Back to Journals » Cancer Management and Research » Volume 18

Research Progress on Copper Metabolism, Cuprotosis, and Their Regulatory Mechanism in Tumor Diagnosis and Treatment
Authors Wang R, Wang F, Xu Y, Sun W, Jiang P, Guo Y, Yu QQ ![]()
Received 19 June 2025
Accepted for publication 1 December 2025
Published 22 January 2026 Volume 2026:18 546059
DOI https://doi.org/10.2147/CMAR.S546059
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Rui Wang,1,* Fei Wang,2,* Yuting Xu,2,* Weiwei Sun,1 Pei Jiang,3 Yujin Guo,4 Qing-Qing Yu5
1School of Stomatology, Jining Medical University, Jining, Shandong, People’s Republic of China; 2Department of Obstetrics and Gynecology, Jining NO.1. People’s Hospital, Jining, Shandong, People’s Republic of China; 3Translational Pharmaceutical Laboratory, Jining NO.1 People’s Hospital, Jining, Shandong, People’s Republic of China; 4Department of Clinical Pharmacology, Jining NO.1 People’s Hospital, Jining, Shandong, People’s Republic of China; 5Clinical Research Center, Jining NO.1. People’s Hospital, Jining, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qing-Qing Yu, Clinical Research Center, Jining NO.1. People’s Hospital, Jining, Shandong, 27200, People’s Republic of China, Email [email protected] Yujin Guo, Department of Clinical Pharmacology, Jining NO.1 People’s Hospital, Jining, Shandong, 27200, People’s Republic of China, Email [email protected]
Abstract: A co-factor of multiple metalloenzymes that promote normal physiological functions, such as the immune system, neurological system, human hematological function, bone health, and antioxidant action, copper is one of the most important trace metals in the human body. The copper content in the human body maintains a relatively constant level; too much copper can cause Wilson’s disease, and too little can cause Menkes disease. Copper is involved in the physiological functions of many body systems and has a very complex transport system with many transporters. In order to offer fresh concepts and a theoretical foundation for cancer treatment, this article will go into great detail about the distinct metabolic pathways of copper and the function of cell death, particularly the function of copper-controlled cell death, especially cuproptosis, in cancer detection and treatment. Cuproptosis, a copper-dependent cell death distinct from necrosis, apoptosis, and ferroptosis, is triggered by excessive intracellular copper binding to lipoylated tricarboxylic acid (TCA) cycle proteins, leading to abnormal protein aggregation and loss of iron-sulfur clusters in respiratory chain complexes. Future research may focus on optimizing the specificity of copper metabolism-related biomarkers (eg, CTR1, SOD1) for early tumor screening, developing targeted copper ionophores/chelators with low systemic toxicity, and exploring the crosstalk between copper metabolism and tumor immunity to establish multi-dimensional diagnosis and treatment strategies.
Keywords: copper metabolism, cuproptosis tumor, regulatory mechanism, tumor diagnosis and treatment
Introduction
The human body primarily obtains adequate copper from daily diets, such as fish, nuts, and legumes, with a recommended daily intake of 0.9 mg for adults. As a catalytic cofactor, copper participates in multiple vital life activities. After digestion in the gastrointestinal tract (mainly the small intestine), copper enters the bloodstream in ionic form, mainly oxidized Cu2+ and reduced Cu+, and these two forms can be interconverted.1 During their interconversion, specific essential proteins and enzymes utilize the redox properties of copper to perform physiological functions (eg, electron transfer in the mitochondrial respiratory chain). According to the results of the study, the level of copper in tumor tissue and serum was significantly increased in cancer patients.2 Based on this, scientists have seen the important role of copper metabolism in tumor development and the importance of copper metabolism in the diagnosis and treatment of tumors. Elevated serum copper levels were detected in 68% of patients with gastrointestinal cancers and this elevation was positively correlated with tumor stage.3 Similarly, in breast cancer, tumor tissue copper concentration was 2.3-fold higher than that in adjacent normal tissue.4 For example, while studying illismo, Tsvetkov et al discovered a copper-dependent death that, unlike cell necrosis,5 apoptosis,6 and autophagic cell death,7 occurs when copper is directly bound to the lipoylated components of the tricarboxylic acid cycle.8 This article reviews the significance of copper metabolism and the regulation of cell death by copper metabolism in cancer research.
Copper Metabolism and Its Regulatory Mechanism
When in the determinants of copper ionophore intacxicity, Tsvetkov et al found that the use of BSO depletes the endogenous intracellular copper chelator glutathione, thereby inducing cell death; At the same time, intracellular isoprenoid levels are significantly increased after treatment with potent copper ionophore pulses,8 indicating that cuproptosis is triggered by excessive intracellular copper accumulation. Excess copper binds directly to acylated proteins involved in TCA intracellularly, eventually leading to abnormal protein aggregation and ultimately leading to the loss of iron-sulfurin in the respiratory chain complex, and cells will be stressed due to changes in proteins, leading to cell death.
Definition of Copper Metabolism and Its Mechanism
Copper metabolism refers to a series of physiological processes such as the absorption, transport, distribution, storage and excretion of copper in the human body.
Copper transporters refer to membrane proteins responsible for copper uptake, intracellular transport, and excretion, such as CTR1 (high-affinity copper importer for Cu+ uptake) and ATP7A/B (copper exporters mediating copper efflux to bile or bloodstream).9 In humans, cellular uptake of copper is primarily provided through the Cu transporter CTR1 (copper transporter 1),9 and in a peptide-based model of CTR1, Thibaut Galler et al found that the trimeric arrangement of the N-terminus of CTR1 enables specific copper uptake in intestinal epithelial cells.10 After entering the bloodstream, it is transported primarily by ceruloplasmin (CP),11 which accounts for at least half of the Cu in plasma,12–14 in contrast to albumin and transcudin, which are the main components of the exchangeable plasma Cu pool. Subsequently, copper ions are transported to the liver through the portal venous system,15 where they are primarily stored in hepatocytes. In addition, excess copper is excreted into bile through the ATPase copper transporter β (ATP7B).16
The mechanism of copper ion action in the organism is mainly regulated through three types of proteins. Combined with the metabolic flow of copper metabolism in the organism, the first category is mainly copper transmembrane transport-related proteins, such as CTR1 which mainly transports Cu+ The second category is proteins that bind and store copper ions, including the natural intracellular copper ion chelators: the MT and glutathione (GSH).17 The third category is the copper ion chaperones,18 the cytoplasmic copper ion chaperone (ATOX1) binds Cu(I) via two cysteamine residues and transports it to the metal binding site of ATP7B.17 Superoxide dismutase (CCS) directly binds copper ions and transports them to superoxide dismutase 1 (SOD1).19 Among other things, SOD1 catalyzes the production of H2O2 from superoxide radicals as a means of maintaining free radical homeostasis in the body, an imbalance of which may lead to cell death.20 The cytochrome c oxidase copper chaperone protein 17 (COX-17) transports Cu2+ to secondary copper-containing proteins including cytochrome c oxidase synthase protein 1 (SCO1), SCO2, and thereafter further to the two subunits of SCO, thus activating the enzyme activity of the respiratory chain.21 COX17 carries the copper ions from the cytoplasm to the inner mitochondrial membrane, and a portion of the copper ion enters the inner mitochondrial membrane by means of SLC25A3; some are involved in constituting the correct spatial conformation of SOD1 (which is located in the membrane gap) and in ensuring the correct function of cytochrome C oxidase in the inner membrane; and the other part is expelled into the cytoplasm. These three proteins, which are closely related to copper metabolism, regulate the normal physiological functions of the organism through different mechanisms, as seen in Figure 1 (eg, Figure 1).
 |
Figure 1 Copper transport and metabolic pathways in the body. Arrows indicate the direction and route governing copper ion transport within the organism, with the proteins positioned above the arrows corresponding to the indispensable transporters and factors involved in this transport cascade. |
Regulatory Mechanisms of Copper Metabolism
Cell death caused by excessive copper accumulation has been a hot topic for many scholars. At present, different scholars have put forward many hypotheses on this kind of problem through experiments, mainly focusing on the signaling pathways involved in the four types of proteins that regulate copper metabolism. Here, we will focus on the signaling pathways regulated by the copper transport proteins CTR1, ATP7A and ATP7B, the copper binding proteins CP, GSH and MT, and the copper chaperones ATOX1, SOD1 and COX-17. The role of these copper metabolism-related proteins is shown in Table 1. (eg, Table 1).
 |
Table 1 Genes Involved in Copper Metabolism |
From Figure 2, it can be seen that different copper metabolism-related proteins regulate the development process of tumors by affecting different pathways. (eg, Table 2) The MAPK (mitogen-activated protein kinase) pathway is a key cell proliferation pathway stimulated by Cu. It is involved in the regulation of various physiological functions such as cell proliferation, death and differentiation in the organism. CTR1, a transporter protein for copper ions, responds to the activation of extracellular Ras signaling pathway and further activates the MAPK pathway,33 thus the MAPK pathway regulated by copper ion imbalance plays an important role in cancer development.
 |
Table 2 Mechanisms of Drug Resistance Associated with Copper Metabolism |
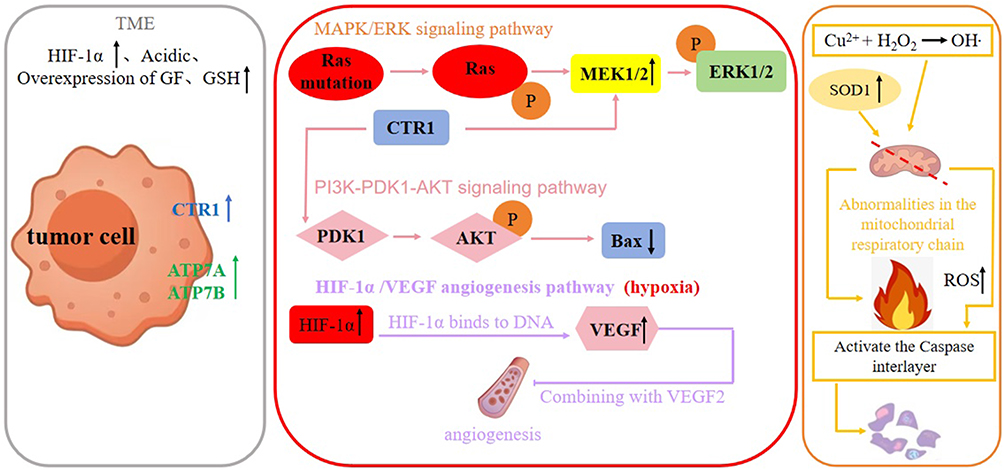 |
Figure 2 Copper metabolism regulates tumorigenicity. This figure depicts copper transporter CTR1-regulated signaling pathways in the tumor microenvironment (TME) and the molecular mechanism by which copper ions induce reactive oxygen species (ROS) to promote tumor cell apoptosis. An upward arrow following a protein or factor indicates promotion, while a downward arrow denotes inhibition. |
Ras is one of the central cellular regulatory molecules located in a variety of signaling and proteins38 and is an upstream protein of the Raf-MEK-ERK pathway.39 CTR1 is an important regulatory protein on the Ras-to-Erk signaling pathway that stimulates the level of phosphorylation of Erk1/2. Erk1/2 is an extracellular signaling-regulated kinase- serine-threonine kinase, whose hyperactivation plays an important role in cancer development.40 Active Ras binds and activates Raf kinases, which phosphorylate and activate the phosphorylation of the serine-threonine MAPK kinases Mek1 and Mek2.33 Through CTR1, copper plays a role in the Ras/MAPK signaling pathway at the binding site of Mek1/2 phosphorylation of Erk1/2. In addition, Mek1 can directly interact with Cu33 to stimulate Erk1/2 phosphorylation. In both mechanisms of action, Mek1 can function as a key kinase regulator of Erk signaling.41
MT is a cysteine-rich, low-molecular metal-binding protein that balances free radicals in the body to reduce stress responses and contains clusters of metal thiolates that bind heavy metals. Its high affinity for heavy metals prevents metal toxicity,42 for example, MT plays a role in Cu(I) binding and detoxification when intracellular copper is overloaded, and there are four isoforms of MT, among which, MT1 and MT2 can chelate SLC31A1 to transport large amounts of copper ions.43 In different types of tumor cells, MT expression varies. In melanoma, nasopharyngeal carcinoma and other tumor cells, the expression is up-regulated;44,45 in hepatocellular carcinoma, the expression is down-regulated.46 The sulfhydryl group of GSH has a strong reducing property, which reduces Cu2+ to Cu+ and promotes its binding to transporter proteins. In addition, GSH can also bind to free copper ions to form GSH-copper complexes, which prevents excessive free copper accumulation in the body and avoids oxidative stress, preventing the body from accumulating excessive free copper and triggering oxidative stress.
Mitochondria are the main site of cellular respiration, and high levels of ROS can cause mitochondrial dysfunction, leading to apoptosis and autophagy.47 Copper-mediated Fenton effect or depletion of antioxidant molecules causes elevated reactive oxygen species (ROS), which is a common metabolite in normal physiological functioning of the human body, and maintains a low to moderate level in the body, and when ROS are persistently elevated, it can be damaging to both normal and tumor cells,48 leading to mitochondrial dysfunction resulting in cell death. The Fenton effect mediated by copper ions generates active free radicals that greatly elevate the concentration of reactive free radicals and promote cell death.49 In the combined treatment of cancer with elesclomol and copper, Gao et al50 found that elesclomol promoted the degradation of ATP7A, leading to the retention of copper in mitochondria and the accumulation of reactive oxygen species (ROS), which promotes the degradation of CTR1 (SLC7A11), enhances oxidative activation of cancer cells, and delays the proliferation of cancer cells. In addition to ATP7A, copper ion chaperones and copper ion binding proteins also regulate cancer cells by regulating ROS and thus cancer cells. For example, CCS binds cytoplasmic copper to transport copper to SOD1,51 which transports copper to the nucleus and activates the transcription factor HIF-1α;52 Atox1 mediates cancer cell metastasis by coordinating copper transport.53 Glutathione (GSH) is a specific antioxidant in the body and its associated antioxidant system reduces cellular damage by reactive free radicals.54 The transport of copper by these proteins maintains the relative stability of copper ion concentration within the cell and, in turn, the relative stability of ROS.
Hyperactivation of the PI3K-PDK1-AKT axis is a central template for many important physiological or pathological metabolisms. It has been shown that the PI3K-AKT signaling pathway is frequently dysregulated in many tumorigenesis,55 so it can be assumed that copper metabolism-associated proteins have an impact on the proper functioning of the PI3K-AKT signaling pathway. Some researchers have found that copper can strongly activate AKT kinase in a transporter protein CTR1-dependent manner, and CTR1 binds to PDK1 to activate AKT kinase and promote AKT phosphorylation; moreover, CTR1 is negatively regulated by Nedd4l-mediated ubiquitination and degradation. Copper activates the PI3K-AKT pathway in a CTR1-dependent manner, and Nedd4l-mediated ubiquitination of CTR1 negatively regulates this process;56 recent studies have further clarified the role of copper ions in PDK1-AKT binding, providing a new target for pathway inhibition.
Mechanism of Cuproptosis
Cuproptosis, a copper-dependent cell death modality first identified by Tsvetkov et al,8 is fundamentally distinct from apoptosis, ferroptosis, and necroptosis. Its core mechanism involves two key steps: first, excessive intracellular copper directly binds to lipoylated components of the tricarboxylic acid (TCA) cycle (eg, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase), leading to abnormal aggregation of these lipoylated proteins;8,17 second, aggregated proteins induce the loss of iron-sulfur (Fe-S) clusters in mitochondrial respiratory chain complexes (eg, Complex I, Complex II), disrupting electron transport and ATP synthesis.8 This dual effect triggers proteotoxic stress and mitochondrial dysfunction, ultimately leading to cell death.
Recent studies have further supplemented the regulatory network of cuproptosis: for example, glutathione (GSH) acts as an endogenous copper chelator to inhibit cuproptosis by reducing free copper ions;54 while copper ionophores (eg, elesclomol) promote cuproptosis by depleting GSH and increasing intracellular copper accumulation.50 Additionally, the copper chaperone ATOX1 was found to inhibit cuproptosis by transporting copper to ATP7B for efflux, and knockdown of ATOX1 significantly enhances the sensitivity of colorectal cancer cells to copper ionophores. These findings indicate that cuproptosis is tightly regulated by copper metabolism-related proteins and provides a new target for tumor therapy.
Abnormal Copper Metabolism Regulates Tumor Occurrence Through Tumor-Related Signaling Pathways
Copper metabolism affects tumorigenesis through three interrelated pathways: regulating tumor-associated signaling pathways, reshaping the tumor microenvironment, and disrupting mitochondrial function. The specific mechanisms are elaborated as follows. Abnormal regulation of copper metabolism, ie, imbalance of copper concentration in the body, is the main cause of tumorigenesis, because the imbalance of copper concentration in the body leads to abnormal function of related proteins, which cannot work correctly within the related signaling pathways, leading to lesions of normal cells or proliferation of tumor cells. Abnormal copper metabolism not only disrupts intracellular redox homeostasis but also interacts with key signaling pathways in the tumor microenvironment, thereby promoting tumorigenesis; the specific regulatory mechanisms are as follows.
Compared with the normal physiological environment, tumors form their special ecological environment called tumor microenvironment (TME) during the development process, which is characterized by hypoxia, PH acidity, high glutathione and overexpression of growth factors, which provides tumor cells with the support needed for tumor growth and also influences the process of tumor development. Copper exhibits abnormal distribution and regulation in the tumor microenvironment (TME). Within the serum of Wilson patients, CP activity was significantly reduced,57 while within the serum of patients with gastrointestina cancers, CP levels showed an elevated trend.58 Copper ions can act as one of the catalysts, catalyzing the overexpression of hydrogen peroxide to mediate the Fenton reaction, generating hydroxyl radicals by which tumor cells are killed.59 Copper is one of the essential trace elements, which is involved in a variety of key physiological processes within cells and can regulate key components of the tumor microenvironment.
Copper ions are cofactors for a number of enzymes including cytochrome c oxidase,60 which plays a key role in the oxidative phosphorylation of cellular respiration, participates in the electron transport chain, and promotes ATP synthesis. Copper is also involved in the regulation of the activity of oxide dismutase (SOD), which scavenges intracellular superoxide anion radicals and is essential for maintaining intracellular redox balance.
In tumor cells, elevated concentrations of copper have been detected, and the tumorigenesis in which copper metabolism is involved is mainly accomplished through the promotion of tumor angiogenesis61 and the generation of excess ROS. Abnormalities in the expression of copper metabolism proteins lead to hyperactivation or aberrant inhibition of the relevant pathways resulting in the proliferation of tumor cells.
Copper metabolism is associated with the more tumor-associated signaling pathways. The MAPK signaling pathway is closely associated with the development of several cancer types.62 In particular, 40–50% of melanomas and other common tumors such as thyroid cancer have mutations in the BRAF gene, resulting in constitutive activation of MAPK channels.63 MAPK mediates intracellular signaling that is relevant to a wide range of cellular activities, including cell proliferation, differentiation, survival, death, and transformation.64–66There are four signaling families that comprise the MAPK pathway: the MAPK/ERK family, BMK-1, JNK and p38 signaling family; three kinases: MAP3K, MAPKK, and MAPK. JNK and p38 signaling are mainly associated with cellular stress and apoptosis, whereas the ERK/MAPK signaling pathway is closely related to cell proliferation.37,67,68 JNK/p38 MAPK can induce caspase−8-mediated cleavage of the pro-apoptotic pure BH3 protein Bid, which ectopically translocates the C-terminus of Bid, aggregates to mitochondria and induces the release of cytochrome c, which further induces cysteine asparaginase activation.69 In addition, p38 and JNK can also regulate the autophagic program, where activation of receptor-interacting proteins (serine-threonine kinases) and Jun amino-terminal kinases induce cell death in the form of autophagy, and autophagic death requires the genes ATG7 and beclin1 and is induced by caspase-8 inhibition.70 Hypersensitive response and sustained activation within the MAPK pathway are responsible for cell death through a positive feedback loop.71 The release of cytochrome c from mitochondria induces the activation of caspase-9 and subsequent caspase-3, which in turn hydrolyzes Bal-2 and promotes the release of more caspase-9 and caspase-3 from cytochrome c through positive feedback regulation.72 Hydrolysis of the Bid protein of caspase-8 produces t-Bid, which goes on to induce the release of cytochrome c that which in turn promotes protein hydrolysis and activation of caspase-8, closing the vicious circle.73
The HIF-1 (hypoxia-inducible factor-1) pathway is a major regulator of angiogenesis and is synergistically associated with VEGF and PIGF.74 Under hypoxic conditions, HIF accumulation is directly up-regulated mainly by VEGF,75 which induces the expression of related receptors,23 disrupting oxygen homeostasis, and when oxygen homeostasis is disrupted, VEGF binds related receptors and stimulates capillary growth.76 Copper ions regulate the activity of HIF-1α in the tumor microenvironment (TME), promoting VEGF expression and angiogenesis. Beyond this, Li et al found that copper metabolism disorders could upregulate the expression of M2-type macrophage markers (eg, CD206, IL-10) in TME by activating the STAT3 signaling pathway, thereby inhibiting the anti-tumor immune response of T cells. This finding further clarifies the crosstalk between copper metabolism and TME immune regulation.77 If copper ions accumulate in excess, HIF-1α is overexpressed and tissue hypoxia is enhanced, which promotes angiogenesis and thus the process of tumor cell development.
As a redox sensor, CTR1 plays an important role in promoting vascular proliferation, and its main angiogenic growth factor is VEGF2. Upon inhibition of CTR1, the levels of VEGF-induced p-MEK1/2, p-ERK1/2, p-p38MAPK, and p-Akt were reduced,78 and angiogenesis was inhibited. Rong et al also demonstrated that in ovarian cancer cells that Cu complexes significantly induced apoptosis in tumor cells, showing significant anti-ovarian cancer and anti-angiogenic effects.79 Reactive oxygen species can also act as a signaling molecule to promote angiogenesis.80 NGF (neurotrophic growth factor) affects the extent of transport and localization of the transport proteins CTR1 and CCS and is able to bind to Cu to activate a kinase cascade reaction that induces Erk1/2 phosphorylation and promotes the MAPK signaling pathway.81 In addition, restoration of ATP4B expression significantly inhibits cell proliferation, cell viability, migration, invasion, tumorigenicity, and induction of apoptosis, and 34 of the differential proteins in ATP4B, such as Bax, Bid, and Bcl-2, are involved in mitochondrial function and structure, and are closely related to energy production and apoptosis.82 The disappearance of the cristae within the mitochondria is an important marker of dysfunction in respiratory chain organisms.83
In confirming the mechanism of action of ATP4B on mitochondria, Y. Pan et al found that ectopic expression of ATP4B decreased the membrane potential of mitochondria, ATP production was reduced, intracellular ROS increased, and mitochondria showed vacuolated and almost completely depleted inner cristae.84 Excess reactive oxygen species in mitochondria have been shown to be effective in killing cancer cells85 and can activate intrinsic apoptotic pathways.86 Second, tumor immune escape is an important way for tumor cells to evade effective killing by the immune system. Programmed cell death ligand-1 (PD-LI) on the surface of tumor cells binds to programmed cell death receptor-1 (PD-1) on the surface of T cells, triggering apoptosis of tumor-reactive T cells, decreasing CD8+ T cell-mediated tumor cytotoxicity, and promoting immune escape,87 which facilitates tumor angiogenesis, and induces tumor cell metastasis and migration.
Due to metabolic abnormalities in tumor cells, tumor cells tend to have low ROS levels, and in copper metabolism CP can transport copper to other parts of the tissue, but also act as a scavenger of free radicals and superoxide ions to maintain stable ROS levels. In the cytoplasm CCS transports copper to superoxide dismutase, and SOD1 is an important antioxidant enzyme in copper metabolism, and it has been shown that SOD1 deficiency attenuates mammary tumorigenesis.88 SOD1 integrates signals from oxygen and glucose to inhibit cellular respiration and promotes glycolysis,89 and the role of SOD1 in tumor cells maintains ROS at a relatively low level and promotes tumor cell growth. In tumor cells, SOD1 acts to maintain ROS at relatively low levels, promoting tumor cell proliferation and tumor development.
In summary, in the abnormal copper metabolic pathway, copper proteins promote tumorigenesis through the regulation of signaling pathways such as MAPK and the concentration of ROS in the body. The level of intracellular copper-related proteins and the manifestation of various abnormal physiological functions caused by them may provide evidence for the diagnosis and treatment of many tumors.
The Role of Copper Metabolism and Its Regulatory Mechanisms in Tumor Diagnosis
Copper metabolism-regulated cell death provides many important evidences for tumor diagnosis. In terms of biomarkers, copper metabolism-related proteins can be used as potential tumor diagnostic markers, for example, copper transporter protein CTR1, etc. The up-regulation of CTR1 expression meets the needs of tumor cell proliferation. Since tumor metabolism differs from normal cells, detection of copper ion concentration can also be evidence for tumor diagnosis. Tumor cells resist when copper metabolism is abnormal within the cell, and detection of the expression levels and modification status of proteins associated with copper death may indicate tumor development or progression.
For example, IL-24 is a tumor suppressor with spectrally specific anti-tumor effects, which can promote apoptosis, inhibit tumor angiogenesis, and increase tumor sensitivity to radiotherapy and chemotherapy to exert anti-tumor effects; anti-apoptotic (Bcl-2) and pro-apoptotic (Bcl-XL) genes are also important proteins in anti-tumor resistance. IL-24 down-regulates anti-apoptotic gene expression and increases the expression of pro-apoptotic genes, thereby affecting normal cell growth.90 This process creates dysfunction in mitochondria and accelerates the process of apoptosis. Therefore, intracellular IL-24 protein content and Bcl-2 and Bcl-XL genes might be important clues for tumor diagnosis and treatment. Among multiple cell death modalities, LC-ll/LC3-I and Bax can be used as marker genes to determine the process of cell death development. Duan et al demonstrated that LC-ll/LC3-I and Bax expression were up-regulated in the additive group and p-MAPK and p-ERK protein expression was down-regulated in the study of Huayu xiaoxing compound containing drug, indicating that the drug induces cellular death.91 IAV has been shown to induce a variety of programmed cell deaths, and HUANG Jiawang et al demonstrated that IAV can induce cell death by activating the HIF-1α/iNOS/VEGF signaling pathway.92 In the developmental process of cell death, many scholars have seen the important role of MAPK and its related signaling pathway. Detecting the expression level of key proteins in the signaling pathway can be an important basis in clinical tumor diagnosis.
In ovarian cancer cells, the expression of ERK1/2, AKT, FAK and VEGFR2 proteins and their phosphorylated proteins p-ERK1/2, p-AKT, p-FAK and p-VEGFR2 in the VEGF/VEGFR2 signaling pathway is up-regulated, and the phosphorylation level of the related proteins and the level of angiogenesis can be detected to assess the level of tumor development. In addition to detecting phosphorylation levels, K. Elsnerova et al assessed the prognosis and progression of ovarian cancer by performing real-time PCR to evaluate the stability of six potential reference genes reflecting the expression levels of 12 proteins such as ATB7A, ATP11B, and others.93 Through immunohistochemistry (IHC), tumor immune cell analysis, and cancer-related pathway analysis, Y. Li et al assessed the progression, analysis, and infiltration of tumor cells by detecting the expression of copper metabolism-related proteins and genes.94 Among the mechanisms of protein regulation of tumor cells discussed earlier, SOD1 plays an important role in tumors, and detection of SOD1 level can reflect the level of intracellular ROS at a certain level.
Abnormal levels of copper metabolism-related proteins provide important evidence for tumor diagnosis, in addition to this, abnormal levels of copper metabolism-related proteins can also be an important target for tumor diagnosis, and through the targeted regulation of certain proteins, the relevant signaling pathways can be stabilized to inhibit the process of tumor development.
The Role of Copper Metabolism and Its Regulatory Mechanism in Tumor Therapy
Given the crucial role of copper metabolism in tumor cells and the continuous advancement of related research, targeting copper metabolism in tumor cells has become a popular therapeutic entry point. Identifying key pathways related to copper regulation is a critical prerequisite for developing targeted drugs for M. L. Turski et al pointed out that the MAPK signaling pathway is a key cell proliferation pathway stimulated by copper, which may serve as a direct target for potent copper chelation-based cancer chemotherapeutic drugs.33 Currently, the core of tumor therapeutic strategies designed around copper metabolism lies in two categories of targeted drugs: drug optimization and cuproptosis-mediated combination therapy. The integration of nanotechnology has further expanded the potential applications of cuproptosis research.
Core Categories of Copper-Targeted Tumor Therapeutic Drugs
Copper-targeted drugs interfere with the survival and proliferation of tumor cells by regulating intracellular copper ion levels in tumor cells. Currently, they are mainly divided into two categories: “copper chelators” (reducing intracellular copper) and “copper ionophores” (which increase intracellular copper). Although their mechanisms of action are opposite, both drugs target copper metabolism to exert antitumor effects, and the application of some drugs provides references for the treatment of copper metabolism disorders.
Copper Chelators: Inhibiting Tumors by Reducing Intracellular Copper
Copper chelators bind to copper ions in the body, thereby reducing the amount of copper available to tumour cells and inhibiting tumour growth, angiogenesis, and associated signalling pathways. A plethora of copper chelators with antineoplastic properties have been the focus of extensive research in recent times,13 including
•Tetrathiomolybdate (TM): The compound known as tetrathiomolybdate (TM) is of particular interest in this study. The anticancer properties of the substance in question were first proposed by Brewer and Merajver.94 It has been demonstrated that TM can inhibit the activity of NF-κB, thereby reducing the expression of VEGF, bFGF, IL-1α, IL-6, and IL-8.95 Furthermore, TM has been shown to downregulate HIF-1α, thus decreasing the expression of pro-angiogenic genes.94 In addition to these effects, TM has been demonstrated to inhibit the activities of SOD1, ATOX1, and ATP7A, thereby comprehensively interfering with the copper metabolism of tumour cells.
•D-penicillamine (D-pen): The process of tumour growth is reduced by the depletion of copper ions. The effects of the drug include the inhibition of tumour angiogenesis and the expression of intercellular adhesion molecules,96 as well as the suppression of LOX activity and the reduction of VEGF expression, thus hindering cancer progression.97
•Trientine: It has been demonstrated that the polyamine structure of the substance in question is capable of reducing the production of IL-8 in liver cancer98 and of inhibiting the expression of CD31.99 A substantial body of research has been dedicated to the study of drugs associated with copper metabolism in clinical practice. The mechanisms of action and applicable symptoms of these drugs are outlined in Table 3. The detection of copper chelators in plasma is contingent on the utilisation of high-sensitivity methodologies. Recent advancements in liquid chromatography-tandem mass spectrometry (LC-MS/MS) have enhanced the precision of trientine quantification.100
 |
Table 3 Mechanism of Action of Copper Chelators and Copper Ion Carriers and Progress in Clinical Trials |
It is noteworthy that copper chelators can also be utilised in the treatment of diseases caused by copper metabolism disorders, such as Wilson’s disease (an autosomal recessive genetic disorder), which is caused by mutations in the ATP7B gene encoding a transporter ATPase.101 This mutation leads to excessive accumulation of copper in the liver, brain, and eyes, resulting in the formation of golden-brown to green-brown annular copper deposits around the cornea. However, treatment with copper chelators can eventually eliminate corneal copper discolouration.102 Furthermore, tretinoin has been shown to counteract excessive copper accumulation in the body by forming stable complexes and reducing copper transport and uptake.103 In April 2022, the FDA approved TETA-4HCl for adult patients with stable Wilson’s disease who have undergone copper-depletion therapy and are tolerant to penicillamine104 (as per Figure 2, eg, Figure 2).
Copper Ionophores: Inducing Tumor Cell Death by Increcytotoasing Intracellular Copper
Copper ionophores (also known as copper ionophore molecules) are small molecules that facilitate the transmembrane entry of copper ions into cells. The induction of cuproptosis is achieved through the augmentation of the intracellular copper concentration. Common ionophores in current use include disulfiram (DSF), elesclomol, and everolimus, and their anti-cancer effects are entirely dependent on copper ions.105
•Disulfiram (DSF): It has been demonstrated that this compound is capable of forming complexes with metal ions, thereby inhibiting proteasome activity in cancer cells. In addition, it has been shown to target and regulate gene expression and cell signalling pathways, ultimately inducing apoptosis and inhibiting the proliferation and metastasis of cancer cells.106
•Elesclomol: The mechanisms by which this occurs are not yet fully elucidated, but it has been demonstrated to target mitochondrial metabolism, inducing oxidative stress and inhibiting the tricarboxylic acid cycle (TCA),8 thereby suppressing tumour progression.
The mechanisms of action of these ionophores also include DNA interaction and reactive oxygen species (ROS) generation,107 and their research has also benefited from the therapeutic experience of copper metabolism disorders, such as Menkes disease (an X-linked recessive copper transport disorder), which is caused by different mutations in the copper-transporting ATPase ATP7A. A plethora of small-molecule copper complexes have been employed in the treatment of this disease, yielding a range of outcomes.108 These studies serve as a valuable reference point for comprehending copper metabolism within tumour contexts.
Structural Optimization of Copper-Targeted Drugs
Despite the evidence that traditional copper-targeted drugs have antitumour potential, systemic toxicity and insufficient tumour targeting remain significant obstacles to their clinical application. Recent studies have focused on optimising the molecular structure of drugs to enhance their specific binding and delivery efficiency to tumour cells in order to address these issues. For instance, researchers have achieved substantial enhancement in the tumour-targeting efficacy of drugs through the modification of the ligand structures of copper chelators.109 In vitro experiments have demonstrated that the delivery efficiency of the optimised drugs is 30–50% higher than that of the original formulations. This modification strategy provides a new direction for reducing the systemic toxicity of copper-targeted drugs and improving their clinical safety.109
Novel Strategies for Cuproptosis-Mediated Combination Therapy
The in-depth study of cuproptosis has led to a new research focus on combination therapy based on copper metabolism.
For instance, Wang et al108 constructed chiral copper-amino acid nanoparticles capable of simultaneously depleting glutathione (GSH) in tumour cells and converting Cu2+ to Cu+. The present design has been demonstrated to induce cuproptosis in addition to enhancing the efficacy of photothermal therapy (PTT) through the augmentation of intracellular reactive oxygen species (ROS) levels. In a murine model of colorectal cancer, this combined strategy achieved a tumour inhibition rate of 89.7%, which was significantly higher than that of PTT alone (56.2%).
Moreover, recent preclinical studies have demonstrated that the combination of copper chelators with immune checkpoint inhibitors (ICIs) can reverse ICI resistance. For instance, tetrathiomolybdate (TM) has been shown to reduce regulatory T cell (Treg) infiltration in the tumour microenvironment (TME) by decreasing copper lev els, thereby enhancing the antitumor activity of anti-PD-1 antibodies.22 These findings suggest that copper metabolism-based combination therapy has broad application prospects for overcoming tumour drug resistance and improving treatment efficacy.
Application of Nanotechnology in Copper Metabolism-Mediated Tumor Therapy
In addition to the structural optimisation of drugs and the development of cuproptosis-mediated combination therapy, advances in nanotechnology have provided more efficient technical support for copper metabolism-mediated tumour therapy. The utilisation of nanomaterials confers several advantages, including the capacity to circumvent systemic damage and the selective targeting of tumour cells, thereby facilitating the regulation of copper metabolism at the specific site of action.22 The combination of these modalities with other therapeutic interventions can yield combination therapy, thereby enhancing therapeutic efficacy.
The Following Specific Applications are Worthy of Note
•Cuproptosis-mediated combination therapy: Researchers have devised combination strategies that utilise nanomaterials. For instance, Wang et al employed the chirality of nanomaterials to bind excess GSH in tumour cells and convert Cu2+ to Cu+. It has been demonstrated that by increasing the concentration of ROS, a significant increase in the death of tumour cells was observed, thereby achieving the effect of inhibiting tumour cell growth.24
•Optimization of controlled release and local therapy: The copper-containing nanocapsules designed by Liao et al have been shown to achieve the controlled release of copper ions when exposed to ionising radiation,25 thereby optimising the precision of local anti-tumour therapy.
•Synergy with other therapeutic approaches: Beyond enhancing targeting, nanomaterials have the potential to be integrated with chemotherapy, photothermal therapy and sonodynamic therapy to improve the efficacy of copper-dependent cell death.25 Furthermore, the employment of Cu nanoparticles has been demonstrated to enhance the efficacy of conventional chemotherapeutic agents by generating ROS and enhancing drug stability, thereby underscoring their potential in overcoming tumour drug resistance.26
•Supporting chemodynamic therapy (CDT): In CDT, copper ions act as catalysts to promote the accumulation of H2O2 and the elimination of GSH in tumour cells, thereby catalyzing the intracellular Fenton reaction and specifically killing tumour cells.26 The employment of nanomaterials ensures the precise delivery of copper ions, thereby enhancing catalytic efficiency.
Summary and Prospects
In conclusion, the tumour therapeutic strategy targeting copper metabolism regulation has formed a comprehensive research system encompassing “copper-targeted drugs (chelators + ionophores) - drug structural optimization - cuproptosis-mediated combination therapy - nanotechnology empowerment.” The system has been shown to inhibit tumour growth and progression by regulating intracellular copper levels, interfering with copper-dependent signalling pathways, or inducing cuproptosis.
As research into the regulatory mechanisms of copper metabolism in diseases progresses, cuproptosis has emerged as a promising therapeutic avenue for tumours and has provided novel insights into the treatment of other copper metabolism-related diseases. In the future, it is anticipated that copper metabolism-mediated therapeutic strategies will be more widely applied in clinical practice. This will be achieved by further optimising drug targeting, exploring more combination therapy regimens, and advancing the integration of nanotechnology.
Conclusion
In summary, cell death mediated by copper metabolism has an inextricable role in tumorigenesis, screening, diagnosis and treatment. Despite the continuous development of tumor diagnostic techniques, they still face certain limitations. The specificity and sensitivity of tumor markers need to be improved. More and more scholars are focusing on copper ions as a therapeutic target to achieve inhibition of tumor cells. Improving tumor diagnosis accuracy not only relies on biological markers but also requires attention to patients’ psychological status; studies have shown that personalized copper-targeted therapy can reduce anxiety in patients with advanced tumors.27 These researches on the mechanism of copper metabolism provides a theoretical basis for the study of anticancer drugs.
In the future, with the advancement of biotechnology such as single-cell RNA sequencing (scRNA-seq) for copper metabolism-related gene profiling, spatial metabolomics for in situ detection of intracellular copper levels, and intelligent nano-drug delivery systems (eg, chiral copper-amino acid nanoparticles) for targeted copper ion delivery, it is expected to achieve high-sensitivity detection of cuproptosis-related biomarkers (eg, CTR1, SOD1), optimize the combination regimen of copper-targeted drugs (eg, TM combined with immune checkpoint inhibitors), and establish a “copper metabolism-TME-immune response” integrated diagnosis and treatment model. These advances will further improve the specificity and accuracy of early tumor diagnosis, prolong the overall survival of patients with advanced tumors, and enhance their quality of life.
Acknowledgments
This article was supported by Grant No.CX2024082Z from Jining Medical University, Grant No.2023YXNS037 from Jining City Key Research and Development Program, Grant No.CXPJJH122001-2243 from Hubei Chen Xiaoping Science and Technology Development Foundation, Grant No. 202301060260 from Medicine and Health Projects of Shandong Province.
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63(5):797s–15. doi:10.1093/ajcn/63.5.797
2. Denoyer D, Masaldan S, Fontaine LA, et al. Targeting copper in cancer therapy: “Copper that cancer”. Metallomics. 2015;7(11):1459–1476. doi:10.1039/C5MT00149H
3. Weinlich R, Oberst A, M BH, et al. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–136. doi:10.1038/nrm.2016.149
4. Boz A, Evliyaoğlu O, Yildirim M, et al. The value of serum zinc, copper, ceruloplasmin levels in patients with gastrointestinal tract cancers. Turk J Gastroenterol. 2005;16(2):81–84.
5. Pan Q, Kleer CG, Van Golen KL, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62(17):4854–4859.
6. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070
7. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
8. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
9. Öhrvik H, Thiele DJ. The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J Trace Elem Med Biol. 2015;31:178–182. doi:10.1016/j.jtemb.2014.03.006
10. Galler T, Lebrun V, Raibaut L, et al. How trimerization of CTR1 N-terminal model peptides tunes Cu-binding and redox-chemistry. Chem Commun. 2020;56(81):12194–12197. doi:10.1039/D0CC04693K
11. Ramos D, Mar D, Ishida M. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11(3):e0149516. doi:10.1371/journal.pone.0149516
12. Sato M, Gitlin JD. Mechanisms of copper incorporation during the biosynthesis of human ceruloplasmin. J Biol Chem. 1991;266(8):5128–5134. doi:10.1016/S0021-9258(19)67764-1
13. Hirano K, Ogihara T, Ogihara H, et al. Identification of apo- and holo-forms of ceruloplasmin in patients with Wilson’s disease using native polyacrylamide gel electrophoresis. Clin Biochem. 2005;38(1):9–12. doi:10.1016/j.clinbiochem.2004.09.008
14. Middleton RB, Linder MC. Synthesis and turnover of ceruloplasmin in rats treated with 17 beta-estradiol. Arch Biochem Biophys. 1993;302(2):362–368. doi:10.1006/abbi.1993.1224
15. Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134(21). doi:10.1242/jcs.240523
16. Hernandez S, Tsuchiya Y, García–Ruiz JP, et al. ATP7B copper-regulated traffic and association with the tight junctions: copper excretion into the bile. Gastroenterology. 2008;134(4):1215–1223. doi:10.1053/j.gastro.2008.01.043
17. Xie J, Yang Y, Gao Y, et al. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22(1):46. doi:10.1186/s12943-023-01732-y
18. Kim B-E, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4(3):176–185. doi:10.1038/nchembio.72
19. Casareno RLB, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem. 1998;273(37):23625–23628. doi:10.1074/jbc.273.37.23625
20. Dong X, Zhang Z, Zhao J, et al. The rational design of specific SOD1 inhibitors via copper coordination and their application in ROS signaling research. Chem Sci. 2016;7(9):6251–6262. doi:10.1039/C6SC01272H
21. Nývltová E, Dietz JV, Seravalli J, et al. Coordination of metal center biogenesis in human cytochrome c oxidase. Nat Commun. 2022;13(1):3615. doi:10.1038/s41467-022-31413-1
22. Doguer C, Ha JH, Collins JF. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr Physiol. 2018;8(4):1433–1461. doi:10.1002/j.2040-4603.2018.tb00047.x
23. Das A, Ash D, Fouda AY, et al. Cysteine oxidation of copper transporter CTR1 drives VEGFR2 signalling and angiogenesis. Nat Cell Biol. 2022;24(1):35–50. doi:10.1038/s41556-021-00822-7
24. Tadini-Buoninsegni F, Smeazzetto S. Mechanisms of charge transfer in human copper ATPases ATP7A and ATP7B. IUBMB Life. 2017;69(4):218–225. doi:10.1002/iub.1603
25. Huang G-J, Huang GJ, Wu M. Experimental study on the interrelationship between metallothionein and copper antitumor effects. Biophysical Journal. 1997;73(3):1424–1429. doi:10.1016/S0006-3495(97)78174-8
26. Bu-Meng Chen WU, Yue-Ling JIN, Shin-Qin YE, et al. Sex differences in serum copper and zinc levels as well as SOD activity and GSH content in rats with stress gastric ulcer. Laboratory Animals Comparative Med. 2005;02:96–8+109.
27. Mangini V, Belviso BD, Nardella MI, et al. Crystal structure of the human copper chaperone ATOX1 bound to zinc ion. Biomolecules. 2022;12(10):1494. doi:10.3390/biom12101494
28. Arnesano F, Banci L, Bertini I, et al. Characterization of the binding interface between the copper chaperone Atx1 and the first cytosolic domain of Ccc2 ATPase. J Biol Chem. 2001;276(44):41365–41376. doi:10.1074/jbc.M104807200
29. Jiang S J Z. Cu+ transport system. Chemistry Life. 2003;1:41–44.
30. Seguin A, Jia X, M EA, et al. The mitochondrial metal transporters mitoferrin1 and mitoferrin2 are required for liver regeneration and cell proliferation in mice. J Biol Chem. 2020;295(32):11002–11020. doi:10.1074/jbc.RA120.013229
31. Steelman LS, Chappell WH, Abrams SL. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3(3):192–222. doi:10.18632/aging.100296
32. Li T, Peng J, Zeng F, et al. Association between polymorphisms in CTR1, CTR2, ATP7A, and ATP7B and platinum resistance in epithelial ovarian cancer. Int J Clin Pharmacol Ther. 2017;55(10):774–780. doi:10.5414/CP202907
33. Turski ML, Brady DC, Kim HJ, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32(7):1284–1295. doi:10.1128/MCB.05722-11
34. Luo J, Yao J-F, Deng X-F. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin αvβ3 and activating FAK/PI3K/AKT signaling. J Exp Clin Cancer Res. 2018;37(1):23. doi:10.1186/s13046-018-0694-6
35. Song GQ, Wu P, Dong X-M, et al. Elemene induces cell apoptosis via inhibiting glutathione synthesis in lung adenocarcinoma. J Ethnopharmacol. 2023;311. doi:10.1016/j.jep.2023.116409
36. Song GQ, Wu P, Dong XM, et al. Elemene induces cell apoptosis via inhibiting glutathione synthesis in lung adenocarcinoma. J Ethnopharmacol. 2023;311:116409.
37. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi:10.1038/nrm2308
38. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi:10.1038/nrc2109
39. Zhou B, Der CJ, Cox AD. The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol. 2016;58:60–69. doi:10.1016/j.semcdb.2016.07.012
40. Guo YJ, Pan WW, Liu SB, et al. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
41. Catalanotti F, Reyes G, Jesenberger V, et al. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16(3):294–303.
42. Klaassen CD, Liu J, Diwan BA. Metallothionein protection of cadmium toxicity. Toxicol Appl Pharmacol. 2009;238(3):215–220. doi:10.1016/j.taap.2009.03.026
43. Juárez-Rebollar D, Rios C, Nava-Ruíz C, et al. Metallothionein in brain disorders. Oxid Med Cell Longev. 2017;2017(1):5828056. doi:10.1155/2017/5828056
44. Jayasurya A, Bay BH, Yap WM, et al. Proliferative potential in nasopharyngeal carcinoma: correlations with metallothionein expression and tissue zinc levels. Carcinogenesis. 2000;21(10):1809–1812. doi:10.1093/carcin/21.10.1809
45. Weinlich G, Eisendle K, Hassler E, et al. Metallothionein - overexpression as a highly significant prognostic factor in melanoma: a prospective study on 1270 patients. Br J Cancer. 2006;94(6):835–841. doi:10.1038/sj.bjc.6603028
46. Datta J, Majumder S, Kutay H, et al. Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3-kinase signaling cascade. Cancer Res. 2007;67(6):2736–2746. doi:10.1158/0008-5472.CAN-06-4433
47. Kang Z, Qiao N, Liu G, et al. Copper-induced apoptosis and autophagy through oxidative stress-mediated mitochondrial dysfunction in male germ cells. Toxicol In Vitro. 2019;61:104639. doi:10.1016/j.tiv.2019.104639
48. Aboelella NS, Brandle C, Kim T, et al. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers. 2021;13(5):986. doi:10.3390/cancers13050986
49. Hao Y-N, Zhang W-X, Gao Y-R, Wei Y-N, Shu Y, Wang J-H. State-of-the-art advances of copper-based nanostructures in the enhancement of chemodynamic therapy. J Mater Chem B. 2021;9(2):250–266. doi:10.1039/D0TB02360D
50. Gao W, Huang Z, Duan J, et al. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15(12):3527–3544. doi:10.1002/1878-0261.13079
51. Wang J, Luo C, Shan C, et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem. 2015;7(12):968–979. doi:10.1038/nchem.2381
52. Feng W, Ye F, Xue W, et al. Copper regulation of hypoxia-inducible factor-1 activity. Mol Pharmacol. 2009;75(1):174–182. doi:10.1124/mol.108.051516
53. Blockhuys S, Zhang X, Wittung-Stafshede P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc Natl Acad Sci U S A. 2020;117(4):2014–2019. doi:10.1073/pnas.1910722117
54. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217(7):2291–2298. doi:10.1083/jcb.201804161
55. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. doi:10.1016/j.cell.2017.04.001
56. Guo J, Li B, Zhang Q, et al. Highly accessible atomically dispersed Fe‐nx sites electrocatalyst for proton‐exchange membrane fuel cell. Adv Sci. 2021;8.
57. Scheinberg IH, Gitlin D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease). Science. 1952;116(3018):484–485. doi:10.1126/science.116.3018.484
58. Tang Z, Liu Y, He M, et al. Chemodynamic therapy: tumour microenvironment-mediated Fenton and Fenton-like reactions. Angew Chem Int Ed Engl. 2019;58(4):946–956. doi:10.1002/anie.201805664
59. Gitlin JD. Wilson disease. Gastroenterology. 2003;125(6):1868–1877. doi:10.1053/j.gastro.2003.05.010
60. Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 2020;9(1):198. doi:10.3390/cells9010198
61. Dankner M, Rose AA, Rajkumar S, et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37(24):3183–3199. doi:10.1038/s41388-018-0171-x
62. Mccubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signaling. 2006;8(9–10):1775–1789. doi:10.1089/ars.2006.8.1775
63. Torii S, Yamamoto T, Tsuchiya Y, et al. ERK MAP kinase in G 1 cell cycle progression and cancer. Cancer Science. 2006;97(8):697–702. doi:10.1111/j.1349-7006.2006.00244.x
64. Dhillon AS, Hagan S, Rath O, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
65. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–869. doi:10.1152/physrev.2001.81.2.807
66. Khokhlatchev AV, Canagarajah B, Wilsbacher J, et al. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93(4):605–615. doi:10.1016/S0092-8674(00)81189-7
67. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi:10.1038/35065000
68. Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304(5676):1500–1502. doi:10.1126/science.1096645
69. Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;21(7):2346. doi:10.3390/ijms21072346
70. Kirsch DG, Doseff A, Chau BN, et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274(30):21155–21161. doi:10.1074/jbc.274.30.21155
71. Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 2002;9(10):1046–1056. doi:10.1038/sj.cdd.4401065
72. Zimna A, Kurpisz M. Hypoxia‐inducible factor‐1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int. 2015;2015(1):549412. doi:10.1155/2015/549412
73. Li Y, Zhao J, Chen S. Copper metabolism disorders reshape tumor microenvironment via macrophage polarization. Commun Biol. 2024;7(1):824. doi:10.1038/s42003-024-06488-9
74. Fan R, Wei JC, Xu BB, et al. A novel chiral oxazoline copper(ii)-based complex inhibits ovarian cancer growth in vitro and in vivo by regulating VEGF/VEGFR2 downstream signaling pathways and apoptosis factors. Dalton Trans. 2023;52(33):11427–11440. doi:10.1039/D3DT01648J
75. Tang X, Wang JJ, Wang J, et al. Endothelium-specific deletion of Nox4 delays retinal vascular development and mitigates pathological angiogenesis. Angiogenesis. 2021;24(2):363–377. doi:10.1007/s10456-020-09757-3
76. Tomasello B, Bellia F, Naletova I, et al. BDNF- and VEGF-responsive stimulus to an NGF mimic cyclic peptide with copper ionophore capability and Ctr1/CCS-driven signaling. ACS Chem Neurosci. 2024;15(9):1755–1769. doi:10.1021/acschemneuro.3c00716
77. Pan Y, Wang X, He Y, et al. Tumor suppressor ATP4B serve as a promising biomarker for worsening of gastric atrophy and poor differentiation. Gastric Cancer. 2021;24(2):314–326. doi:10.1007/s10120-020-01128-7
78. Cheung KS, Chan EW, Wong AY, et al. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: a population-based study. Gut. 2018;67(1):28–35. doi:10.1136/gutjnl-2017-314605
79. Pelicano H, Feng L, Zhou Y, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278(39):37832–37839. doi:10.1074/jbc.M301546200
80. Soberanes S, Urich D, Baker CM, et al. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284(4):2176–2186. doi:10.1074/jbc.M808844200
81. Peng Y, Zhang Z, Yang G, et al. N6-methyladenosine reader protein IGF2BP1 suppresses CD8+ T cells-mediated tumor cytotoxicity and apoptosis in colon cancer. Apoptosis. 2024;29(3):331–343. doi:10.1007/s10495-023-01893-7
82. Gomez ML, Shah N, Kenny TC, et al. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene. 2019;38(29):5751–5765. doi:10.1038/s41388-019-0839-x
83. Reddi AR, Culotta VC. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell. 2013;152(1–2):224–235. doi:10.1016/j.cell.2012.11.046
84. Lijun X, Shuping W, Chen C, et al. Effect of heating iopivol contrast agent in CT enhancement scanning by electronic thermostat. Shanghai Nurs. 2021;21(01):38–40.
85. D Kaixuan, C Shan, D Gaopi, et al. Mechanism of autophagic death of trophoblasts induced by targeting the MAPK pathway with resolving blood stasis and eliminating crimes compound. Shizhen Guojian. 2024;35(09):2156–2160.
86. H Jia-Wang, M Xin-Yue, F Zhi-Ying, et al. Mechanism of iron death induced by influenza A virus in lung epithelial cells through activation of HIF-1α/iNOS/VEGF signaling pathway. Microbiology Letters. 2024;51(01):306–322.
87. Elsnerova K, Mohelnikova-Duchonova B, Cerovska E, et al. Gene expression of membrane transporters: importance for prognosis and progression of ovarian carcinoma. Oncol Rep. 2016;35(4):2159–2170. doi:10.3892/or.2016.4599
88. Li Y, Yu Z. Pan-cancer analysis reveals copper transporters as promising potential targets. Heliyon. 2024;10(17):e37007. doi:10.1016/j.heliyon.2024.e37007
89. Brewer GJ, Dick RD, Grover DK. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin Cancer Res. 2000;6(1):1–10.
90. Denoyer D, Clatworthy SA, Cater MA. Copper complexes in cancer therapy. Met Ions Life Sci. 2018;18:469–506.
91. Kim KK, Abelman S, Yano N, et al. Tetrathiomolybdate inhibits mitochondrial complex IV and mediates degradation of hypoxia-inducible factor-1α in cancer cells. Sci Rep. 2015;5(1):14296. doi:10.1038/srep14296
92. Crowe A, Jackaman C, Beddoes KM, et al. Rapid copper acquisition by developing murine mesothelioma: decreasing bioavailable copper slows tumor growth, normalizes vessels and promotes T cell infiltration. PLoS One. 2013;8(8):e73684. doi:10.1371/journal.pone.0073684
93. Mammoto T, Jiang A, Jiang E, et al. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am J Pathol. 2013;183(4):1293–1305. doi:10.1016/j.ajpath.2013.06.026
94. Moriguchi M, Nakajima T, Kimura H, et al. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin‐8 production. Int J Cancer. 2002;102(5):445–452. doi:10.1002/ijc.10740
95. Yoshiji H, Yoshii J, Ikenaka Y, et al. Inhibition of renin–angiotensin system attenuates liver enzyme-altered preneoplastic lesions and fibrosis development in rats. J Hepatol. 2002;37(1):22–30. doi:10.1016/S0168-8278(02)00104-6
96. Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21. doi:10.1038/s41572-018-0018-3
97. Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14(1):103–113. doi:10.1016/S1474-4422(14)70190-5
98. Kirk FT, Munk DE, Swenson ES. Effects of tetrathiomolybdate on copper metabolism in healthy volunteers and in patients with Wilson disease. J Hepatol. 2024;80(4):586–595. doi:10.1016/j.jhep.2023.11.023
99. Li Y. Copper homeostasis: emerging target for cancer treatment. IUBMB Life. 2020;72(9):1900–1908. doi:10.1002/iub.2341
100. Cater MA, Pearson HB, Wolyniec K, et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem Biol. 2013;8(7):1621–1631. doi:10.1021/cb400198p
101. Jiao Y, Hannafon BN, Ding WQ. Disulfiram’s anticancer activity: evidence and mechanisms. Anticancer Agents Med Chem. 2016;16(11):1378–1384. doi:10.2174/1871520615666160504095040
102. Denoyer D, Masaldan S, La Fontaine S, et al. Targeting copper in cancer therapy:’Copper That Cancer’. Metallomics. 2015;7(11):1459–1476.
103. Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol. 2011;7(1):15–29. doi:10.1038/nrneurol.2010.180
104. Zhang L, Li M, Wang H. Copper metabolism targeted therapy for solid tumors: mechanisms and clinical progress. Pharmaceuticals. 2023;16(1):37. doi:10.3390/ph16010037
105. M MJ, Anderson G, Bottorff JL, et al. Exploring empathy: a conceptual fit for nursing practice? Image J Nurs Sch. 1992;24(4):273–280. doi:10.1111/j.1547-5069.1992.tb00733.x
106. Choukaife H, Seyam S, Alallam B, et al. Current advances in chitosan nanoparticles based oral drug delivery for colorectal cancer treatment. Int J Nanomed. 2022;17:3933–3966. doi:10.2147/IJN.S375229
107. Wang S, Zhao Y, Yao S, et al. Chirality of copper-amino acid nanoparticles determines chemodynamic cancer therapeutic outcome. Small. 2024;20(28):e2309328. doi:10.1002/smll.202309328
108. Azarian M, Ramezani Farani M, C WC, et al. Advancements in colorectal cancer treatment: the role of metal-based and inorganic nanoparticles in modern therapeutic approaches. Pathol Res Pract. 2024;264:155706. doi:10.1016/j.prp.2024.155706
109. Huang JW, Ma, XY, Feng, ZY et al, et al. Mechanism of iron death induced by influenza A virus in lung epithelial cells through activation of HIF-1α/iNOS/VEGF signaling pathway Microbiology Letters. 2024 51 101 306–322.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.