Back to Journals » Biologics: Targets and Therapy » Volume 8
Resistant mutations in CML and Ph+ALL – role of ponatinib
Authors Miller GD, Bruno BJ, Lim C
Received 14 June 2014
Accepted for publication 19 July 2014
Published 20 October 2014 Volume 2014:8 Pages 243—254
DOI https://doi.org/10.2147/BTT.S50734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Geoffrey D Miller, Benjamin J Bruno, Carol S Lim
Department of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, University of Utah, Salt Lake City, Utah, USA
Abstract: In 2012, ponatinib (Iclusig®), an orally available pan-BCR-ABL tyrosine kinase inhibitor (TKI) developed by ARIAD Pharmaceuticals, Inc., was approved by the US Food and Drug Administration for use in resistant or intolerant chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL). Ponatinib is the only approved TKI capable of inhibiting BCR-ABL with the gatekeeper T315I kinase domain mutation, known to be the cause for 20% of resistant or relapsed CML cases. In 2013, ponatinib sales were temporarily suspended due to serious side effects seen in nearly 12% of the patient population. These side effects are thought to stem from the potent nature and pan-activity of this TKI. ARIAD Pharmaceuticals, Inc. has since been permitted to resume sales and marketing of ponatinib to a limited patient population with an expanded black box warning. In the following review, the use of ponatinib in CML and Ph+ALL will be discussed. Mechanisms of resistance in CML are discussed, which provide insight and background into the need for this third generation TKI, followed by the molecular design and pharmacology of ponatinib, which lead to its success as a therapeutic. Finally, the efficacy, safety, and tolerability of ponatinib will be highlighted, including summaries of the important clinical trials involving ponatinib as well as its current place in therapy.
Keywords: BCR-ABL, T315I, Ph+ALL, PACE trial, EPIC trial, ARIAD, compound mutations
A Letter to the Editor has been received and published for this article.
Management issues in CML and Ph+ALL
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia (Ph) chromosome, which results from a chromosomal translocation between chromosomes 9 and 22. This genetic aberration in turn forms the fusion oncoprotein tyrosine kinase BCR-ABL, whose kinase activity is responsible for the constitutive activation of growth, survival, and proliferation signaling pathways seen in CML cells.1–3 The end result is an expansion of functionally normal cells of the myeloid lineage causing a leukemia phenotype. Inhibition of the BCR-ABL kinase domain results in leukemic cell death and often successful management of the disease.4
The Ph chromosome (and thus, BCR-ABL) is also present in some cases of acute lymphoblastic leukemia referred to as Ph chromosome-positive acute lymphoblastic leukemia (Ph+ALL), which is a subset of leukemia characterized by an expansion of immature cells from the lymphocyte lineage. Unlike CML, only 20%–30% of adult cases and 3%–5% of childhood cases contain the Ph chromosome and subsequent BCR-ABL protein.5 Specifically, presence of the Ph chromosome in ALL correlates with a less favorable prognosis.6 Whereas in CML the leukemic cells are normally dependent on BCR-ABL signaling for growth and survival, solely inhibiting the BCR-ABL tyrosine kinase in Ph+ALL is not sufficient to inhibit leukemic cell growth.7 Thus, more broad, intense therapies are needed to eradicate Ph+ALL.
Current treatments of CML and Ph+ALL
In 2001, imatinib mesylate (Gleevec®), a small molecule targeting the adenosine triphosphate (ATP)-binding region of the BCR-ABL tyrosine kinase domain, was US FDA approved for first-line treatment of CML.8–10 While the introduction of this drug aided in improving 10-year survival rates to 80% to 90%,11 not all patients responded to imatinib therapy.9 In many of these cases, resistance was due to the presence or acquisition of mutations in the kinase domain that prevented imatinib binding and activity. Thus, second and third generation tyrosine kinase inhibitors (TKIs) were designed and developed to overcome this resistance or intolerance. Dasatinib (Sprycel®), a dual-Src family kinase inhibitor, and nilotinib (Tasigna®), a more potent BCR-ABL inhibitor, were approved for CML therapy in patients who were intolerant or nonresponsive to imatinib in 2006 and 2007, respectively. Each of these inhibitors was designed to inhibit a broader range of kinases, thereby exhibiting greater activity against a number of kinase domain mutations that could not be inhibited by imatinib.4
Currently, newly diagnosed CML patients in the chronic phase have the option of beginning treatment on imatinib, dasatinib, or nilotinib. A majority of patients will initially respond to one of these treatments. However, in nearly one-third of patients, first-line TKI treatment will eventually become ineffective, forcing patients to start on another TKI.12 Most patients will convert to either nilotinib or dasatinib. However, for some patients, especially those possessing the T315I kinase domain mutation, ponatinib (Iclusig®) will be the ideal TKI. Additionally, omacetaxine, a protein synthesis inhibitor, has been approved recently for relapsed CML patients. While this drug shows a marginal success rate, significant side effects (including, for example, myelosuppression, bleeding, and hyperglycemia) are seen in 99% of patients. Such side effects are likely due to the nonspecificity of the molecule making it undesirable for many CML patients, and thus will not be further discussed.13 Table 1 shows some of the approved therapies for CML, including their mode of action, usage indications, and common side effects.
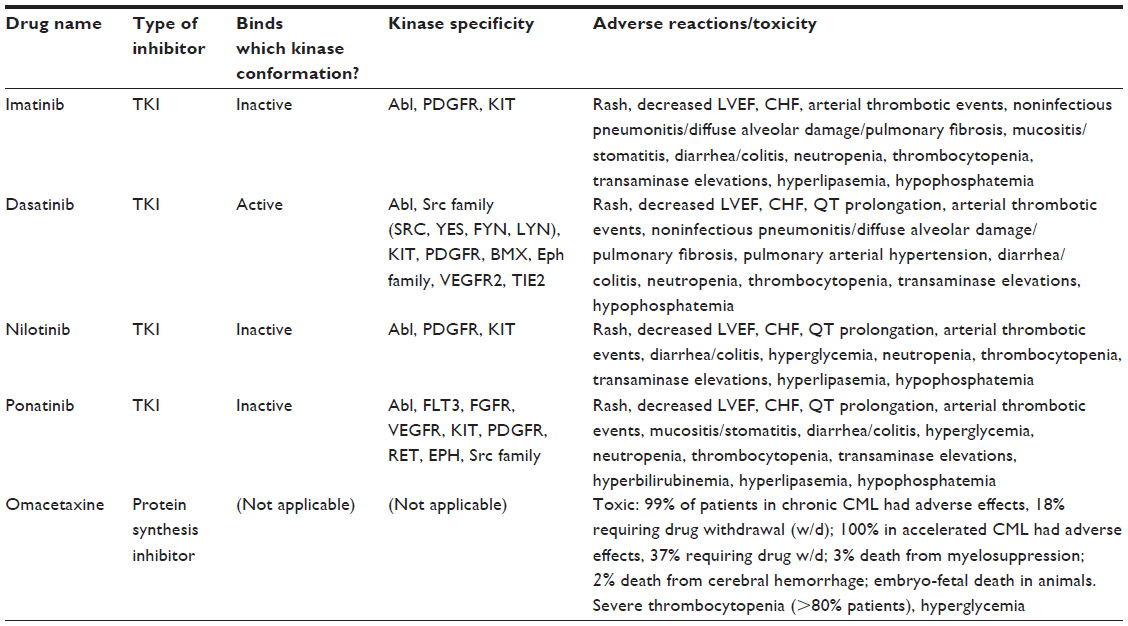 | Table 1 Select approved drugs for CML therapy, kinase specificity, and notable adverse effects and toxicity. |
While the treatment of Ph+ALL includes the use of the aforementioned TKIs, the overall treatment strategy differs from CML. Current frontline treatment of childhood Ph+ALL includes chemotherapy with a rigorous induction phase, often using cyclophosphamide, vincristine, adriamycin, and dexamethasone (known as hyper-CVAD) in combination with a TKI such as imatinib or dasatinib.14 Even if remission is achieved, the only possibility of a cure is hematopoietic stem cell transplantation. Most patients who do not undergo hematopoietic stem cell transplantation eventually relapse.14 Depending on the molecular status of BCR-ABL transcripts during follow-up, patients may be administered a TKI as maintenance therapy.
In the following sections, the mechanism of resistance to TKIs will be discussed, as well as the role of ponatinib in these scenarios. Additionally, the pharmacology and mode of action of ponatinib will be discussed to describe how and why ponatinib is often effective against both native and mutant BCR-ABL. Finally, the efficacy and safety of ponatinib will be discussed, including a summary of various clinical trials; and the current place and future use of ponatinib in therapy will be described and speculated. The majority of this review will focus on the potential of ponatinib in the treatment of CML. Although CML and Ph+ALL involve different treatment strategies, overlapping issues arise in TKI-resistant CML and BCR-ABL inhibition in Ph+ALL.
Mechanisms of mutational resistance in CML and Ph+ALL
It is well documented how the introduction of imatinib (and other TKIs) has profoundly changed the therapeutic management of CML and Ph+ALL.15–17 Despite these successes, it is estimated that one-third of CML patients on first-line imatinib therapy will require other therapy due to imatinib intolerance or resistance.12 BCR-ABL independent resistance represents another mechanism of TKI resistance, which will not be discussed in this review. For excellent reviews on this topic, two other papers provide excellent topical summaries.18,19
Resistance to TKIs can be classified into two types: primary resistance and secondary resistance. Primary resistance refers to the lack of initial response, presumably due to the presence of baseline mutations in the kinase domain. On the other hand, secondary resistance refers to the loss of an established response, often due to the prolonged exposure to the TKI.20 Whether it is primary or secondary, the most common and well understood mechanism of resistance against all TKIs is the appearance or development of point mutations in the BCR-ABL kinase domain.19 In patients with primary resistance, many leukemic cells harbor mutations from the onset of disease. Conversely, a clonal model of resistance is often used to describe secondary resistance from a molecular standpoint. Here, nonresistant cells are first eliminated by TKI therapy giving surviving resistant clones a selective advantage for future outgrowth. The observation that sequential treatment with TKIs often leads to enhanced TKI resistance supports this theory.21,22 In a subset of patients who were treated sequentially with imatinib and dasatinib but did not reach disease remission, new BCR-ABL mutations were observed after dasatinib treatment, which had not been recognized after imatinib treatment. These double mutants, as described in the “Emergence of compound mutations” section, can confer resistance to multiple drugs and enhance BCR-ABL oncogenicity.21
Importantly, the kinase domain mutations that are responsible for resistance occur at strategic locations, which prevent TKI activity. When these residues are mutated, they inhibit TKI binding but still allow ATP binding and kinase activity.23 Imatinib, dasatinib, and nilotinib each bind the BCR-ABL kinase in a characteristic manner (Table 1). Due to these binding specifics, resistance profiles specific to each TKI will exist. In some cases, mutations will occur directly in the kinase active site, sterically hindering TKI binding. In other cases, mutations will occur at residues that affect the overall three-dimensional structure of the kinase. Because imatinib and nilotinib bind BCR-ABL in its inactive conformation (Table 1), mutations may occur in resistant patients that destabilize or prevent adoption of the inactive conformation of the kinase. Failure to adopt this conformation prevents the binding and activity of imatinib and nilotinib.18,21 Most importantly, clinically relevant mutations in the BCR-ABL kinase domain all occur on residues such that the integrity of the tyrosine kinase itself is not eliminated. Specifically, more than 55 residues showing varying levels of resistance to imatinib have been identified.18 It should be noted that while many of the studied kinase domain mutations are observed in CML, mutations in BCR-ABL also appear in Ph+ALL with similar effects.
Emergence of compound mutations
In a 2013 study, 1,700 patient samples with BCR-ABL kinase domain mutations were analyzed using direct DNA sequencing. In this set of samples, 11.4% showed two or more mutations in the kinase domain.24 Double mutants in the BCR-ABL kinase domain can exist in two forms: compound or polyclonal. Compound mutations, which account for 70.2% of double mutations in the BCR-ABL kinase domain, refer to multiple mutations in the same BCR-ABL molecule.24 The other 29.8% of double mutations are represented as polyclonal mutants, where mutations occur in multiple BCR-ABL molecules within a single cell. As previously mentioned sequential treatments with TKIs can lead to the emergence of multiple mutations in a single patient. Not surprisingly, the composition of the compound mutations noted in this study reflected the TKI treatment history, where one or more mutational components were associated with a typical clinical or in vitro resistance profile to the specific TKIs.24 Altogether, 30 different compound mutations were observed, with the T315I mutation being the most frequently observed mutation among all compound mutants. It has been suggested that the best way to prevent the onset of compound mutations is to initially treat with a drug that can “collectively suppress” all single point mutations. A molecule such as ponatinib, which was shown to inhibit all clinically important kinase domain mutations including T315I during preclinical evaluation, has the potential to achieve this feat. It is unlikely, however, that ponatinib will be used for first-line therapy at its current dosing regimen.21,25
Pharmacology of ponatinib
Although dasatinib and nilotinib have shown efficacy against many of the clinically relevant mutations for which imatinib is ineffective, neither has shown the ability to inhibit BCR-ABL with the T315I mutation (BCR-ABLT315I).26 Thus, the need arose for a new TKI that not only inhibits BCR-ABLT315I but also inhibits native BCR-ABL and other commonly seen BCR-ABL mutations. In order to design a molecule that could meet these criteria, it is important to understand the structural features of the BCR-ABL kinase domain containing the T315I mutation.
Effects of the T315I mutation
Although the T315I mutation does not disturb the overall structure of the BCR-ABL protein, it affects the topology of the ATP binding region.27 Current TKIs are unable to inhibit this mutant form of the enzyme for two main reasons. The T315I mutant form of BCR-ABL lacks a threonine residue, which provides a hydroxyl group critical for hydrogen bonding with first and second generation TKIs. This hydrogen bond has been described to drive both the potency and specificity of these ABL inhibitors.27,28 The hydrogen bond disappears when the threonine is replaced with an isoleucine (Ile) residue. Additionally, residue 315 is located directly in front of the hydrophobic pocket, which is why it is referred to as the “gatekeeper” residue. When mutated to Ile, the bulkier Ile side chain protrudes further into the enzyme active site. This initiates a steric clash subsequently blocking the entrance of a TKI into the hydrophobic pocket while still allowing access to ATP. For these reasons, first and second generation TKIs fail to inhibit BCR-ABLT315I.25,27,29
Design of ponatinib
Overcoming both losses of an active-site hydrogen bond and the steric hindrance caused by the T315I mutation were the main focus during the design process of ponatinib.30 While still binding to the kinase active site of BCR-ABL, the overall design of the ponatinib molecule was still based on the ATP-mimetic template provided by the previous TKIs imatinib, nilotinib, and dasatinib. The structure of ponatinib is shown in Figure 1. The five critical regions of the molecule and the structure–activity relationships of each region are described in the following sections.
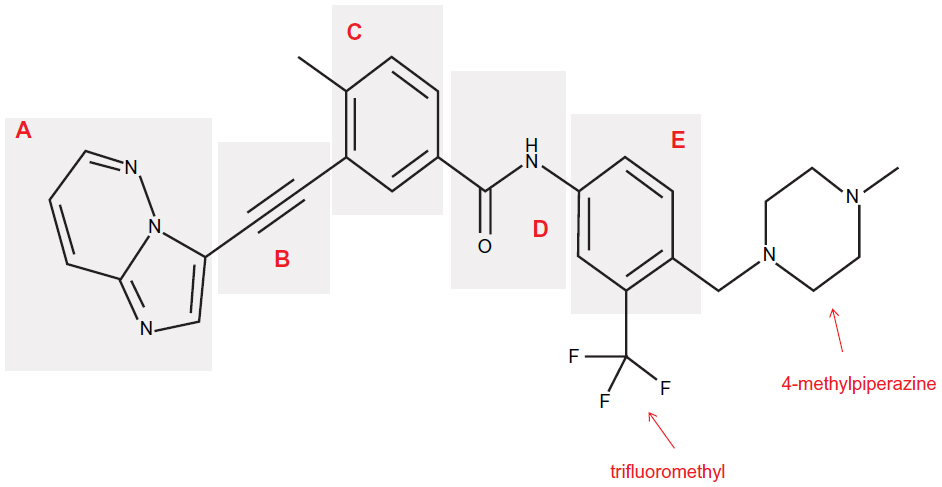 | Figure 1 Chemical structure of ponatinib. |
Hinge region
The hinge region (Figure 1A) was optimized to improve binding affinity, lipophilicity, and overall oral bioavailability. The final product resulted in a 6-5 fused ring system with an imidazole[1,2]pyridazine chemical moiety that occupies the adenine-binding pocket of the kinase domain.30 Chemically, the imidazole moiety forms hydrogen bonds with the hinge of the enzyme pocket (Figure 2) while the pyridazine ring interacts hydrophobically with F382 of the DFG region and Y253 in the P-loop of BCR-ABL (Figure 2). Finally, the lack of atomic substitutions and substituents in this fused ring system has led to optimal lipophilicity and oral bioavailability.29
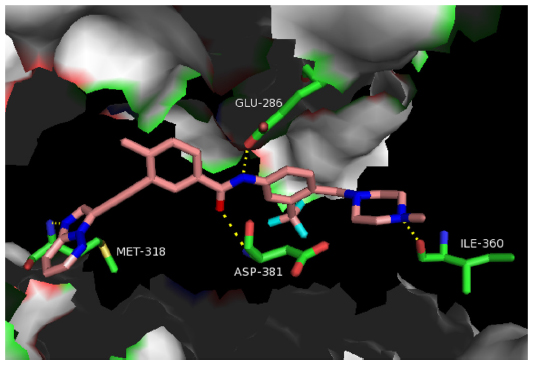 | Figure 2 Hydrogen bonding pattern between ponatinib and BCR-ABLT315I. |
Ethynyl linker
Ponatinib differs from previous BCR-ABL TKIs due to the presence of a triple bond ethynyl linker (Figure 1B), which connects the hinge region and A ring. Most importantly, this ethynyl linker allows ponatinib to span the bulky isoleucine side chain present in BCR-ABLT315I (Figure 2). Structurally, this Ile side chain protrudes into the active site in front of the hydrophobic pocket. Previous TKIs could not span this side chain and thus could not dock into the hydrophobic pocket. Structure–activity studies revealed that the presence of the triple bond allows a ten-fold increase in potency when compared to similar molecular designs containing single or double bonded systems.29 Vinyl and acetylene linkages were also attempted during the design process. While vinyl and acetylene linkages also showed promise, the final ethynyl linkage showed superior chemical and pharmacological stability.
A ring
The A ring of ponatinib (Figure 1C) binds in the hydrophobic pocket of the BCR-ABL enzyme located just behind the gatekeeper residue. Existing as a methylphenyl moiety, the A ring appeared very sensitive to any substitutions or chemical modifications and thus, was not changed.29,30
Linker 2
Linker 2 (Figure 1D) chemically exists as an amide linkage with the purpose of connecting the A and B rings, spatially aligning the molecule with the structure of the BCR-ABL.29 Hydrogen bonds are formed between the linker carbonyl group and residue D381, and between the NH group and residue E286 (Figure 2).28
B ring
The B ring (Figure 1E) consists of a trifluoromethyl ring with a 4-methylpiperazine substituent. This B ring binds into the aspartate-phenylalanine-glycine (DFG)-out pocket of the enzyme active site. The addition of these particular substituents has allowed for optimization of maximal BCR-ABLT315I inhibition due to the formation of critical hydrogen bonds between the piperazine group and the enzyme backbone, notably at residue I360 (Figure 2).28 Specifically, methylpiperazine increases potency and molecular recognition, while piperazine itself is a solubilizing group, improves permeability, and decreases lipophilicity and protein binding.
Taken together, the optimization process of the five regions listed above led to the final design of ponatinib. Although the overall structure remains similar in many ways to other approved TKIs, the ability to potently inhibit native BCR-ABL, BCR-ABLT315I, and many other mutant forms of BCR-ABL made ponatinib a highly versatile drug with great potential moving into clinical evaluation.
Efficacy, safety, and tolerability of ponatinib
Preclinical testing of ponatinib
The design and preclinical testing of ponatinib began in 2006 as the need for a CML therapy capable of inhibiting many resistant forms of BCR-ABL arose (Figure 3). Initial preclinical testing involved demonstrating that ponatinib could both decrease autophosphorylation of the BCR-ABL kinase as well as inhibit the proliferation of cells dependent on BCR-ABL. Namely, these in vitro experiments utilized K562 (BCR-ABL positive human cells) and Ba/F3 (pro-B mouse cells), which stably transduced to express BCR-ABL with a wild-type or mutant tyrosine kinase domain. From these studies, it was found that ponatinib inhibits both wild-type and mutant BCR-ABL kinases with IC50 (50% inhibitory concentration of the drug) values between 0.37–2.0 nM.25 Further, cell proliferation assays demonstrated inhibition with IC50 values ranging from 0.3–0.5 nM for non-mutated and up to 36 nM for mutant BCR-ABL. It was also found that a dose of 40 nM was able to completely suppress the emergence of BCR-ABL resistant mutants in vitro, although efficacy may be limited particularly in examples with the E255V point mutation (IC50 of 36 nM). Finally, ponatinib also inhibited the activity of members of the vascular endothelial growth factor (VEGF), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR) families at similar doses, but it showed IC50 values more than 1,000-fold higher for inhibition of insulin receptor and members of the cyclin kinase and aurora kinase families.25
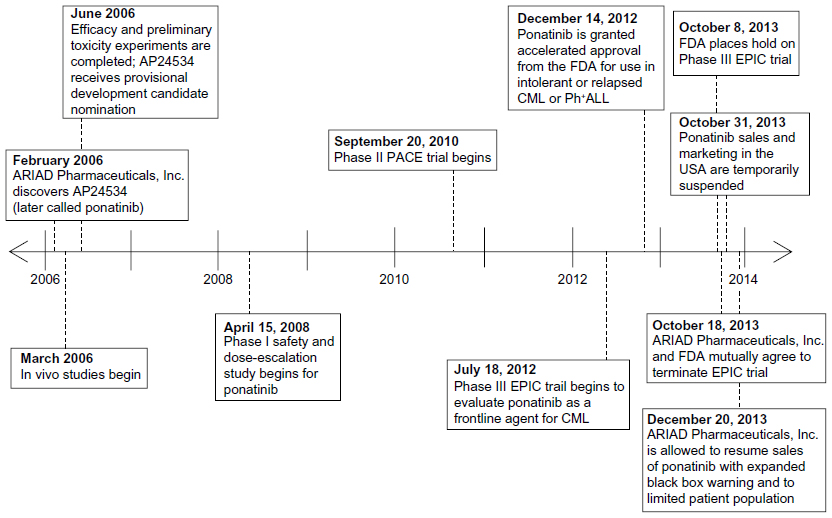 | Figure 3 Timeline of ponatinib events from discovery to present day. |
Based on this information, researchers proceeded to a xenograft mouse model (Figure 3) as is standard for in vivo CML studies. Cells harboring native BCR-ABL and BCR-ABLT315I were subcutaneously injected into the right flank in female mice, and these mice were given ponatinib at doses of 10, 30, and 50 mg/kg. The 10 mg/kg and 30 mg/kg doses were able to suppress tumor growth, while the 50 mg/kg dose was able to cause 96% reduction in the mean tumor volume.25 Pharmacokinetic studies of the 30 mg/kg dose revealed mean plasma levels of 782, 561, and 8 nM at 2, 6, and 24 hours posttreatment, respectively. No pharmacokinetic data for the 50 mg/kg dose was reported. This demonstrated that plasma concentrations of ponatinib effective against mutated forms of BCR-ABL could be sustained for 24 hours using the 30 mg/kg dose.
Moving ponatinib into clinical trials
The first Phase I trial involving ponatinib enrolled CML and Ph+ALL patients who were resistant or had relapsed while they were on the standard of care therapy. The goal of the study was to determine the maximum tolerated or recommended daily dose of ponatinib, while also investigating the safety, anti-leukemic activity, and pharmacokinetic/pharmacodynamic profile of the drug.31 Eighty-one patients were enrolled, 65 of whom had Ph+ disease. Daily doses of 2–60 mg were tested. No dose-limiting toxic effects were observed up to the 30 mg/day dose, while the 60 mg/day dose led to toxicity in six out of 19 patients. Adverse effects included elevation of pancreatic enzymes, clinical pancreatitis, fatigue, and increases in liver enzymes aspartate transaminase (AST) and alanine transaminase (ALT) (Table 1). Pharmacokinetic studies demonstrated a linear relationship between dose and peak blood concentration. For the 30 mg/day dose, the half-life was 22 hours; and the trough blood concentration was above 40 nM, which is the minimal concentration thought to be required to prevent the emergence of BCR-ABL mutants resistant to ponatinib. The anti-leukemic activity of the drug was also promising; 42/43 (98%) of patients with chronic phase (CP)-CML (Table 2) had a complete hematologic response (see Table 3 for clinical terms); 31/43 (72%) had a major cytogenetic response (MCyR), 27/43 (63%) had a complete cytogenetic response (CCyR), and 19 patients had a major molecular response (MMR). Further, all 12 patients with CP-CML and the T315I mutation had a complete hematologic response, and 8/12 (67%) had a MMR. Despite the fact that the a dose of 30 mg/day showed excellent results in the Phase I trial, researchers proceeded to Phase II study using the 45 mg/day dose to ensure coverage of any and all BCR-ABL TKI resistant clones.31–33
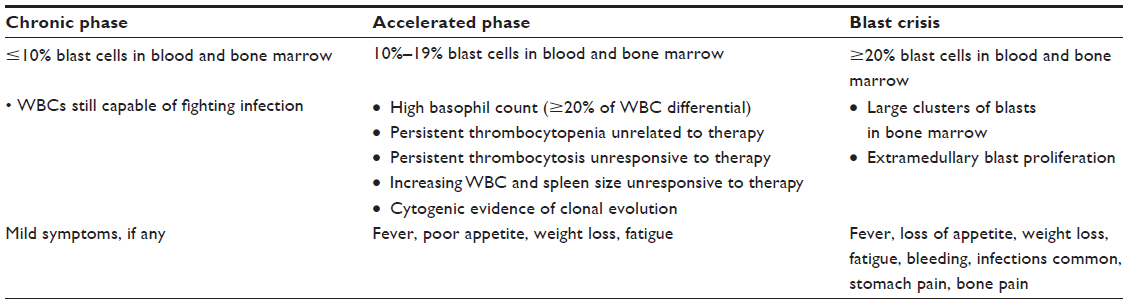 | Table 2 World Health Organization (WHO) definitions and descriptions of CML stages, and commonly seen symptoms |
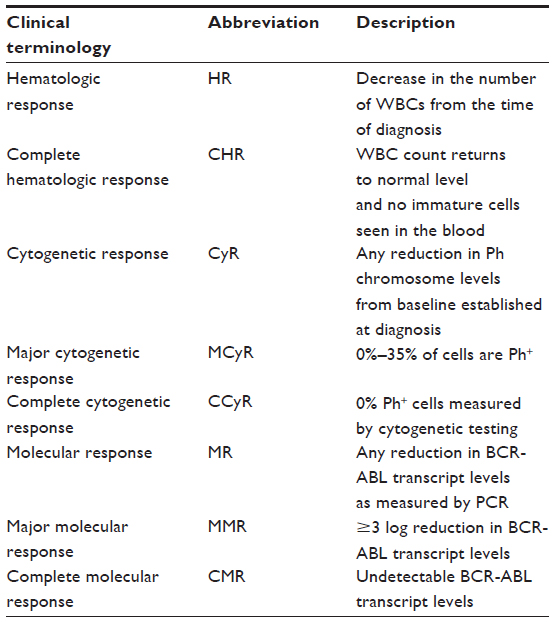 | Table 3 Clinical terminology describing patient response in CML |
The Phase II Ponatinib Ph+ALL and CML Evaluation (PACE) trial enrolled 449 patients: 267 with CP-CML, 83 with accelerated phase (AP)-CML, and 94 with blast crisis (BC)-CML or Ph+ALL (Table 2).32 Patients were eligible for the study if they were resistant or had unacceptable side effects to dasatinib or nilotinib, or harbored the T315I mutation. The trial showed that 37% of patients had received two TKIs, and 55% had received three or more. Of those who had received dasatinib or nilotinib, 88% were resistant. At 12 months after the initiation of ponatinib therapy, 56% of patients with CP-CML had a MCyR, 46% had CCyR, and 34% had MMR (Table 3). Interestingly, higher response rates were reached for those with the T315I mutation, but the presence of this mutation was determined not to be a significant prognostic factor.34 Six patients had an unsustained MCyR, but no change in mutation status was seen in those with unsustained responses. In patients with AP-CML and BC-CML (Table 2) or Ph+ALL, rates of major hematologic response at 6 months were 39% and 31%, respectively. Importantly, no single mutation conferring resistance was seen.
Responses were observed for each of 15 mutations present at baseline for CP-CML patients, and 26 unique mutations in total.35 Response rates were similar for those with and without mutant BCR-ABL, with MCyR occurring in 56% of all patients, 49% of patients with native BCR-ABL, 64% for those with one mutation, 62% for those with two or more mutations, and 74% for those with the T315I mutant.35 Though no single point mutation has shown resistance against ponatinib, compound mutations developed in 21 patients, mainly those in BC-CML or Ph+ALL. While ponatinib should still inhibit a number of compound mutants based on their in vitro cell proliferation IC50 values, a select few clinically reported compound mutants, including G250E/T315I, E255K/T315I, and E255V/T315I (IC50 values of 49 nM, 106 nM, and 425 nM, respectively22), have shown increased resistance. Because these compound mutants appear only infrequently, and based on these results from the clinical trials described above, ponatinib was granted accelerated FDA approval in December 2012 for use in resistant or intolerant CML or Ph+ALL or in patients harboring BCR-ABLT315I (Figure 3).36
Safety profile of ponatinib
Minimal safety concerns were initially reported. Sixty-seven percent of patients had dose interruptions, with 55% having at least one dose reduction. Serious nonhematologic adverse drug events (ADEs) included pancreatitis, abdominal pain, and myocardial infarction (MI). Serious hematologic ADEs included thrombocytopenia, anemia, neutropenia, and pancytopenia. Cardiovascular, cerebrovascular, and peripheral vascular events were observed in 7.1%, 3.6%, and 4.9% of the population, respectively, although it was speculated these were minimally related to the treatment. Serious arterial thrombotic events were seen in 8.9% of patients on ponatinib, predominantly in patients with either documented ischemic conditions or with one or more risk factors at baseline.
An analysis of the cardiovascular risk profile of ponatinib was performed with the results of the PACE trial.37 There were 14 cases of MI, five of coronary artery disease, and two of angina (21 in total). At the start of the study, 10/21 had active cardiovascular disease, and five had valvular or pericardial disease. Further, 81% of these patients had at least two cardiovascular risk factors, while 95% had at least one. At the time of the drug’s approval, the label noted that 8% of patients had cardiovascular, cerebrovascular, and peripheral vascular thrombosis including fatal MIs and strokes.38 However, at a 2.7-year follow-up of the Phase I study, 48% of patients reported adverse vascular events. At a follow-up of the Phase II study (median time of 1.3 years), 24% of patients had an MI, stroke, limb ischemia, or stenosis of the vessels of the heart, extremities or brain requiring revascularization.38 It is unlikely that these events can be fully explained by underlying cardiovascular risk, as numbers this high were not seen in late phase trials of other TKIs. Further, adverse events occurred as soon as two weeks into therapy and even in patients with no cardiovascular risk factors.38
As the increased cardiovascular risks associated with ponatinib were uncovered, ponatinib was removed from the market in October 2013.39 Additionally, the Evaluation of Ponatinib versus Imatinib in Chronic Myeloid Leukemia (EPIC) trial, a Phase III trial investigating the use of ponatinib versus imatinib for first-line use, was halted following this decision. The FDA and ARIAD Pharmaceuticals, Inc. “mutually agreed the trial should be terminated” due to the now understood increased cardiovascular risks associated with ponatinib.40 Ponatinib has since been put back on the market for a narrowed patient population: treatment of adults with T315I-positive CML (any phase) and Ph+ALL, and treatment of adult patients for whom no other TKI therapy is indicated.38 A timeline highlighting important events in the discovery and approval process of ponatinib is provided in Figure 3.
One question that still remains concerning TKI therapy is whether or not the therapy can ever be discontinued without resulting in disease recurrence. The Evaluation of the Persistence of the Complete Molecular Remission After Stopping Imatinib Chronic Myeloid Leukemia (STIM) was undertaken to determine if those with the deepest level of disease suppression could successfully be taken off of imatinib therapy without disease recurrence.41 Briefly, patients on imatinib for at least 3 years and in complete molecular response (CMR) (deepest response) for at least 2 years were eligible for the study. They were taken off imatinib therapy and monitored for a median follow-up time of 17 months. Fifty-four of 100 patients had a molecular relapse, and 46 were relapse-free at a median follow up time of 14 months. The majority of relapses (35/54) occurred in the first 3 months, and only one patient relapsed after 7 months. Males, those with a good prognosis (based on the Sokal scale for CML prognosis,42) and those with imatinib therapy duration of more than 50 months were more likely to remain in CMR. Encouragingly, all of the patients who relapsed remained sensitive to imatinib therapy, and the median time to CMR return was 3 months. Imatinib therapy infrequently leads to sustained CMR for the 2 years required for acceptance into this study. The cumulative incidence of CMR on imatinib therapy was 0.5% at 1 year and 8.3% at 5 years in newly diagnosed CP-CML patients.43 Studies with the second-generation TKI nilotinib showed that patients on nilotinib were more likely to be in CMR at 24 months than those on imatinib (22.1% versus 8.7%).44 The PACE trial results indicated that 15% of patients who entered treatment with CP-CML and 23% of patients with the T315I mutation were in CMR at 12 months.32 Therefore, patients on ponatinib may be more likely to successfully discontinue treatment than those taking imatinib.
Conclusion and place in therapy for ponatinib
Ponatinib is currently FDA-approved for T315I-positive CML (in any phase) and T315I-positive Ph+ALL, or any phase of CML or Ph+ALL for which no other TKIs are indicated.45 As it has only been available on the market since December 2012, more time is required to sufficiently analyze the effect that ponatinib will have on long-term CML and Ph+ALL therapy. It is speculated that more potent inhibition of BCR-ABL earlier in therapy with drugs like ponatinib may lead to decreased development of resistance.12,32 Regardless of the time of therapy, detection and quantitative monitoring of patients on ponatinib for potentially resistant (poly)mutant subclones could be performed using newer methods including ligase-dependent polymerase chain reaction (PCR)46 or long-range next generation sequencing.47 The toxicity profile of ponatinib, however, precludes its use alone as a frontline agent.48 To circumvent this unwanted toxicity, one possibility may include using ponatinib in combination with other therapeutic agents to allow for dose-lowering of ponatinib. Our lab has taken a multidomain targeting approach of concurrently inhibiting both the tyrosine kinase domain (with ponatinib) and the N-terminal coiled-coil domain of BCR-ABL with a rationally designed peptidomimetic.49 Using this therapeutic strategy, we have shown the ability to lower the dose of ponatinib necessary for BCR-ABL inhibition,50 which would likely result in a safer toxicity profile.33 This coiled-coil dimerization inhibitor has in fact shown activity by itself against the BCR-ABLT315I,50 as well as the BCR-ABL E255V/T315I compound mutant (Woessner et al., unpublished data, 2014), which is not targetable by any current TKI, and could be used as an alternative to ponatinib. Other strategies include concurrently targeting BCR-ABL along with proteins and signaling pathways important to leukemia progression including MUC-1 (Mucin-1, cell surface associated),51 Alox5 (arachidonate 5-lipoxygenase),51 STAT3 (signal transducer and activator of transcription 3),52 and Hedgehog,53 to name a few. It is also expected that combination targeting with ponatinib may result in reducing the emergence of resistance in cells, which could lead to greater long-term success.19,54 Lastly, it has also been reported that dosing of potent drugs such as ponatinib with a high risk of ADEs should be lowered to the minimum effective dose rather than the maximum tolerable dose, which in this case turned out to be toxic in clinical trials.33
While ponatinib is the only approved TKI that targets BCR-ABLT315I, other drugs in development capable of targeting this resistant form of BCR-ABL in either CML or Ph+ALL include: rebastinib (formerly DCC-2036), an ABL switch control inhibitor;55,56 HG-7-85-01, a multi-targeted kinase inhibitor;57 and AT9283, a multi-targeted kinase inhibitor.58,59 For more comprehensive lists and discussions of ATP-competitive, allosteric, and other inhibitors of BCR-ABL, other reviews focus specifically on these topics.4,60 Additionally, drugs are being developed to target CML stem cells by attacking other signaling pathways.18,61 One example includes SL-401, a therapeutic targeting the IL-3 receptor that is overexpressed on CML stem cells.62,63
Due to the broad-spectrum activity of ponatinib for tyrosine and other kinase families (Table 1),64–68 the drug is also being examined for use in other cancers and malignancies. As of March 2014, ponatinib was either being used in or currently suspended from six clinical trials for cancers and malignancies other than CML or Ph+ALL via inhibition of FGFR (fibroblast growth factor receptor), EGFR (epidermal growth factor receptor), ALK (anaplastic lymphoma kinase), and RET (rearranged during transfection).69–74 Ultimately, ponatinib may prove to be useful in combination with other agents that robustly target BCR-ABL (multidomain targeting),50 or other pathways involved in cancer progression besides BCR-ABL (for example, in combination with STAT3 inhibitors52) for a synthetically lethal approach for full eradication of CML or Ph+ALL.
Disclosure
The authors have no conflicts of interest in this work.
References
Bartram CR, de Klein A, Hagemeijer A, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306(5940):277–280. | |
Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. | |
Ren R. The molecular mechanism of chronic myelogenous leukemia and its therapeutic implications: studies in a murine model. Oncogene. 2002;21(56):8629–8642. | |
Woessner DW, Lim CS, Deininger MW. Development of an effective therapy for chronic myelogenous leukemia. Cancer. 2011;17(6):477–486. | |
Jeha S, Coustan-Smith E, Pei D, et al. Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2014;120(10):1514–1519. | |
Thomas DA. Philadelphia chromosome positive acute lymphocytic leukemia: a new era of challenges. Hematology/the Education Program of the American Society of Hematology. Hematology Am Soc Hematol Educ Program. 2007:435–443. | |
Lee HJ, Thompson JE, Wang ES, Wetzler M. Philadelphia chromosome-positive acute lymphoblastic leukemia: current treatment and future perspectives. Cancer. 2011;117(8):1583–1594. | |
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England Journal of Medicine. 2001;344(14):1031–1037. | |
Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. The New England Journal of Medicine. 2006;355(23):2408–2417. | |
Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England Journal of Medicine. 2001;344(14):1038–1042. | |
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol. 2014;89(5):547–556. | |
Shami PJ, Deininger M. Evolving treatment strategies for patients newly diagnosed with chronic myeloid leukemia: the role of second-generation BCR-ABL inhibitors as first-line therapy. Leukemia. 2012;26(2):214–224. | |
Prescribing Information for Synbrio (omacetaxine mepesuccinate). 2012. | |
Ottmann OG, Pfeifer H. Management of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL). Hematology Am Soc Hematol Educ Program. 2009:371–381. | |
Deininger M, O’Brien SG, Guilhot F, et al. International Randomized Study of Interferon Vs STI571 (IRIS) 8-Year Follow up: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Treated with Imatinib. Blood [abstract]. 2009;114. | |
Jabbour E, Kantarjian HM, Saglio G, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood. 23, 2014;123(4):494–500. | |
Larson RA, Hochhaus A, Hughes TP, et al. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012;26(10):2197–2203. | |
O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nature reviews. Cancer. 2012;12(8):513–526. | |
Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Molecular Cancer. 2010;9:75. | |
Soverini S, Branford S, Nicolini FE, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leukemia Research. 2014;38(1):10–20. | |
Shah NP, Skaggs BJ, Branford S, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. The Journal of Clinical Investigation. 2007;117(9):2562–2569. | |
Eide CA, Zabriskie MS, Adrian LT, et al. Resistance profiling of BCR-ABL compound mutations linked to tyrosine kinase inhibitor therapy failure in chronic myeloid leukemia [abstract]. Blood. 2011;118(21):1416. | |
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature reviews. Cancer. 2009;9(1):28–39. | |
Khorashad JS, Kelley TW, Szankasi P, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489–498. | |
O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. | |
O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Research. 2005; 65(11):4500–4505. | |
Zhou T, Parillon L, Li F, et al. Crystal structure of the T315I mutant of AbI kinase. Chemical Biology and Drug Design. 2007;70(3):171–181. | |
Buffa P, Romano C, Pandini A, et al. BCR-ABL residues interacting with ponatinib are critical to preserve the tumorigenic potential of the oncoprotein. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2014;28(3):1221–1236. | |
Zhou T, Commodore L, Huang WS, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534):lessons for overcoming kinase inhibitor resistance. Chemical Biology And Drug Design. 2011;77(1):1–11. | |
Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-y l)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. Journal of Medicinal Chemistry. 2010;53(12):4701–4719. | |
Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. The New England Journal of Medicine. 2012;367(22):2075–2088. | |
Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. The New England Journal Of Medicine. 2013;369(19):1783–1796. | |
Quintas-Cardama A. Ponatinib in Philadelphia chromosome-positive leukemias. The New England Journal of Medicine. 2014; 370(6):577. | |
Mauro MJ, Cortes JE, Kim DW. Multivariate analyses of the clinical and molecular parameters associated with efficacy and safety in patients with chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) treated with ponatinib in the PACE trial [Abstract 3747]. Blood. 2012;120. | |
Deininger M, Cortes JE, Kim DW. Impact of baseline mutations on response to ponatinib and end of treatment mutation analysis in patients with chronic myeloid leukemia [Abstract 7001]. American Society of Clinical Oncology. 2013;31(Suppl). | |
Chustecka Z. FDA Approves Ponatinib for Rare Leukemias. Medscape Medical News. December 14, 2012. | |
Khoury HJ, Cortes JE, Kim DW. Analysis of the cardiovascular risk profile of Ph+ leukemia patients treated with ponatinib [Abstract 7048]. American Society of Clinical Oncology. 2013;31(Suppl). | |
Prasad V, Mailankody S. The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA. 2014;311(4):353–354. | |
Mulcahy N. Leukemia Drug Ponatinib (Iclusig) Pulled From Market. Medscape Medical News. October 31, 2013. | |
Ariad Ends Phase III Leukemia Drug Trial. Genetic Engineering and Biotechnology News. October 18, 2013. | |
Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035. | |
Bonifazi F, De Vivo A, Rosti G, et al. Testing Sokal’s and the new prognostic score for chronic myeloid leukaemia treated with alpha-interferon. Italian Cooperative Study Group on Chronic Myeloid Leukaemia. Br J Haematol. 2000;111(2):587–595. | |
de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol. 2008;26(20):3358–3363. | |
Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12(9):841–851. | |
ICLUSIG® (ponatinib) tablets for oral use. [prescribing information]. Cambridge: ARIAD Pharmaceuticals, Inc.; 2013. | |
Preuner S, Mitterbauer G, Mannhalter C, et al. Quantitative monitoring of BCR/ABL1 mutants for surveillance of subclone-evolution, -expansion, and -depletion in chronic myeloid leukaemia. Eur J Cancer. 2012;48(2):233–236. | |
Kastner R, Zopf A, Preuner S, et al. Rapid identification of compound mutations in patients with Philadelphia-positive leukaemias by long-range next generation sequencing. Eur J Cancer. 2014;50(4):793–800. | |
Frankfurt O, Licht JD. Ponatinib-a step forward in overcoming resistance in chronic myeloid leukemia. Clin Cancer Res. 2013;19(21):5828–5834. | |
Dixon AS, Miller GD, Bruno BJ, et al. Improved coiled-coil design enhances interaction with Bcr-Abl and induces apoptosis. Mol Pharm. 2012;9(1):187–195. | |
Miller GD, Woessner DW, Sirch MJ, Lim CS. Multidomain Targeting of Bcr-Abl by Disruption of Oligomerization and Tyrosine Kinase Inhibition: Toward Eradication of CML. Mol Pharm. 2013;10(9):3475–3483. | |
Woessner DW, Lim CS. Disrupting BCR-ABL in combination with secondary leukemia-specific pathways in CML cells leads to enhanced apoptosis and decreased proliferation. Mol Pharm. 2013;10(1):270–277. | |
Eiring AM, Kraft IL, Page BDG, et al. BP5-087, a Novel STAT3 Inhibitor, Combines With BCR-ABL1 Inhibition To Overcome Kinase-Independent Resistance In Chronic Myeloid Leukemia. Presented at: 55th ASH Annual Meeting and Exposition; December 7-10, 2013; New Orleans, LA. Abstract. | |
Dao KH, Tyner JW. Next-generation medicine: combining BCR-ABL and Hedgehog-targeted therapies. Clin Cancer Res. 2013;19(6):1309–1311. | |
Gozgit JM, Squillace RM, Wongchenko MJ, et al. Combined targeting of FGFR2 and mTOR by ponatinib and ridaforolimus results in synergistic antitumor activity in FGFR2 mutant endometrial cancer models. Cancer Chemother Pharmacol. 2013;71(5):1315–1323. | |
Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71(9):3189–3195. | |
Clinical Trial NCT00827138: Study Safety and Preliminary Efficacy of DCC-2036 in Patients With Leukemias (Ph+ CML With T315I Mutation). ClinicalTrials.gov. 2013. | |
Weisberg E, Choi HG, Ray A, et al. Discovery of a small-molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFRalpha, Kit, and Src kinases: novel type II inhibitor of gatekeeper mutants. Blood. 2010;115(21):4206–4216. | |
Clinical Trial NCT01431664:AT9283 in Treating Young Patients With Relapsed or Refractory Acute Leukemia. ClinicalTrials.gov. 2014. | |
Tanaka R, Squires MS, Kimura S, et al. Activity of the multitargeted kinase inhibitor, AT9283, in imatinib-resistant BCR-ABL-positive leukemic cells. Blood. 2010;116(12):2089–2095. | |
Hantschel O, Grebien F, Superti-Furga G. The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer Res. 2012;72(19):4890–4895. | |
Chomel JC, Turhan AG. Chronic myeloid leukemia stem cells in the era of targeted therapies: resistance, persistence and long-term dormancy. Oncotarget. 2011;2(9):713–727. | |
Brooks C, Macri V, Albini A, Bergstein I, Rowinsky E. SL-401, a Targeted Therapy Directed To The Interleukin-3 Receptor (IL-3R), Possesses Preclinical Cytotoxic Activity Against Chronic Eosinophilic Leukemia. Blood. 2013;21(122). | |
Konopleva M. SL-401, a targeted therapeutic directed at the interleukin-3 receptor, inhibits the growth of leukemia stem cells from patients with CML. Presented at: 15th International Conference on CML; September 26, 2013; Estoril, Portugal. | |
De Falco V, Buonocore P, Muthu M, et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab. 2013;98(5):E811–E819. | |
Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11(3):690–699. | |
Gozgit JM, Wong MJ, Wardwell S, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther. 2011;10(6):1028–1035. | |
Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia. 2013;27(1):32–40. | |
Smith CC, Lasater EA, Zhu X, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121(16):3165–3171. | |
Clinical Trial NCT01761747:Ponatinib for Squamous Cell Lung and Head and Neck Cancers. ClinicalTrials.gov. 2014. | |
Clinical Trial NCT01888562: Ponatinib in the Treatment of FGFR Mutation Positive Recurrent or Persistent Endometrial Carcinoma. ClinicalTrials.gov. 2014. | |
Clinical Trial NCT01935336: Study of Ponatinib in Patients With Lung Cancer Preselected Using Different Candidate Predictive Biomarkers. ClinicalTrials.gov. 2014. | |
Clinical Trial NCT01813734: Ponatinib in Advanced NSCLC w/RET Translocations. ClinicalTrials.gov. 2014. | |
Clinical Trial NCT01838642: Ponatinib for Advanced Medullary Thyroid Cancer. ClinicalTrials.gov. 2014. | |
Clinical Trial NCT01874665: A Phase 2 Trial of Ponatinib in Patients With Metastatic and/or Unresectable Gastrointestinal Stromal Tumor. ClinicalTrials.gov. 2014. | |
Rose SE, Shakespeare W. Personal correspondence with ARIAD Pharmaceuticals, Inc. June 9, 2014. | |
Clinical Trial NCT00660920: Safety Study of AP24534 to Treat Chronic Myelogenous Leukemia (CML) and Other Hematological Malignancies. ClinicalTrials.gov. 2013. | |
Clinical Trial NCT01207440: PONATINIB for Chronic Myeloid Leukemia (CML) Evaluation and Ph+ Acute Lymphoblastic Leukemia (ALL) (PACE). ClinicalTrials.gov. 2013. | |
Clinical Trial NCT01650805: Ponatinib in Newly Diagnosed Chronic Myeloid Leukemia (CML) (EPIC). ClinicalTrials.gov. 2014. | |
Inman S. Late-stage ponatinib study discontinued. OncLive. Epub October 18, 2013. | |
Nelson R. Ponatinib (Iclusig) returns to market, with a few caveats. Medscape Medical News. December 20, 2013. | |
Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nature Reviews Cancer. 2013;13(8):559–571. | |
Stages of Chronic Myeloid Leukemia [webpage on the Internet]. National Cancer Institute; 2014 [updated June 27, 2014]. Available from: http://www.cancer.gov/cancertopics/pdq/treatment/CML/Patient/page2. Accessed September 19, 2014. | |
Chronic Myeloid Leukaemia [webpage on the Internet]. Patient; 2014 [updated July 19, 2012]. Available from: http://www.patient.co.uk/doctor/chronic-myeloid-leukaemia-pro#. Accessed September 19, 2014. | |
How is chronic myeloid leukemia classified? [webpage on the Internet]. American Cancer Society; 2014 [updated February 10, 2014]. Available from: http://www.cancer.org/cancer/leukemia-chronicmyeloidcml/ detailedguide/leukemia-chronic-myeloid-myelogenous-staging. Accessed July 14, 2014. | |
Clopton J. Feeling Sick With CML: Coping With Later Phases. WebMD. 2014. | |
Staging for chronic myeloid leukemia (CML) [webpage on the Internet]. London: Cancer Research UK; 2014. Available from: http://www.cancerresearchuk.org/about-cancer/type/cml/treatment/staging-for-chronic-myeloid-leukaemia. Accessed July 14, 2014. | |
Kantarjian H, Schiffer C, Jones D, Cortes J. Monitoring the response and course of chronic myeloid leukemia in the modern era of BCR-ABL tyrosine kinase inhibitors: practical advice on the use and interpretation of monitoring methods. Blood. 2008;111(4):1774–1780. | |
GLEEVEC (imatinib mesylate) tablets for oral use. [prescribing information]. East Hanover: Novartis Pharmaceuticals Corporation; 2014. | |
SPRYCEL (dasatinib) tablet for oral use. [prescribing information]. Princeton: Bristol-Meyers Squibb Company; 2010. | |
TASIGNA (nilotinib) Capsules for oral use. [prescribing information]. East Hanover: Novartis Pharmaceuticals Corporation; 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.