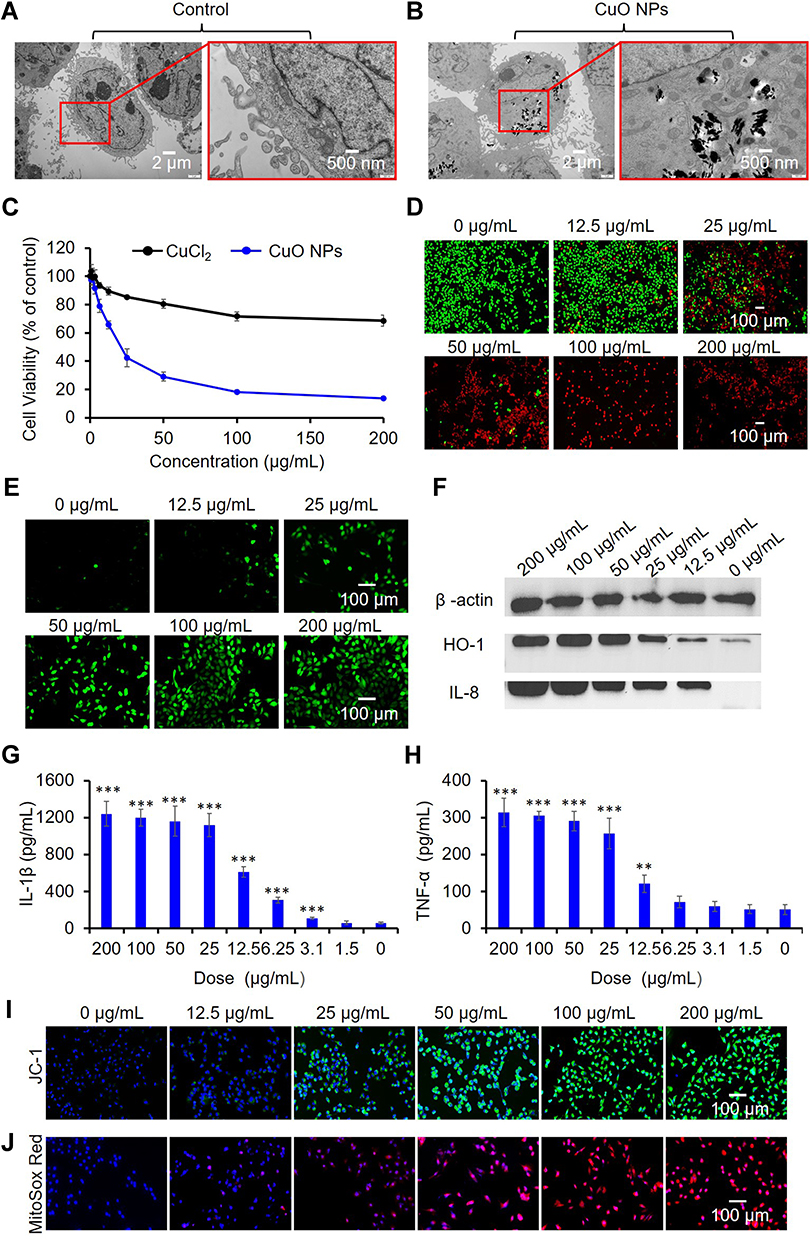 |
Figure 4 Cytotoxicity and underlying mechanism of CuO NPs in AC16 cells. (A) TEM images of AC16 cells before exposure and (B) after exposure to 25 μg/mL CuO NPs for 6 h. (C) Effects of Cu ions (CuCl2) and CuO NPs on the viability of AC16 cells. Cells were exposed to serial concentrations (0, 0.4, 0.8, 1.5, 3.1, 6.25, 12.5, 25, 50, 100, and 200 μg/mL) Cu ions or CuO NPs, and cell viability was assessed using the CCK-8 assay. (D) Live/dead staining of AC16 cells. The live and dead cells were stained with calcein AM (green) and PI (red), respectively. (E) CLSM images of AC16 cells to detect intracellular ROS (DCF, green). (F) Western blots of HO-1 and IL-8 expression. (G and H) ELISA analysis of IL-1β and TNF-α production. ** p < 0.01, *** p < 0.001 vs control. CLSM images showing (I) mitochondrial membrane depolarization (JC-1, green) and (J) elevated superoxide generation (Mitosox Red, red). Cell nuclei were stained with DAPI (blue). From D to J, AC16 cells were treated with CuO NPs at 200, 100, 50, 25, 12.5 and 0 μg/mL for 6 h. |
Taken together, the CuO NPs-induced escalation in the hierarchical oxidative stress from Tier 1 to Tier 2 and Tier 3 might underlie the cytotoxic effects of CuO NPs in both BEAS-2B and AC16 cells. The increase in the production of mitochondrial superoxide could be due to the generation of radical HO• on the surface of CuO NPs.
Acute Proinflammatory and Profibrogenic Effects of CuO NPs in the Lungs of Spontaneously Hypertensive Rats
The induction of proinflammatory cytokines (Tier 2 response) by CuO NPs in BEAS-2B cells suggested that CuO NPs might induce lung inflammation in vivo. We applied intratracheal instillation to observe the pulmonary effects of CuO NPs (2 mg/kg body weight) in SHRs, and animals were sacrificed 24 or 72 h after instillation. We collected BALF from the right lung and used it to evaluate the number of neutrophil cells and levels of cytokines. Neutrophils are characteristic cell markers of lung inflammation. Figure 5A shows that CuO NPs induced a statistically significant increase in neutrophil counts after exposure for 24 h. The neutrophil cell counts decreased to a certain degree after exposure to CuO NPs for 72 h but were still significantly higher than those in the PBS control group. Because myeloperoxidase (MPO) plays a key role in the function of neutrophils and is found in abundance, the activity of MPO can be used as a direct indicator of the number of neutrophils and an indirect indicator of lung injury. We detected that the activity of MPO was consistent with the neutrophil counts in BALF, as shown in Figure 5B. The levels of proinflammatory cytokines in BALF, including those of IL-8, TNF-α, and IL-1β, were significantly higher at 24 h than 72 h post-exposure (Figures 5C–E). The release of chemokines, such as IL-1β and TGF-β1, by injured epithelial cells attracts macrophages. TGF-β1 is a strong stimulator of fibrosis. Figure 5F shows a sharp increase in the level of TGF-β1 24 h after exposure to CuO NPs, followed by a decline in the level of TGF-β1, which was still significantly higher than that in the control 72 h post-exposure. As a sensitive marker of oxidative stress, the level of HO-1 in the BALF displayed a similar tendency (Figure 5G). As an increase in the levels of macrophage inflammatory protein 2 (MIP-2) often precedes the increase in the number of neutrophils, MIP-2 might be used to evaluate the recruitment of inflammatory cells. We found that the level of MIP-2 in the BALF was increased by approximately 1-fold after 24–72 h exposure to CuO NPs compared with that in the control (Figure 5H). These results indicated a rapid response of proinflammatory mediators after exposure to CuO NPs. We also observed an elevation in the levels of ET-1 in BALF at 24 h post-exposure (Figure 5I), suggesting endothelial dysfunction in pulmonary vessels after exposure to CuO NPs. We further visualized these characteristic features of acute lung inflammation and fibrosis using hematoxylin and eosin (H&E) (Figure 5J) and Masson’s trichrome (Figure 5K) staining of left lung sections. We observed noticeable accumulation of CuO NPs in the alveoli 24 h post-exposure (black arrows), which was accompanied by infiltration of neutrophils (multinuclear; red arrows), and mononuclear leukocytes (likely lymphocytes or macrophages; green arrows). We especially detected that the infiltration of inflammatory cells was greatly accelerated at 72 h, with the numbers of mononuclear leukocytes being significantly increased (Figure 5J). Masson’s trichrome staining of lung tissues further indicated that exposure of rats to CuO NPs via the respiratory tract caused serious lung fibrosis, as shown by the blue colored collagen (Figure 5K) compared with that in the control. All these in vivo results suggested that CuO NPs exerted acute proinflammatory and profibrogenic effects on the lungs, in consistency with our in vitro observations.
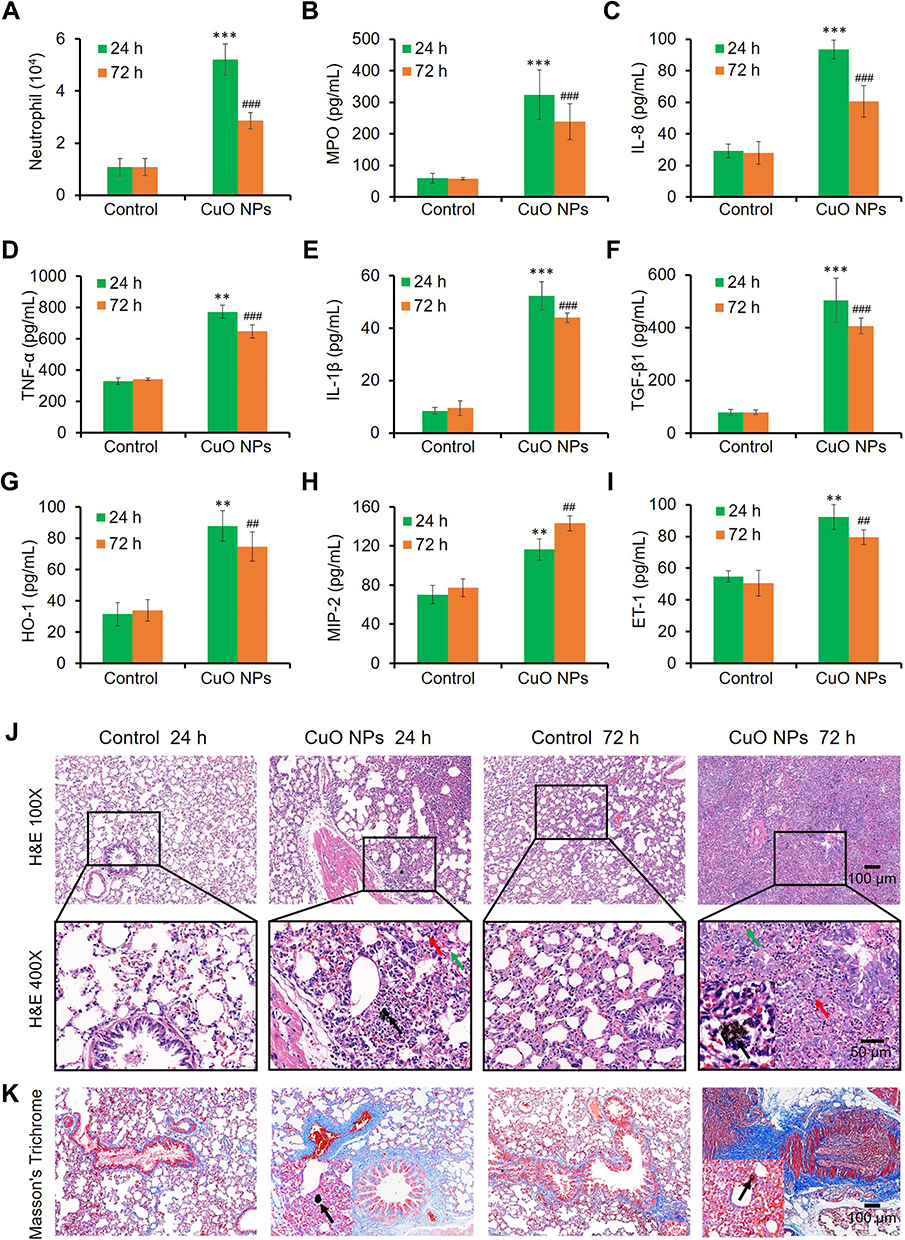 |
Figure 5 Acute pulmonary inflammation and fibrosis in SHRs induced by CuO NPs. (A) Neutrophil counts in the BALF. (B–I) MPO, IL-8, TNF-α, IL-1β, TGF-β1, HO-1, MIP-2, and ET-1 levels in the BALF, respectively. ** p < 0.01, *** p < 0.001 vs control at 24 h post-exposure. ##p < 0.01, ###p < 0.001 vs control at 72 h post-exposure. (J) H&E stains and (K) Masson’s trichrome stains of the left lung sections. Red arrows indicated neutrophils, green arrows indicated lymphocytes and/or macrophages, black arrows indicated CuO NPs. |
CuO NPs Induced Liver and Kidney Inflammation and Fibrosis in Spontaneously Hypertensive Rats
We also separated the sera from the blood of SHRs to perform biochemical assays. We found that intratracheal instillation of CuO NPs significantly and time-dependently increased the serum levels of alanine transferase (ALT) and aspartate transferase (AST) in SHRs compared with those in control SHRs (Figures 6A and B), suggesting that CuO NPs induced liver injury. We also evaluated indicators of kidney function, including the serum levels of creatinine (CRE) and blood urea nitrogen (BUN). We noticed a significant increment in the levels of CRE and BUN after exposure to CuO NPs compared with that in control (Figures 6C and D), suggesting that CuO NPs also induced kidney injury. We further evaluated liver and kidney inflammation and fibrosis features using H&E and Masson’s trichrome staining. In particular, H&E staining of liver tissues revealed that the liver of control animals showed a normal structure featured with radial arrangement of hepatocytes around the central vein (CV). However, exposure to CuO NPs induced serious liver injury, as manifested by liver structural disturbance, potential focal necrosis (disruption of cellular structure and absence of nuclei; circled areas), and CV congestion (green arrows) (Figure 6E) in a time–dependent manner. Likewise, Masson’s trichrome staining of liver tissues revealed the presence of remarkable fibrosis (blue color in interstitial areas) and smaller vein congestion (red color inside veins) (Figure 6E).
 |
Figure 6 Liver and kidney injuries induced by CuO NPs. (A) Serum alanine transferase (ALT) activity. (B) Serum aspartate transferase (AST) activity. (C) serum creatinine (CRE) level. (D) Blood urea nitrogen (BUN) level. * p < 0.05, ** p < 0.01 vs control at 24 h post-exposure. #p< 0.05, ##p < 0.01 vs control at 72 h post-exposure. (E) H&E and Masson’s trichrome stains of liver tissues. (F) H&E and Masson’s trichrome stains of kidney tissues. Blue arrows indicate smaller vein congestion, green arrows indicate CV congestion, black arrows indicate glomerular swelling. |
We observed that CuO NPs also induced acute kidney injury, as manifested by the time-dependent glomerular congestion and congestion of other smaller vessels (black arrows), and narrowing of the renal capsule, suggesting glomerular swelling especially at 72 h after exposure (blue arrows), as indicated by H&E staining (Figure 6F). Masson’s trichrome staining of kidney tissues revealed the presence of interstitial fibrosis (blue color), glomerular and smaller vein congestion (red color inside blood vessels), and potential cell swelling especially at 72 h after exposure (Figure 6F).
CuO NPs Induced Moderate Cardiac Inflammation and Fibrosis in Spontaneously Hypertensive Rats
As shown in our in vitro experiments, CuO NPs induced an abundant production of cytokines in AC16 cells, suggesting that CuO NPs might cause cardiac inflammation. We therefore performed in vivo experiments to examine the potential hazardous effect of CuO NPs on the hearts of SHRs. To this end, we measured the levels of serum biomarkers of inflammation, including CRP and TNF-α. We detected elevated levels of CRP and TNF-α after intratracheal instillation of CuO NPs for 24 and 72 h compared with those in control animals, as shown in Figure 7A and B. This finding indicated that CuO NPs induced moderate cardiac injury and fibrosis. In addition, we found that the serum levels of ICAM-1, ET-1, ACE, and plasma levels of vWF were also significantly elevated after exposure to CuO NPs for 72 h (Figures 7C–F), suggesting that respiratory exposure to CuO NPs induced systemic inflammation and endothelial injuries, potentially including damage to the coronary arteries. ET-1 is the most potent vasoconstrictive substance and plays a key role in the pathogenesis of hypertension. As an important target in the management of hypertension, ACE regulates blood pressure under normal conditions. Overexpression of ACE is known to raise blood pressure, resulting in damages in the heart, brain, and kidneys. vWF is an important factor in the blood coagulation signaling and plays important roles in body hemostasis and blood coagulation. Upon stress or tissue damage, the blood level of vWF increases. The time-dependent increases in the levels of ET-1, ACE, and vWF in response to exposure to CuO NPs suggested a trend of formation of cardiovascular lesions, including endothelial dysfunction, hypertension, and thrombosis. We further confirmed these cardiovascular injury features using H&E and Masson’s Trichrome staining of the myocardium. Accordingly, H&E staining mainly revealed the presence of increased perivascular and interstitial fibrosis in the myocardium after exposure to CuO NPs from 24 to 72 h (Figure 7G). We also observed coronary vein congestion (yellow arrows) and coronary arterial white thrombus (blue arrows) in the myocardium (Figure 7G). Likewise, Masson’s trichrome staining showed similar changes to those observed with H&E staining, especially cardiac fibrosis and coronary congestion (Figure 7H). Overall, we noticed that the cardiac injury induced by CuO NPs was less severe than that in the lungs, liver, and kidneys. Nonetheless, the increased blood levels of CRP, TNF-α, ICAM-1, ET-1, ACE, and vWF might suggest systematic inflammation, endothelial injury, and prothrombosis to some extent at the molecular level.
 |
Figure 7 Moderate cardiac injury induced by CuO NPs in SHRs. Serum levels of some biochemical parameters related to inflammation, coagulation, cell junction and vasomotor were measured. (A) CRP. (B) TNF-α. (C) ICAM-1. (D) ET-1. (E) ACE. (F) vWF. * p < 0.05, ** p < 0.01 vs control at 24 h post-exposure. #p < 0.05, ##p < 0.01 vs control at 72 h post-exposure. (G) H&E and (H) Masson’s trichrome stains of the myocardial injuries manifested with inflammation (blue color), coronary vein congestion and susceptible coagulation (yellow arrows), and fibrosis (blue color). |
Conclusion
This study demonstrated that respiratory exposure to CuO NPs induced biotoxicities at the cellular and organismal levels. We found that these underlying mechanisms were correlated with hierarchical oxidative-stress mechanisms, involving the generation of ROS, upregulation of HO-1, mitochondrial dysfunction, and production of proinflammatory and profibrogenic cytokines. In vivo, intranasal exposure of SHR animals to CuO NPs for 24–72 h induced multiple organ injuries, including in the lungs, heart, liver, and kidneys. These organ injuries were more serious in the lungs, liver, and kidneys than in the heart, as manifested by acute inflammation and fibrosis. This study provided a biosafety warning that special attention should be paid to the professional or medical application of CuO NPs, and also suggested that individuals with existing cardiovascular diseases, such as hypertension, are very susceptible to exposure to CuO NPs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (22007063, 82170523), Shanxi Medical Key Science and Technology Project Plan of China (2020XM01), Natural Science Foundation of Shanxi (20210302124302) and partially by Shanxi “1331 Project” Quality and Efficiency Improvement Plan (1331KFC). The authors would like to thank Chengdu Lilai Biotechnology Co., Ltd. for their technical assistance. We would like to thank Editage (www.editage.cn) for English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Donaldson K, Stone V, Seaton A, MacNee W. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ Health Perspect. 2001;109(Suppl 4):523–527. doi:10.1289/ehp.01109s4523
2. Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N Engl J Med. 2000;343(24):1742–1749. doi:10.1056/NEJM200012143432401
3. Elder A, Couderc JP, Gelein R. Effects of on-road highway aerosol exposures on autonomic responses in aged, spontaneously hypertensive rats. Inhal Toxicol. 2007;19(1):1–12. doi:10.1080/08958370600985735
4. Rückerl R, Phipps RP, Schneider A. Ultrafine particles and platelet activation in patients with coronary heart disease–results from a prospective panel study. Part Fibre Toxicol. 2007;4(1):1–14. doi:10.1186/1743-8977-4-1
5. Liu LJS, Box M, Kalman D. Exposure assessment of particulate matter for susceptible populations in Seattle. Environ Health Perspect. 2003;111(7):909–918. doi:10.1289/ehp.6011
6. Gauderman WJ, Avol E, Gilliland F. The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med. 2004;351(11):1057–1067. doi:10.1056/NEJMoa040610
7. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today. 2015;10(4):487–510. doi:10.1016/j.nantod.2015.06.006
8. Bochenkov VE, Sergeev GB. Adsorption, catalysis, and reactions on the surfaces of metal nano-oxides. Catal Ind. 2010;2(1):1–10. doi:10.1134/S2070050410010010
9. Ebrahimnia-Bajestan E, Niazmand H, Duangthongsuk W, Wongwises S. Numerical investigation of effective parameters in convective heat transfer of nanofluids flowing under a laminar flow regime. Int J Heat Mass Transfer. 2011;54(19):4376–4388. doi:10.1016/j.ijheatmasstransfer.2011.05.006
10. Gabbay J, Borkow G, Mishal J, Magen E, Zatcoff R, Shemer-Avni Y. Copper oxide impregnated textiles with potent biocidal activities. J Ind Text. 2006;35(4):323–335. doi:10.1177/1528083706060785
11. Batley GE, Kirby JK, McLaughlin MJ. Fate and risks of nanomaterials in aquatic and terrestrial environments. Acc Chem Res. 2013;46(3):854–862. doi:10.1021/ar2003368
12. Zhao Z, Ukidve A, Krishnan V. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv Drug Delivery Rev. 2019;143:886–902.
13. Reinosa JJ, Romero JJ, Jaquotot P, Bengochea MA, Fernández JF. Copper based hydrophobic ceramic nanocoating. J Eur Ceram Soc. 2012;32(2):277–282. doi:10.1016/j.jeurceramsoc.2011.08.013
14. Zhang D-W, Yi T-H, Chen C-H. Cu nanoparticles derived from CuO electrodes in lithium cells. Nanotechnology. 2005;16(10):2338–2341. doi:10.1088/0957-4484/16/10/057
15. Borkow G, Sidwell RW, Smee DF. Neutralizing viruses in suspensions by copper oxide-based filters. Antimicrob Agents Chemother. 2007;51(7):2605–2607. doi:10.1128/AAC.00125-07
16. Din MI, Arshad F, Hussain Z, Mukhtar M. Green adeptness in the synthesis and stabilization of copper nanoparticles: catalytic, antibacterial, cytotoxicity, and antioxidant activities. Nanoscale Res Lett. 2017;12(1):638. doi:10.1186/s11671-017-2399-8
17. Larsen ST, Jackson P, Poulsen SS. Airway irritation, inflammation, and toxicity in mice following inhalation of metal oxide nanoparticles. Nanotoxicology. 2016;10(9):1254–1262. doi:10.1080/17435390.2016.1202350
18. Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311(5761):622–627. doi:10.1126/science.1114397
19. Yuan X, Zhang X, Sun L, Wei Y, Wei X. Cellular toxicity and immunological effects of carbon-based nanomaterials. Part Fibre Toxicol. 2019;16(1):18. doi:10.1186/s12989-019-0299-z
20. Leso V, Fontana L, Iavicoli I. Nanomaterial exposure and sterile inflammatory reactions. Toxicol Appl Pharmacol. 2018;355:80–92. doi:10.1016/j.taap.2018.06.021
21. Karlsson HL, Gustafsson J, Cronholm P, Möller L. Size-dependent toxicity of metal oxide particles—A comparison between nano- and micrometer size. Toxicol Lett. 2009;188(2):112–118. doi:10.1016/j.toxlet.2009.03.014
22. Ahamed M, Siddiqui MA, Akhtar MJ, Ahmad I, Pant AB, Alhadlaq HA. Genotoxic potential of copper oxide nanoparticles in human lung epithelial cells. Biochem Biophys Res Commun. 2010;396(2):578–583. doi:10.1016/j.bbrc.2010.04.156
23. Fahmy B, Cormier SA. Copper oxide nanoparticles induce oxidative stress and cytotoxicity in airway epithelial cells. Toxicol in Vitro. 2009;23(7):1365–1371. doi:10.1016/j.tiv.2009.08.005
24. Ahamed M, Akhtar MJ, Alhadlaq HA, Alrokayan SA. Assessment of the lung toxicity of copper oxide nanoparticles: current status. Nanomedicine. 2015;10(15):2365–2377. doi:10.2217/nnm.15.72
25. Hanagata N, Zhuang F, Connolly S, Li J, Ogawa N, Xu M. Molecular responses of human lung epithelial cells to the toxicity of copper oxide nanoparticles inferred from whole genome expression analysis. ACS Nano. 2011;5(12):9326–9338. doi:10.1021/nn202966t
26. Tao X, Wan X, Wu D, Song E, Song Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A.1 macrophage. J Hazard Mater. 2021;411:125134. doi:10.1016/j.jhazmat.2021.125134
27. Stern ST, McNeil SE. Nanotechnology safety concerns revisited. Toxicol Sci. 2008;101(1):4–21. doi:10.1093/toxsci/kfm169
28. Lai X, Zhao H, Zhang Y. Intranasal delivery of copper oxide nanoparticles induces pulmonary toxicity and fibrosis in C57BL/6 mice. Sci Rep. 2018;8(1):4499. doi:10.1038/s41598-018-22556-7
29. Yokohira M, Hashimoto N, Yamakawa K. Lung carcinogenic bioassay of CuO and TiO(2) nanoparticles with intratracheal instillation using F344 male rats. J Toxicol Pathol. 2009;22(1):71–78. doi:10.1293/tox.22.71
30. Rani VS, Kumar AK, Kumar CP, Reddy ARN. Pulmonary toxicity of copper oxide (CuO) nanoparticles in rats. J Med Sci. 2013;13:571–577. doi:10.3923/jms.2013.571.577
31. Yaqub A, Anjum KM, Munir A. Evaluation of acute toxicity and effects of sub-acute concentrations of copper oxide nanoparticles (CuO-NPs) on hematology, selected enzymes and histopathology of liver and kidney in Mus musculus. Indian J Anim Res. 2017;52:92–98.
32. Assadian E, Zarei MH, Gilani AG, Farshin M, Degampanah H, Pourahmad J. Toxicity of copper oxide (CuO) nanoparticles on human blood lymphocytes. Biol Trace Elem Res. 2018;184(2):350–357. doi:10.1007/s12011-017-1170-4
33. Manna P, Ghosh M, Ghosh J, Das J, Sil PC. Contribution of nano-copper particles to in vivo liver dysfunction and cellular damage: role of IκBα/NF-κB, MAPKs and mitochondrial signal. Nanotoxicology. 2012;6(1):1–21. doi:10.3109/17435390.2011.552124
34. Mohammadyari A, Razavipour ST, Mohammadbeigi M. Exploring vivo toxicity assessment of copper oxide nanoparticle in Wistar rats. J Biol Today World. 2014;3:124.
35. Lei R, Wu C, Yang B. Integrated metabolomic analysis of the nano-sized copper particle-induced hepatotoxicity and nephrotoxicity in rats: a rapid in vivo screening method for nanotoxicity. Toxicol Appl Pharmacol. 2008;232(2):292–301. doi:10.1016/j.taap.2008.06.026
36. Stanaway JD, Afshin A, Gakidou E, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392(10159):1923–1994.
37. Erdely A, Hulderman T, Salmen R. Cross-talk between lung and systemic circulation during carbon nanotube respiratory exposure. potential biomarkers. Nano Lett. 2009;9(1):36–43. doi:10.1021/nl801828z
38. Li Z, Hulderman T, Salmen R. Cardiovascular effects of pulmonary exposure to single-wall carbon nanotubes. Environ Health Perspect. 2007;115(3):377–382. doi:10.1289/ehp.9688
39. Mills KT, Bundy JD, Kelly TN. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134(6):441–450. doi:10.1161/CIRCULATIONAHA.115.018912
40. Kodavanti UP, Schladweiler MC, Ledbetter AD. The spontaneously hypertensive rat as a model of human cardiovascular disease: evidence of exacerbated cardiopulmonary injury and oxidative stress from inhaled emission particulate matter. Toxicol Appl Pharmacol. 2000;164(3):250–263. doi:10.1006/taap.2000.8899
41. Garcia-Canton C, Minet E, Anadon A. Metabolic characterization of cell systems used in in vitro toxicology testing: lung cell system BEAS-2B as a working example. Toxicol In Vitro. 2013;27(6):1719–1727. doi:10.1016/j.tiv.2013.05.001
42. Feng L, Yang X, Asweto CO. Low-dose combined exposure of nanoparticles and heavy metal compared with PM (2.5) in human myocardial AC16 cells. Environ Sci Pollut Res Int. 2017;24(36):27767–27777. doi:10.1007/s11356-017-0228-3
43. Bagate K, Meiring JJ, Gerlofs-Nijland ME. Vascular effects of ambient particulate matter instillation in spontaneous hypertensive rats. Toxicol Appl Pharmacol. 2004;197(1):29–39. doi:10.1016/j.taap.2004.02.005
44. Cao Q, Zhang S, Dong C. Pulmonary responses to fine particles: differences between the spontaneously hypertensive rats and Wistar Kyoto rats. Toxicol Lett. 2007;171(3):126–137. doi:10.1016/j.toxlet.2007.05.007
45. Vanhoutte PM. Endothelial dysfunction and atherosclerosis. Eur Heart J. 1997;18(Suppl):19–29. doi:10.1016/S0195-668X(97)90005-1
46. Badr K, Wainwright CL. Inflammation in the cardiovascular system: here, there and everywhere. Curr Opin Pharmacol. 2004;4(2):107–109. doi:10.1016/j.coph.2004.01.004
47. Wang Z, von Dem Bussche A, Kabadi PK, Kane AB, Hurt RH. Biological and environmental transformations of copper-based nanomaterials. ACS Nano. 2013;7(10):8715–8727. doi:10.1021/nn403080y
48. Weidinger A, Kozlov AV. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules. 2015;5(2):472–484. doi:10.3390/biom5020472
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.