Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Research Progress on Animal Models of Chronic Obstructive Pulmonary Disease
Authors Zhang Y, Liu Z, Wan R, Zhu Y, Su M, Chen G, Zhao K, Pang Q, Li X
Received 19 February 2026
Accepted for publication 23 June 2026
Published 17 July 2026 Volume 2026:21 604242
DOI https://doi.org/10.2147/COPD.S604242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Yi Zhang,1,2,* Zhizhong Liu,1,* Ruilin Wan,1 Yihe Zhu,1 Miao Su,1 Guangshun Chen,1 Kunpeng Zhao,1 Qi Pang,2 Xueling Li2
1Clinical College of Traditional Chinese Medicine, Gansu University of Chinese Medicine, Lanzhou, People’s Republic of China; 2Lanzhou General Petrochemical Hospital (The Fourth Affiliated Hospital of Gansu University of Chinese Medicine), Lanzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qi Pang, Email [email protected] Yi Zhang, Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) is a long-term respiratory condition with high rates of morbidity and mortality. Its pathophysiology is complex, and currently, there are no effective clinical treatments that can fully reverse or halt the disease’s progression. Animal models play a significant role in elucidating the pathogenesis of COPD, dynamic pathological processes, and evaluating potential therapeutic strategies, serving as a key to advancing in-depth research on this disease. However, existing modeling methods still face many limitations in accurately simulating the clinical symptoms, pathophysiological features, and progression of human disease, and there is a lack of standardized guidelines and industry consensus. This paper systematically reviews methods for constructing COPD animal models reported in the literature by reviewing 5,071 relevant articles from CNKI, PubMed, and Wanfang Database published in the past decade. The main models include cigarette smoke exposure, protease-induced, genetically modified, harmful substance exposure, and various combined factor models. A comprehensive review and comparison of their construction principles, procedures, pathological features, advantages, and limitations are provided. This review aims to serve as a valuable reference for COPD researchers and clinicians in the construction of animal models of COPD.
Keywords: chronic obstructive pulmonary disease, animal models, pathological mechanisms, model reference
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a complex condition characterized by persistent, progressively worsening airflow limitation. According to the latest data from The Lancet’ s 2024 Global Burden of Disease study, although the age- standardized mortality rate from COPD has decreased slightly, it remains a major global public health issue due to its high prevalence and significant disability rate. It is estimated that, by 2060, more than 5.4 million people will die annually from COPD and related diseases. The disease burden of COPD is particularly severe in Asia, accounting for over 56% of global COPD-related deaths.1 Its pathological features include chronic bronchitis and emphysema, with clinical signs such as cough, sputum production, and increasing dyspnea.2
Air pollution, including fine particulate matter (PM2.5), along with cigarette smoking, may account for 50% of the risk factors for COPD. The pathogenesis of COPD is complex and not yet fully understood. It involves chronic airway inflammation characterized by infiltration of neutrophils, macrophages, and lymphocytes, an imbalance between oxidative stress and antioxidant defenses, dysregulation of the protease- antiprotease system, genetic factors such as α1- antitrypsin deficiency, apoptosis, and abnormal repair processes.3 This complexity makes it difficult for animal models to fully recapitulate the entire characteristics of human COPD. Therefore, developing animal models that can accurately reproduce key disease features, align with the pathogenesis, with good reproducibility and clinical relevance has become a core aspect in advancing in-depth research on COPD.4 Animal models not only offer a controlled platform for understanding molecular and cellular mechanisms of COPD, but also serve as essential tools for evaluating therapeutic approaches. We have observed that the animal models used in COPD-related research literature include full-disease models, phenotype-specific models, exacerbation models, and mechanism-focused models. This paper systematically reviews the various animal models currently widely used in COPD research, focusing on their modeling methods, specific operational procedures, and applicability scope, aiming to provide a useful reference for researchers and clinical practitioners working on COPD.
Methods for Establishing Animal Models of Chronic Obstructive Pulmonary Disease
Cigarette Smoke Exposure
Cigarette smoke exposure modeling methods simulate core pathogenesis by repeatedly exposing animals to cigarette smoke over extended periods, triggering a series of pathophysiological changes similar to human COPD. The main mechanism of modeling relies on the combined effects of thousands of chemicals in the smoke—such as reactive oxygen species, aldehydes, nicotine, and tar—which cause oxidative stress damage to airways and alveoli, persistent inflammatory responses, and increased protease activity.5 Key variables in modeling operations include:
Animal Selection: Involving Mice,6–8 guinea pig,9–11 Snow Weasel,12,13 dog,14,15 fruit flies,16 etc. The sex was biased toward males, with no justification provided for this choice. It is also noteworthy that In a Chinese database analysis by Li L et al of 176 COPD animal model studies, 79.55% used male animals.17 Martinez FJ et al also noted that male animals are typically selected for COPD animal models to circumvent the interference of the female estrous cycle.18
Cigarette type: Commercial cigarettes used in the studies.6,7 (such as the Red Flag Canal and Red Tower Hill) or standard research cigarettes.9,11 (such as the 3R4F from the University of Kentucky). The 3R4F is preferred in international research because of its standardized composition (about 9.4 mg of tar and 0.73 mg of nicotine per cigarette), which makes it easier to compare results across different laboratories.
Exposure system: Usually uses a full-body exposure chamber or a nasal inhalation system or nasal inhalation system.8,12 Exposing the entire body simplifies manipulation but may cause fur to absorb contaminants; nasal inhalation (eg, modeling ferrets) more directly mimics human inhalation patterns.
Exposure parameter: smoke concentration determined by smoking chamber volume and number of cigarettes burned per session8 or total particulate matter concentration or CO concentration.12 Along with daily exposure frequency, weekly exposure days, and total exposure duration, these factors collectively influence the severity and phenotypic traits of the model. For example, the mouse model cycle lasts 56 days, and it can extend up to 420 days.14,15 The longer the interval, the more evident the emphysema and decline in lung function typically become.
The choice of model evaluation metrics also varies. Liu et al,6 documented observation records of general mouse conditions such as body weight and coat luster. Lung tissue underwent hematoxylin and eosin (H&E) staining to examine pathological changes, and alveolar lavage fluid was analyzed to assess the expression of inflammatory factors. Studies by Jiang Yuhang et al,7 and Li et al,8 indicate that, although the overall condition of the mice after modeling was not included among the evaluation criteria, “lung function”—the gold standard for diagnosing COPD clinically—was among the assessment metrics. Raju S V and others.12 In addition to selecting ferrets as experimental animals, expanded the evaluation criteria to include body volume scanning and audio/video synchronized recording for cough monitoring, which better confirmed the presence of symptoms related to chronic bronchitis. Chapman et al,15 while choosing Beagles as the model animals, also measured the heart weight-to-body weight ratio in the evaluation criteria to further assess the incidence of COPD complications. Due to their small size and other features fruit flies have been used by Prange et al,16 When employed as a model organism, evaluation metrics such as lifespan and body fat percentage were used to assess the model (Table 1).
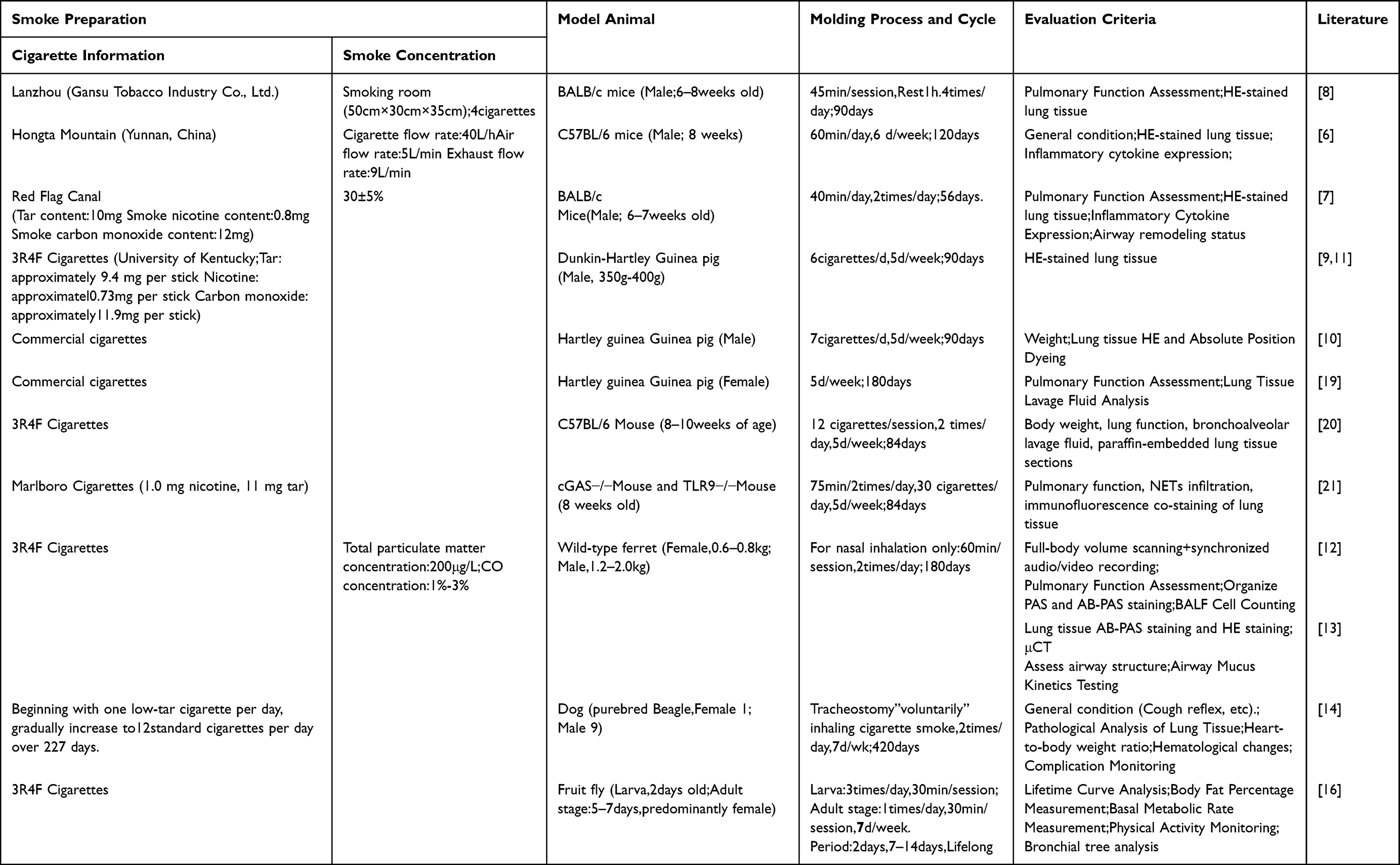 |
Table 1 Cigarette Smoke Exposure |
Applicability
CS exposure is the primary method for modeling the main causes of human COPD. This model shows the strongest link to causes and can mimic the long-term, progressive nature of COPD. It is useful for studying the development of COPD over time, environmental factors, and testing interventions to slow or stop disease progression.
Limitations
The simple cigarette smoke exposure model can simulate chronic disease progression, but it struggles to induce severe, stable inflammation and injury within a short timeframe. Some studies have also pointed out that the simple cigarette smoke model primarily manifests as excessive alveolar dilation, and the degree of inflammatory infiltration may not be as significant as that in the combined model. Additionally, the modeling cycle is lengthy (usually requiring several months), expensive, and individual animals show variable responses to smoke exposure.
Cigarette Smoke Exposure Combined with Lipopolysaccharide Infusion
The combined use of cigarette smoke and bacterial endotoxin lipopolysaccharide (LPS) quickly intensifies the inflammatory response, mimicking an acute exacerbation of chronic obstructive pulmonary disease (AECOPD).22 LPS is a component of the cell wall of Gram-negative bacteria, acting as a pro-inflammatory factor that plays a key role in the development of the airway inflammation. CS-induced epithelial damage and compromised barrier function facilitate access and activation of innate immune cells within the airways and alveoli by subsequent infusion of LPS, producing a “second-hit” effect that rapidly triggers intense neutrophilic inflammation and tissue injury.23 LPS is typically administered via endotracheal instillation or nasal drops. Single or multiple interventions are performed at specific time points during CS exposure (eg, Day 1 and Day 14),24,25 SD or Wistar rats are commonly used animals. Based on the evaluation results, the CS+LPS model shows a more pronounced decline in lung function and denser inflammatory cell infiltration (particularly neutrophils),26 along with a more significant rise in inflammatory mediators in bronchoalveolar lavage fluid (BALF) and earlier signs of airway remodeling within shorter cycles (eg, 28–56 days) (Table 2).
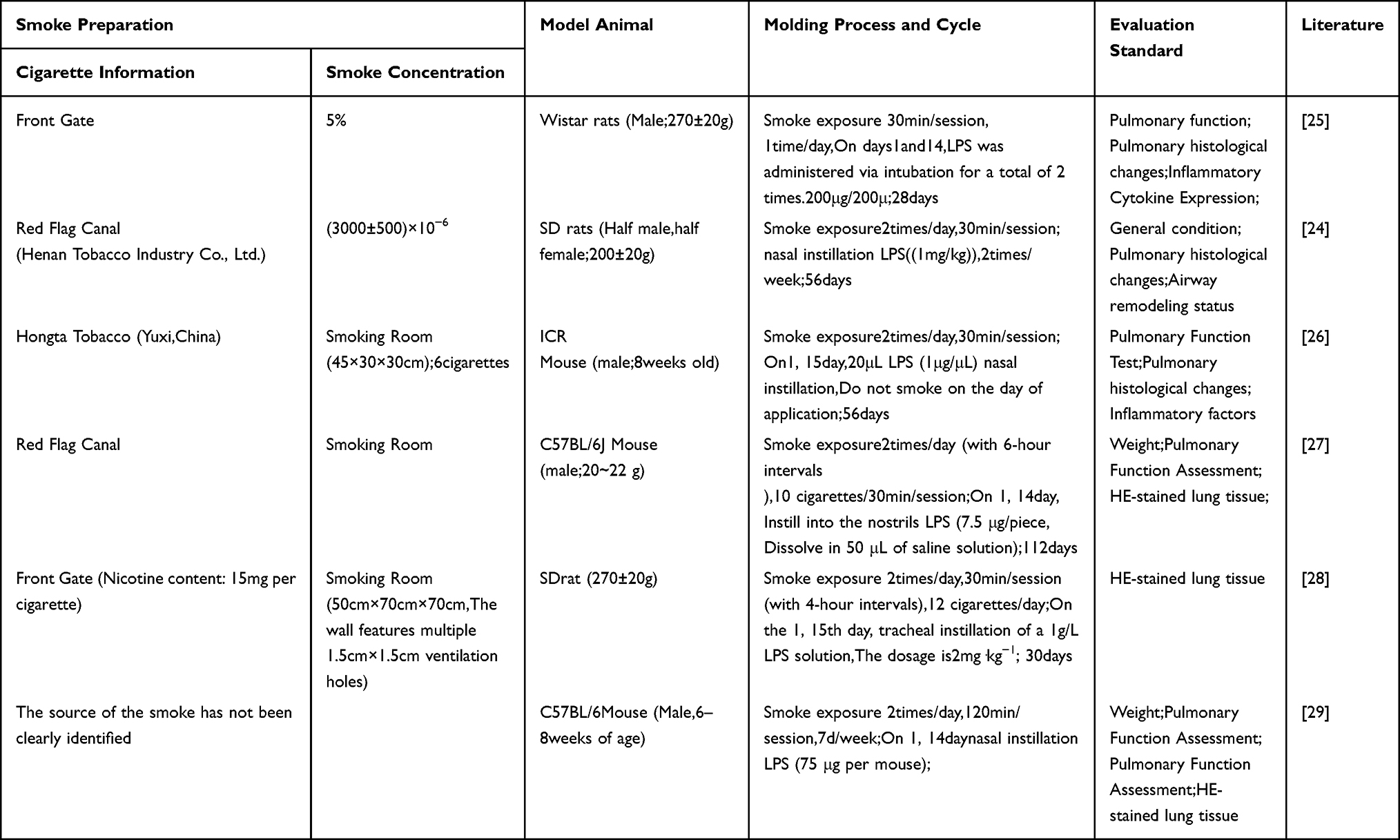 |
Table 2 Cigarette Smoke Exposure Along with Lipopolysaccharide Infusion |
Applicability
This model exhibits more severe inflammation and more pronounced pathological changes. It is suitable for investigating the pathological mechanisms of acute exacerbations in COPD, such as infection-triggered inflammatory cascades and hypermucus secretion, as well as for evaluating drugs designed to control acute inflammation, such as novel anti-inflammatory agents and Mucus Modifier.28,29
Limitations
It is challenging to identify the specific contributions of individual factors in disease development. Animals show poor tolerance, with some individuals prone to severe stress or even death under multiple stimuli.
Cigarette Smoke Exposure Combined with Bacterial/Viral Infections
COPD patients show increased airway colonization by bacteria, and bacterial and/or viral infections are the most common triggers for acute exacerbations.30 The CS co-pathogen infection model, which emphasizes infection-induced complexity, more accurately represents the complex interactions between infection and chronic airway disease seen in clinical settings. Common pathogens include Klebsiella pneumoniae (Kp)31 and influenza viruses (such as H3N2).32 Kp, as a common respiratory pathogen, Liu Shujuan team fields,31 Wang Weiyi team fields.33 Both approaches involve repeated intranasal instillation of Kp bacteria on a CS-pretrauma basis to induce persistent bacterial colonization and chronic infection, leading to more intractable airway inflammation, hypermucus secretion, and airway wall structural changes remodeling; Similarly, Fan Zhengyuan et al,34 selected the CS+Kp method, which only differed in the concentration of the Kp solution compared to the former. Li suyun and others,35 When employing the same modeling method, Wistar rats were selected with progressively increasing smoke exposure frequency. With all other conditions unchanged, the modeling period was 84 days, 28 days longer than in the previous modeling method of the same type. Viral models can show how viral infections worsen COPD by disrupting innate immunity and causing immunopathological effects damage. For example, Jang J H et al,32 Based on CS exposure, a single intranasal instillation of H3N2 influenza virus was used to establish macaque and C57BL/6 mouse models, both with a 42-day cycle. Xie Yuan et al,36 polyinosinic:polycytidylic acid(Poly I:C) was used to simulate viral infection signals.
Repeated intranasal instillations of Poly I:C were given alongside smoke exposure over a 56-day period, successfully establishing a BALB/c mouse model. The cigarette smoke-bacterial/viral co-infection model offers a unique platform for studying the mechanisms behind persistent post-infection chronic inflammation, abnormal lung tissue repair, and increased susceptibility to secondary bacterial infections (Table 3).
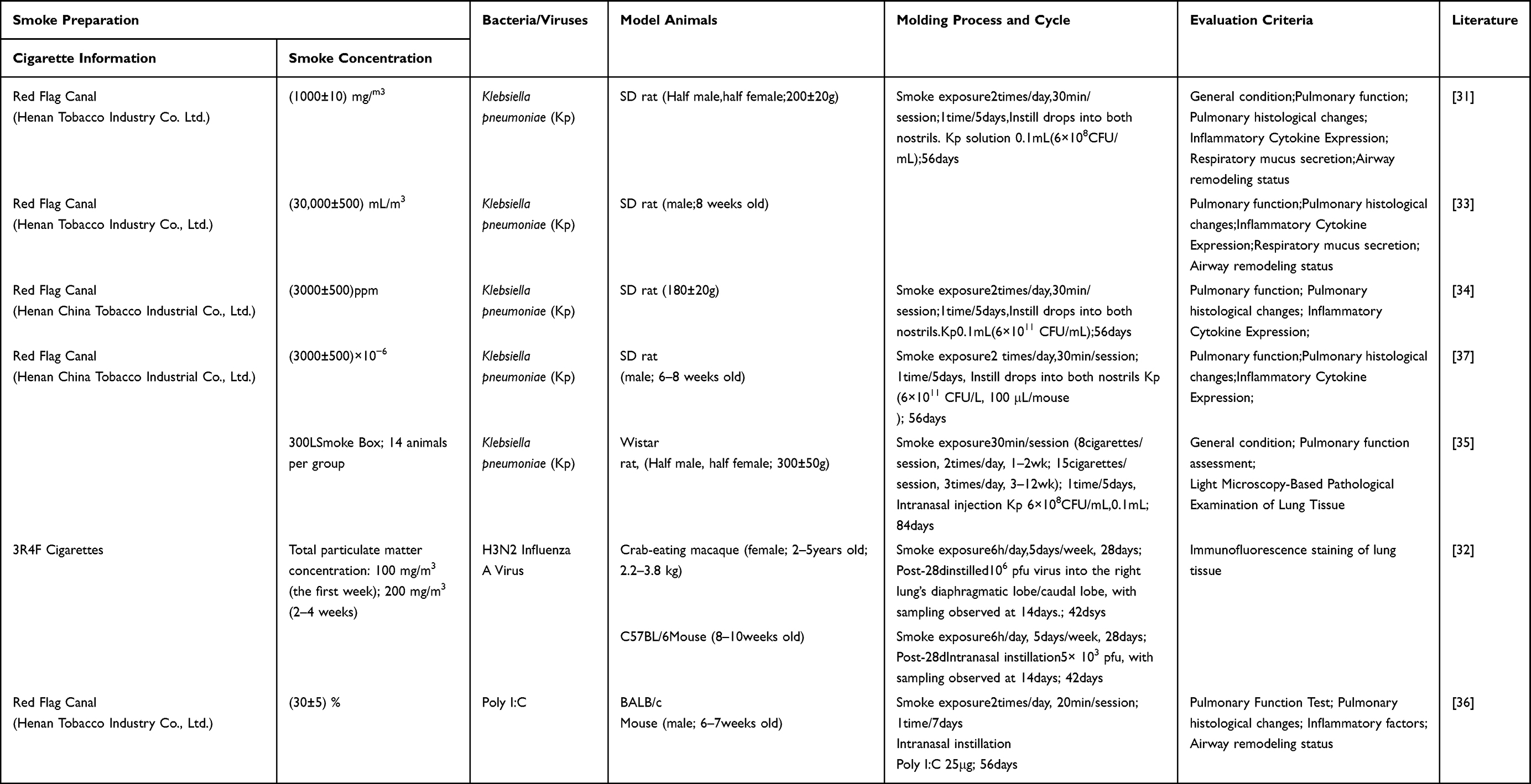 |
Table 3 Cigarette Smoke Exposure Combined with Bacterial/Viral Infections |
Applicability
This model not only captures the features of AECOPD but also accurately reflects the clinical reality where COPD patients frequently experience acute exacerbations caused by infections. It also simulates the clinical progression of incomplete lung function recovery after infection and the “stepwise” decline in disease baseline levels. This makes it a valuable tool for studying the interaction between COPD and infection, as well as for developing combined therapies that target infections or modulate host responses.
Limitations
It needs biosafety facilities for operation and requires strict control over infection dose and timing.
Elastase (ELA) Induction
Proteinase/anti-proteinase imbalance is one of the key theories in the pathogenesis of emphysema.38 Direct instillation of exogenous proteases (such as porcine pancreatic elastase, PPE, or human leukocyte elastase, HLE) into the airways rapidly disrupts the elastic fiber network of the alveolar walls, inducing significant pulmonary emphysema, consistent with the typical clinical manifestations of COPD.39 This model bypasses the chronic inflammatory process and directly targets the pulmonary parenchymal structures. Chan et al,40,41 A single intratracheal instillation of HLE induced marked alveolar space enlargement and pulmonary function alterations (such as increased static compliance) in New Zealand white rabbits within 28 days. Tonon J et al.42 Then select Syrian golden hamsters for a single intravenous infusion of papain (PAP), with significant alterations in alveolar structure detected as early as 40 days post-infusion. In large animals such as sheep, HNE aerosol inhalation has been used to study physiological changes, including mucociliary clearance function, such as by the teams led by Sabater and Abraham.43,44 All chose sheep as their modeling subjects. Typical airway hyperresponsiveness and marked mucociliary dysfunction were detected 14 hours after a single nebulized HNE inhalation. Yang Zhi et al,45 To observe cardiac lesions induced by COPD, miniature pigs were used as experimental subjects. Through alternating multiple instillations of papain (PAP) and trypsin (TRY), typical early COPD symptoms emerged by the fifth week of modeling (Table 4).
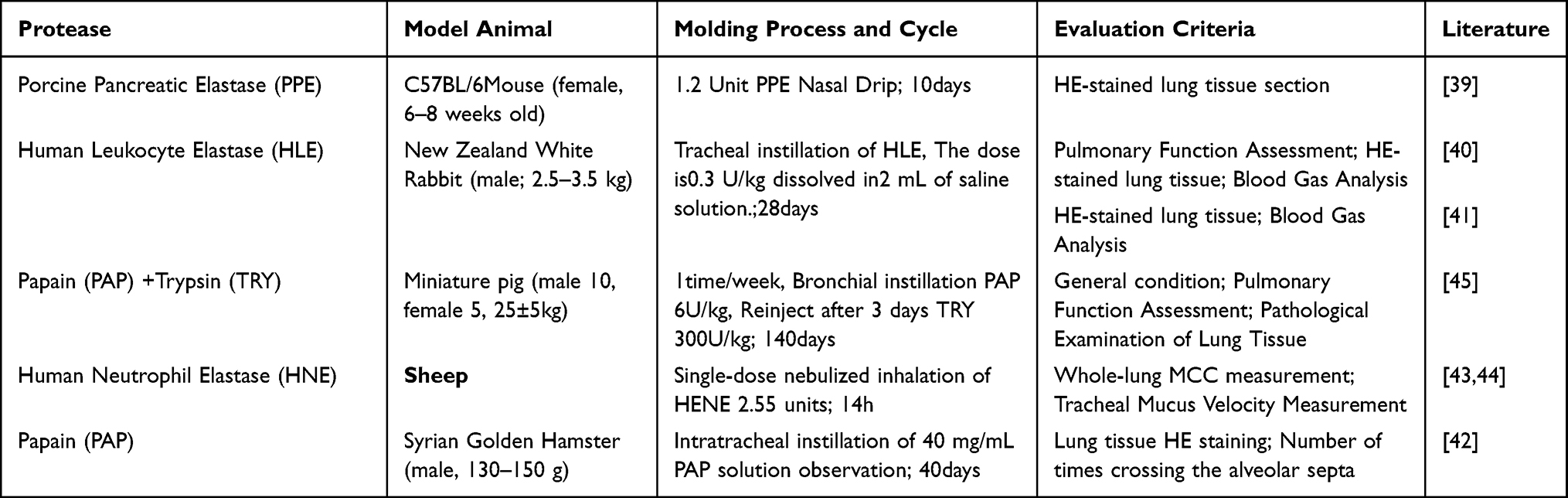 |
Table 4 Elastase (ELA)Induction |
Applicability
Featuring a short cycle, simple operation, stable phenotype, and good consistency, this model efficiently generates significant pulmonary emphysema lesions. Consequently, it is suitable for investigating the cellular and molecular mechanisms underlying the onset and progression of pulmonary emphysema, and is particularly useful for studying elastic fiber degradation and repair. Furthermore, the model is effective for screening and evaluating anti-protease therapies or those that promote alveolar regeneration.
Limitations
This model is essentially an “emphysema model” rather than a complete “COPD model”, lacking the chronic inflammatory processes and airway lesions typical of COPD (such as chronic bronchitis and small airway fibrosis). This model often incorporated as part of a composite model (such as the LPS+PPE model discussed later) and combined with other methods to simulate more comprehensive pathologies.
Genetic Knockout and Transgenic Models
Alpha-1 antitrypsin (AAT) deficiency is the most clearly established genetic risk factor for COPD.38 Genetic models generated using gene-editing technology provide a powerful tool for investigating the role of genetic background in COPD. Borel et al,46 Using CRISPR/Cas9 technology, all five functional Serpina1 genes (encoding AAT) in mice were knocked out in a single step, successfully generating homozygous mice with complete absence of AAT in serum. This model spontaneously develops pulmonary emphysema with increasing age even in the absence of exogenous stimuli (such as smoking), perfectly mimicking hereditary alpha-1 antitrypsin deficiency. Another type of model, such as pIgR−/− mice, their airways lack secretory IgA (SIgA), resulting in a defective mucosal immune barrier.47 These mice do not readily develop disease under conventional conditions, but when exposed to cigarette smoke or specific bacteria, they exhibit exacerbated inflammation and emphysema, revealing the importance of the interaction between genetic susceptibility and environmental exposure (Table 5).
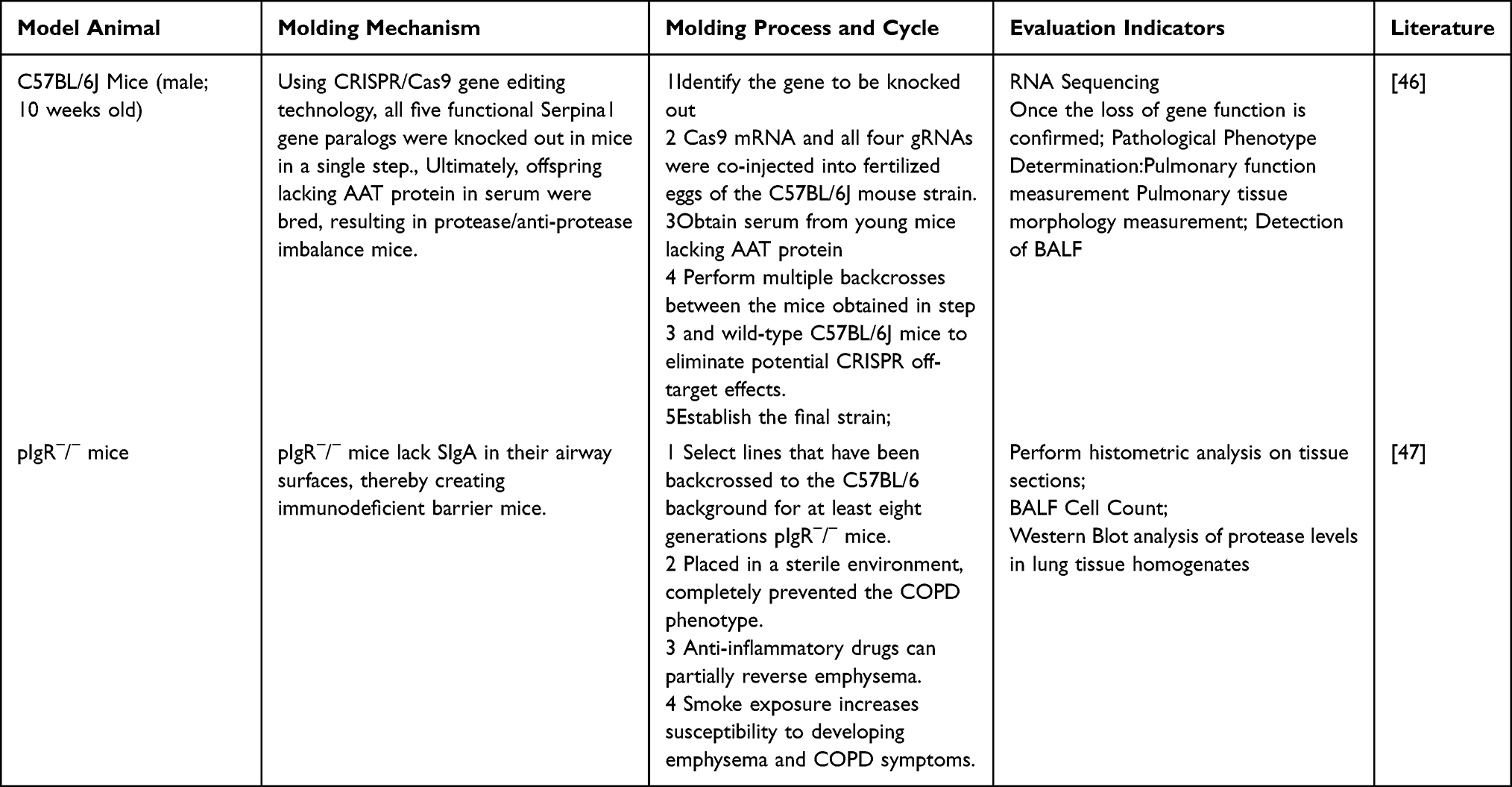 |
Table 5 Knockout and Transgenic Models |
Applicability
Genetic models can elucidate the causal role of specific genes or pathways in disease development and allow for the study of disease natural history without environmental interference. They are crucial for discovering new susceptibility genes, validating drug targets, and developing gene therapies.
Limitations
The breeding and maintenance of gene-edited animals are costly, and certain gene knockouts may cause developmental abnormalities or other complications, necessitating meticulously designed controls; single-gene knockouts may fail to fully replicate the complexity of human COPD; the process is time-consuming, requiring multiple backcrosses to eliminate genetic background interference.
Sulfur Dioxide (SO2) Exposure
SO2 is an irritant gas. High-concentration inhalation can directly damage airway mucosal epithelial cells, triggering acute inflammatory responses, increased mucus secretion, and goblet cell hyperplasia. Long-term exposure may lead to airway wall thickening, fibrosis, and chronic bronchitis-like changes, mimicking the airway pathology characteristics of COPD such as Wagner et al.48 Using SD rats exposed to 20 ppm SO2 throughout the day for 25 days, an early model of COPD was successfully established. In addition to observing pulmonary histopathological changes during evaluation, BrdU assays and AgNOR analysis were employed to assess the proliferative activity and functional status of airway epithelial cells (Table 6).
 |
Table 6 Sulfur Dioxide (SO2) Exposure |
Applicability
Suitable for studying COPD phenotypes characterized by chronic bronchitis, hypersecretion of mucus, and airway inflammation. The model establishment period is relatively short (eg, 25 days), and the procedure is relatively straightforward.
Limitations
SO2 exposure primarily simulates pathological changes induced by chemical irritants, exhibiting weak correlation with major human COPD causative factors (eg, smoking); may induce significant acute injury, which does not fully align with the chronic progressive nature of human COPD; model reproducibility is susceptible to variations in SO2 concentration, exposure duration, and individual animal differences.
Particulate Matter (PM2.5) Induction
Fine particulate matter (PM2.5) can deposit in the alveoli and bronchioles via the respiratory tract, triggering oxidative stress and persistent inflammatory responses. This activates various inflammatory cells and cytokines, leading to lung tissue damage, fibrosis, and airway remodeling.50 Exposure to PM2.5 from different sources can simulate exposure to environmental pollutants, leading to COPD.51 As He et al,52 Expose SD rats to pollution caused by burning cedar sawdust and motorcycle exhaust emissions, respectively, over a period of 7 months. Assessing abnormal lung function and the occurrence of pulmonary fibrosis, Structural remodeling, and elevated expression of inflammatory mediators indicate successful modeling (Table 7).
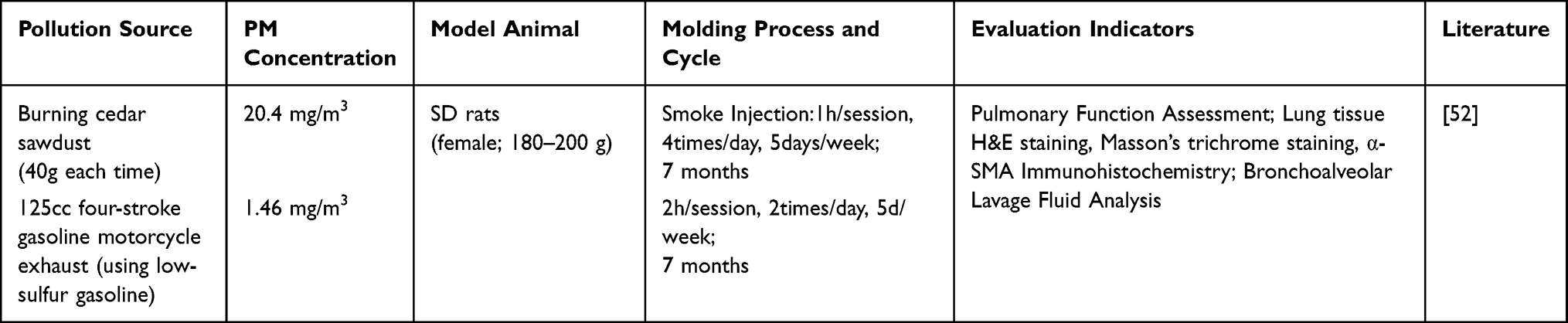 |
Table 7 Particulate Matter (PM2.5) Induced |
Applicability
This is of significant importance for studying COPD unrelated to smoking and for investigating the pathogenesis of COPD associated with environmental air pollution. It is particularly suitable for exploring oxidative stress, immune responses, the inflammatory cascade, and the process of pulmonary fibrosis.
Limitations
The modeling cycle is lengthy, typically requiring several months to establish stable pathology, and is relatively costly. PM2.5 exhibits complex composition, with varying toxic effects among particles from different sources, making model standardization challenging.
Lipopolysaccharide (LPS) Infusion
Intratracheal or intranasal instillation of LPS directly induces a robust neutrophilic inflammatory response, leading to airway mucosal injury, goblet cell hyperplasia, increased mucus secretion, and partial emphysematous changes.53 The mechanism lies in simulating and amplifying the acute exacerbation and chronic inflammatory state of COPD.54 Wang Shanshan and others55 using Wistar rats, a successful model was established by administering two intravascular injections of LPS over a period of 28 days; Liu Junbo and others,56 SD rats were selected, and repeatedly administered LPS via tracheal instillation at a frequency of once per week, successfully establishing the model over 56 days; Similarly, Pera T et al,57 Guinea pigs were selected as experimental subjects and administered LPS via nasal instillation twice weekly for 84 days. In addition to conventional pulmonary function assessments and histopathological observations of lung tissue, Wang Shanshan et al,55 assessed the level of oxidative stress in lung tissue by detecting the function of the MRP1 transporter in pulmonary epithelial cells; Pera T et al,57 By measuring mean alveolar spacing to assess the severity of emphysema, and by measuring pulmonary artery/arteriolar wall thickness to assess the severity of COPD-associated pulmonary vascular remodeling and pulmonary hypertension (Table 8).
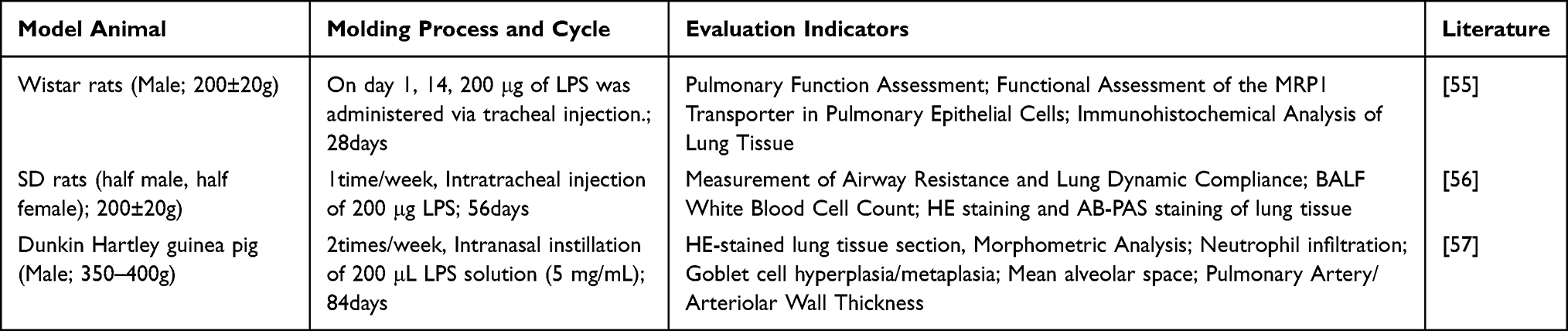 |
Table 8 Lipopolysaccharide (LPS) Infusion |
Applicability
Suitable for simulating bacterial infections or endotoxin-triggered acute exacerbations of COPD, studying airway inflammatory cell infiltration, excessive mucus secretion, and immune response mechanisms. The procedure is straightforward and highly reproducible.
Limitations
The pathology mainly involves acute inflammation, unlike the chronic persistent nature of COPD. Single or multiple LPS stimulations have difficulty fully reproducing pulmonary emphysema and progressive airflow limitation, so they are usually combined with other methods, such as the CS+LPS model or the LPS+PPE model.
Cigarette Smoke Exposure (CS) Combined with Particulate Matter (PM2.5) Induces
The CS+PM2.5 model combines the dual-damage effects of tobacco-related harmful substances and environmental particulate matter, synergistically causing more severe inflammation, oxidative stress, and tissue remodeling. This method more accurately reflects the multifactorial pathogenesis of COPD observed in real-world settings.58 Hisata et al,59 C57/BL6 mice were exposed to cigarette smoke and particulate matter at a concentration of 150 mg/m3 for 2 hours daily, 5 days a week, and lung function was evaluated at 8 months. Lung tissue sections stained with Giemsa revealed pathological changes consistent with COPD (Table 9).
 |
Table 9 Cigarette Smoke Exposure (CS) Combined with Particulate Matter (PM2.5) Induces |
Applicability
Ideal for studying the role of multifactorial interactions in the onset and progression of COPD, especially for assessing lung function decline and pathological changes under combined exposure.
Limitations
Model development is complex and demands strict control of two exposure conditions; the process is time-consuming and requires significant resources.
Forced Swimming + Cigarette Smoke Exposure (CS) + Hypoxia
Long-term hypoxia can worsen pulmonary vascular remodeling, pulmonary hypertension, and right ventricular hypertrophy.60 Combining chronic hypoxia with cigarette smoke exposure effectively mimics the hypoxic conditions commonly seen in COPD patients. Forced swimming triggers a systemic stress response, significantly increasing the body’s oxygen consumption, which closely aligns with the “hypoxia-inflammation” vicious cycle observed in COPD. This makes it suitable for studying COPD complicated by pulmonary heart disease or hypoxemia-related complications. Luan X and others,61 rats were subjected daily to forced swimming (30 minutes), exposure to Lion brand cigarette smoke (20 cigarettes/hour), and hypoxia exposure (10% O2, 7 hours). These procedures were performed six days per week for four weeks. Results showed that model rats experienced a significant decline in lung function and chronic airway inflammation, among other characteristic indicators, indicating that this model can systematically simulate the pathophysiology and molecular features of COPD (Table 10).
 |
Table 10 Forced Swimming + Cigarette Smoke Exposure (CS) + Hypoxia |
Applicability
Suitable for studying the systemic effects of COPD, especially cardiovascular complications and hypoxia-induced inflammatory and remodeling processes.
Limitations
The model is difficult to operate and requires specialized equipment to sustain a stable hypoxic environment; it may indicate late-stage COPD complications rather than early-stage pathology.
Lipopolysaccharide (LPS) Combined with Porcine Pancreatic Elastase (PPE) Infusion
The combined infusion of lipopolysaccharide (LPS) and porcine pancreatic elastase (PPE) induces LPS-mediated airway inflammation, while PPE directly degrades pulmonary elastic fibers, collectively causing significant emphysematous changes, inflammatory cell infiltration, and fibrosis,63 which can mimic the core mechanism of the protease-antiprotease imbalance in COPD. Collie DDS and others field,64 using sheep as experimental animals, sequentially administered PPE and LPS via combined instillation into the same target lung lobe over 70 days to simulate the complex pathology of COPD. The results showed destruction of the alveolar structure, with the emergence of typical COPD emphysema features, whereas chronic LPS stimulation replicated persistent airway inflammation, hypermucous secretion, and fibrotic remodeling (Table 11).
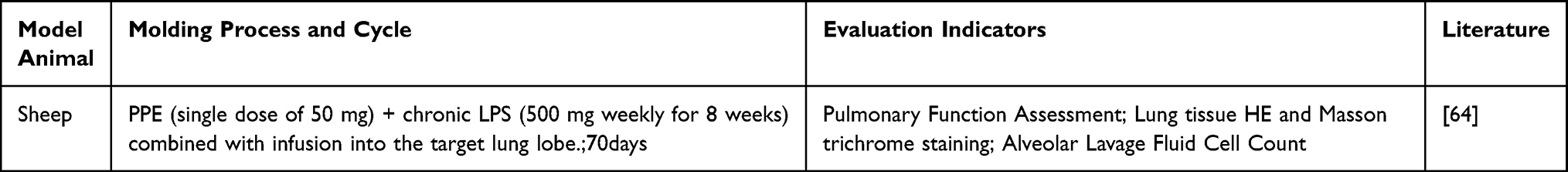 |
Table 11 Lipopolysaccharide (LPS) Combined with Porcine Pancreatic Elastase (PPE) Infusion |
Applicability
Especially suitable for simulating COPD phenotypes characterized by pulmonary emphysema, examining elastin breakdown, alveolar damage, and the interaction between inflammation and proteases. Sheep models, for example, better mimic human lung anatomy and physiology, making them ideal for testing intervention devices or surgical techniques.
Limitations
Demands high technical skill; PPE-related injuries may be too severe to match the slow progression typical of human COPD.
Summary and Discussion
Currently, the establishment of animal models of COPD has formed a technical system primarily based on single-factor induction and multi-factor combined stimulation. The main methods for constructing full-disease models include cigarette smoke (CS) exposure, CS + PM2.5 exposure, CS + lipopolysaccharide (LPS), and CS + bacterial/viral infection. These full-disease models aim to simulate the core manifestations of human COPD as closely as possible, including emphysema, chronic airway inflammation/mucus hypersecretion, accompanied by progressive airflow limitation. Phenotype-specific COPD models are established using various approaches: elastase induction for a dominant emphysema phenotype, SO2 exposure or LPS instillation for notable airway injury, and PM2.5 induction for extensive respiratory system damage. These models aim to recapitulate either a distinct clinical phenotype or the pathological alterations driven by particular etiological triggers. Exacerbation models of COPD are primarily established using CS+LPS, CS+bacterial/viral infection, or LPS instillation. The routine operation of this model is to first establish a chronic disease background and then superimpose stimuli to induce an acute inflammatory response. LPS instillation alone, lacking a chronic background, represents only acute lung injury. The acute exacerbation model superimposes an acute severe inflammatory response on chronic underlying lesions. In addition, there are mechanism-focused models. The main methods for establishing these include gene knockout/transgenic approaches to investigate the genetic pathogenesis of COPD, single LPS instillation to explore the mechanism of acute inflammation, PM2.5 exposure alone to study the toxicological mechanism of particulate matter, and a more specialized combined method involving forced swimming, CS exposure, and hypoxia to simulate the neuro-immune-metabolic crosstalk mechanism. Furthermore, the construction parameters of animal models, such as animal strain selection, gender, cigarette type, exposure system, modeling cycle setting and intervention routes, have not yet formed unified standards, which also affects the comparability of research results and clinical translation potential to a certain extent. At the same time, there is still diversity in the model evaluation criteria. Research purposes are usually used as detection indicators to determine the success of the model, which does not fully comply with the diagnostic criteria for human COPD.
Different modeling methods are selected according to the specific aims of the study. To simulate the natural course of COPD, long-term interventions, or the reversal of emphysematous remodeling, CS exposure is the first choice and is currently regarded as the gold standard for animal models of COPD. On this basis, if a shorter modeling period is desired, alternative methods such as CS + LPS or CS + PM2.5 exposure can also be selected. To model acute exacerbation of COPD or to study anti-inflammatory drug mechanisms, CS + LPS or CS + bacterial/viral infection are appropriate methods, The underlying principle of which is to first establish a background of chronic disease, followed by the superimposition of additional stimuli to trigger an acute inflammatory response. To study a pure emphysema phenotype, the elastase-induced method is an appropriate choice. To study mucus hypersecretion or chronic bronchitis, SO2 exposure or repeated LPS instillation are recommended. To study the pathogenic mechanisms underlying exposure to airborne particulate matter, PM2.5 exposure alone is an appropriate method. To investigate the impact of specific genes on this disease from a genetic perspective, gene knockout or transgenic models can be selected. To study COPD with comorbidities, special models are require. For instance, to study the mechanisms of comorbid conditions such as depression, anxiety, or sleep apnea in COPD, the combined method of forced swimming, CS exposure, and hypoxia can be selected. (Table 12).
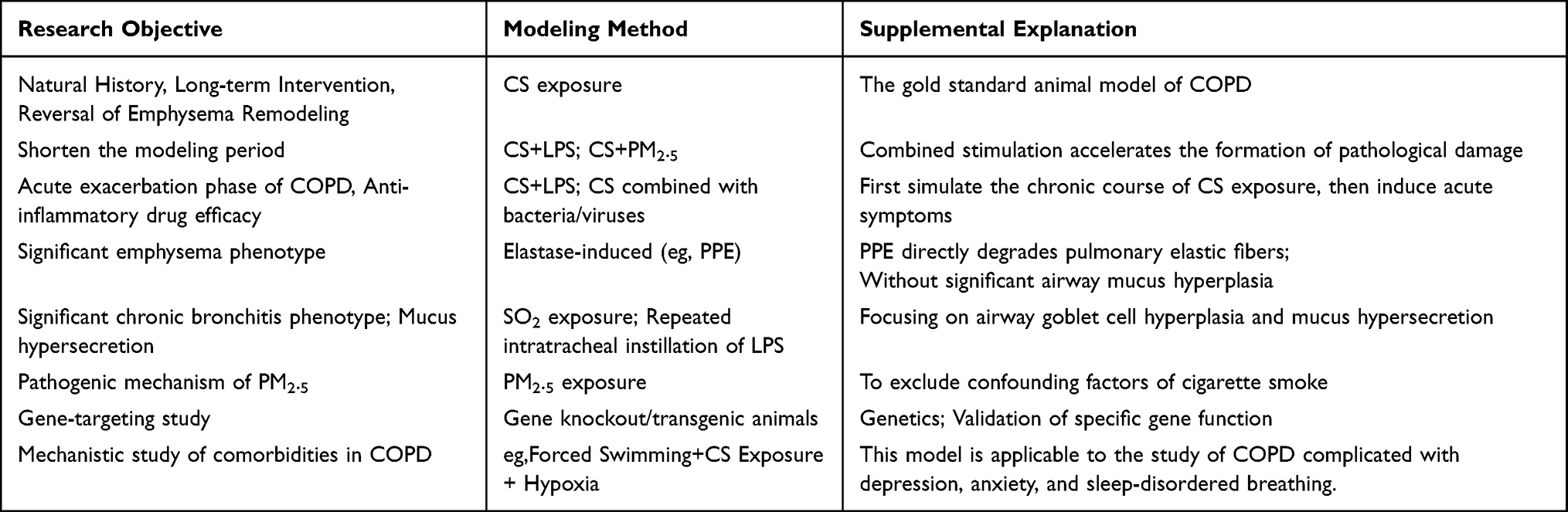 |
Table 12 Recommendations for Modeling Method Selection |
Even when the same modeling method is selected (eg, the classic single cigarette smoke exposure protocol), differences still exist across studies in core parameters such as species, sex, cigarette type, exposure system, and modeling duration, with no further explanation provided. Currently, the sex of animals used for modeling is predominantly male. However, epidemiological studies on COPD have shown that the risk of developing the disease is significantly higher in women than in men. Should this finding be taken into consideration in future animal model preparation, using female animals as the primary choice? As a primary justification for selecting male animals, is the cyclic variation of female hormones precisely the factor that influences the pathogenesis of COPD? In the model evaluation system, it is also necessary to integrate multidimensional assessment methods, including pulmonary function testing, histomorphological analysis, imaging evaluation, and quantitative detection of inflammatory cells and related mediators, in order to systematically validate the reliability and reproducibility of the model at the functional, structural, and molecular levels. These issues will become the key directions for further in-depth research on animal models of COPD, aiming to promote the establishment of animal models that more closely reflect the natural history of the disease and integrate multisystem pathological features.
Although this article provides some reference for COPD researchers in selecting animal modeling methods, due to inherent differences among species in physiological structure, immune response, and repair mechanisms, constructing an animal model that highly simulates the entire pathological progression of human COPD remains a significant challenge.
Abbreviations
COPD, Chronic obstructive pulmonary disease; CS, Cigarette Smoke; LPS, Lipopolysaccharide; AECOPD, Acute Exacerbation of Chronic Obstructive Pulmonary Disease; BALF, Bronchoalveolar Lavage Fluid; Kp, Klebsiella pneumoniae; Poly I:C, Polyinosinic:polycytidylic acid; ELA, Elastase; HLE, Human Leukocyte Elastase; HNE, Human Neutrophil Elastase; PPE, Porcine Pancreatic Elastase; PAP, Papain; TRY, Trypsin; AAT, Alpha-1 Protease Inhibitor; PM2.5, Particulate Matter 2.5.
Acknowledgments
We would like to thank all colleagues in the Department of Traditional Chinese Medicine and the Department of Respiratory Medicine at Lanzhou Petrochemical General Hospital (The Fourth Affiliated Hospital of Gansu University of Chinese Medicine) for their assistance with this project. We also extend our thanks to the staff of the Key Laboratory of Dunhuang Medicine, Ministry of Education, Gansu University of Chinese Medicine, for their support of this project. Additionally, we acknowledge Professor Li Jintian and his team for their contributions to this project. The other members of the team are as follows: Ren Hongyan, Liang Jianqing, Hong Tao, Hu Rong, Guo Qiongqiong, Tian Ping.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China Joint Fund (U23A20502),Open Fund Project of the Key Laboratory of Dunhuang Medicine, Ministry of Education (DHYX25-09),Gansu Provincial Health Industry Scientific Research Project (GSWSKY2023-69) and Gansu University of Chinese Medicine Graduate Research and Innovation Fund (2026CXCY-285).
Disclosure
The authors declare no conflicts of interest.
References
1. Adhikari S, Thapa S, Rattanapan C, Laosee O, Sriram S, Bhatta J. Evaluating the impact of self-management interventions on COPD outcomes in low- and middle-income countries in Asia: a systematic review. Health Qual Life Outcomes. 2025;23(1):81. doi:10.1186/s12955-025-02382-y
2. Li Y, Lu J, Chen Y, et al. The effect of anion inhalation in a mouse model of cigarette smoke-induced chronic obstructive pulmonary disease. COPD J Chronic Obstr Pulm Dis. 2026;23(1):2603725. doi:10.1080/15412555.2025.2603725
3. Zhang W, Sun S, Fan X, He J, Li Q, Jin H. New approaches to treating chronic obstructive pulmonary disease with colla corii asini. Anim Models Exp Med. 2025;9(1):50–16. doi:10.1002/ame2.70077
4. Wang X, Chen G, Bai Q, et al. Simulating cor pulmonale in chronic obstructive pulmonary disease via cigarette smoke exposure and left pulmonary artery ligation in mice. Physiol Rep. 2026;14(1):e70727. doi:10.14814/phy2.70727
5. Zhou JS, Li ZY, Xu XC, et al. Cigarette smoke-initiated autoimmunity facilitates sensitisation to elastin-induced COPD-like pathologies in mice. Eur Respir J. 2020;56(3):2000404. doi:10.1183/13993003.00404-2020
6. Liu MJ, Xu ZP, Guan YQ, et al. Ethyl acetate fraction of thesium chinense turcz. alleviates chronic obstructive pulmonary disease through inhibition of ferroptosis mediated by activating Nrf2/SLC7A11/GPX4 axis. J Ethnopharmacol. 2025;337(Pt 1):118776. doi:10.1016/j.jep.2024.118776
7. Yuhang JIANG, Xiaofeng MEI, Lidan JIA, Yange TIAN, Peng ZHAO.Mechanism of cigarette smoke-induced airway epithelial barrier damage in a mouse model of chronic obstructive pulmonary disease.Chinese. J Pathophysiol. 2022;38(7):1297–1303.
8. Li X, Xu H, Liu K, Shi M, Zeng X, Liu X. LXA4 alleviates inflammation and ferroptosis in cigarette smoke induced chronic obstructive pulmonary disease via the ALX/FPR2 receptor. Int Immunopharmacol. 2025;151:114322. doi:10.1016/j.intimp.2025.114322
9. Paul T, Salazar-Degracia A, Peinado VI, et al. Soluble guanylate cyclase stimulation reduces oxidative stress in experimental chronic obstructive pulmonary disease. PLoS One. 2018;13(1):e0190628. doi:10.1371/journal.pone.0190628
10. Barreiro E, Peinado VI, Galdiz JB, et al. Cigarette smoke-induced oxidative stress: a role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am J Respir Crit Care Med. 2010;182(4):477–488. doi:10.1164/rccm.200908-1220OC
11. Domínguez-Fandos D, Ferrer E, Puig-Pey R, et al. Effects of Aclidinium bromide in a cigarette smoke-exposed Guinea pig model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2014;50(2):337–346. doi:10.1165/rcmb.2013-0117OC
12. Raju SV, Kim H, Byzek SA, et al. A ferret model of COPD-related chronic bronchitis. JCI Insight. 2016;1(15):e87536. doi:10.1172/jci.insight.87536
13. Kaza N, Lin VY, Stanford D, et al. Evaluation of a novel CFTR potentiator in COPD ferrets with acquired CFTR dysfunction. Eur Respir J. 2021;60(1):2101581. doi:10.1183/13993003.01581-2021
14. Auerbach O, Hammond EC, Kirman D, Garfinkel L. Emphysema produced in dogs by cigarette smoking. JAMA. 1967;199(4):241. doi:10.1001/jama.1967.03120040051008
15. Chapman RW. Canine models of asthma and COPD. Pulm Pharmacol Ther. 2008;21(5):731–742. doi:10.1016/j.pupt.2008.01.003
16. Prange R, Thiedmann M, Bhandari A, et al. A drosophila model of cigarette smoke induced COPD identifies Nrf2 signaling as an expedient target for intervention. Aging. 2018;10(8):2122–2135. doi:10.18632/aging.101536
17. Lei LI, Dongliang WU, Shangdian LI, et al. Application analysis of chronic obstructive pulmonary disease model based on data mining. Chin J Comp Med. 2022;32(7):11–17.
18. Martinez FJ, Brochard L, Bush A, Donaldson G, Han MK. Advancing global respiratory health, sleep, and critical care: editorial from the new American journal of respiratory and critical care medicine team. Am J Respir Crit Care Med. 2022;205(1):i–ii. doi:10.1164/rccm.202111-2650ED
19. Churg A, Marshall CV, Sin DD, et al. Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(1):34–43. doi:10.1164/rccm.201103-0468oc
20. Lai HC, Lin TL, Chen TW, et al. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut. 2022;71(2):309–321. doi:10.1136/gutjnl-2020-322599
21. Chen J, Wang T, Li X, et al. DNA of neutrophil extracellular traps promote NF-κB-dependent autoimmunity via cGAS/TLR9 in chronic obstructive pulmonary disease. Signal Transduction Targeted Ther. 2024;9(1):163. doi:10.1038/s41392-024-01881-6
22. Cristaldi M, Buscetta M, Cimino M, et al. Caspase-8 activation by cigarette smoke induces pro-inflammatory cell death of human macrophages exposed to lipopolysaccharide. Cell Death Dis. 2023;14(11):773. doi:10.1038/s41419-023-06318-6
23. Hardaker EL, Freeman MS, Dale N, et al. Exposing rodents to a combination of tobacco smoke and lipopolysaccharide results in an exaggerated inflammatory response in the lung. Br J Pharmacol. 2010;160(8):1985–1996. doi:10.1111/j.1476-5381.2010.00857.x
24. Yinshuang XUAN, Jing MAO, Qingqing BIAN, Tingting SHEN, Ya LI, Suyun LI.Effect of three Tiao-Bu Fei-Shen therapies on intestinal mucosal barrier in rats with stable chronic obstructive pulmonary disease. Chinese J Pathophysiol. 2023;39(4):705–713.
25. Yiping SONG, Dejian CUI, Peiying MAO, Dewen WANG. Establishment of a rat model of chronic obstructive pulmonary disease and the effect of drug intervention. Chinese J Int Med. 2000;(8):712–714.
26. Di M, Niu F, Yang P, et al. Integrated metabolomics and gut microbiota analysis to explore potential mechanism of qi-huo-yi-fei formula against chronic obstructive pulmonary disease. J Pharm Biomed Anal. 2025;252:116511. doi:10.1016/j.jpba.2024.116511
27. Zibo SUN, Jing YAO, Tingyu PAN, et al. Mechanism of Shenji Guben Formula intervention in a mouse model of chronic obstructive pulmonary disease.Chinese. Chin. J. Exp. Tradit. Med. Formulae. 2025. doi:10.13422/j.cnki.syfjx.20252225
28. Hongrong LIN, Nuo XU, Xianqin TANG, et al. Exploring the mechanism of Flos Daturae in improving chronic obstructive pulmonary disease based on network pharmacology and experimental validation. Chin J Exp Traditional Med Formulae. 2025. doi:10.13422/j.cnki.syfjx.20252305
29. Li W, Li Y, Wang Q, et al. Therapeutic effect of phycocyanin on chronic obstructive pulmonary disease in mice. J Adv Res. 2024;66:285–301. doi:10.1016/j.jare.2024.01.009
30. Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med. 2022;10(5):497–511. doi:10.1016/S2213-2600(21)00506-3
31. Shujuan LIU, Ya LI, Zhengyuan FAN, Gaofeng LI, Suyun LI. Ameliorative effect of pomalidomide on airway inflammation and mucus hypersecretion in COPD rats and its mechanism. Med. J. Chin. People’s Lib. Army. 2024;49(1):91–98. doi:10.11855/j.issn.0577-7402.0155.2023.0619.
32. Jang JH, Chand HS, Bruse S, et al. Connective tissue growth factor promotes pulmonary epithelial cell senescence and is associated with COPD severity. COPD. 2016;14(2):228–237. doi:10.1080/15412555.2016.1262340
33. Weiyi WANG, Ruilong LU, Lidan JIA, Miao YANG, Jiansheng LI, Peng ZHAO. Effect of Bu-Fei Yi-Shen Formula on airway remodeling in a rat model of chronic obstructive pulmonary disease. Shizhen Guoyi Guoyao. 2023;34(8):1854–1858. doi:10.3969/j.issn.1008-0805.2023.08.15
34. Zhengyuan FAN, HAN D, Ya LI, Bingyang HAN, Suyun LI. Effect of three Tiao-Bu Fei-Shen therapies on renal injury in rats with chronic obstructive pulmonary disease. Chin J Pathophysiol. 2024;40(9):1688–1699. doi:10.3969/j.issn.1000-4718.2024.09.015
35. Suyun LI, Cuixia QIAO, Jiansheng LI, Yanxia ZHANG, Hongyan ZHOU. Establishment and evaluation of phlegm-dampness syndrome model during acute exacerbation of chronic obstructive pulmonary disease. China J Tradition Chin Med Pharm. 2012;27(3):585–590.
36. Yuan XIE, Xiaofeng MEI, Liuying TAO, Yuhang JIANG, Jiansheng LI, Peng ZHAO. Mechanism of airway epithelial barrier injury in a mouse model of COPD induced by cigarette smoke exposure combined with Poly I:C. Chinese J Pathophysiol. 2024;40(7):1222–1229. doi:10.3969/j.issn.1000-4718.2024.07.009
37. Yanxin WEI, WEI Y, Xinguang LIU, et al. Effect of Tongsai granules on airway epithelial barrier in a rat model of acute exacerbation of chronic obstructive pulmonary disease and role of EGFR/ERK signaling pathway. Chinese J Pathophysiol. 2023;39(12):2204–2213. doi:10.3969/j.issn.1000-4718.2023.12.011
38. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2026. Available from: https://goldcopd.org/2026-gold-report/.
39. Katsoulis O, Toussaint M, Jackson MM, et al. Neutrophil extracellular traps promote immunopathogenesis of virus-induced COPD exacerbations. Nat Commun. 2024;15(1):5766. doi:10.1038/s41467-024-50197-0
40. Chan CS, Lin YS, Lin YK, et al. Atrial arrhythmogenesis in a rabbit model of chronic obstructive pulmonary disease. Transl Res J Lab Clin Med. 2020;223:25–39. doi:10.1016/j.trsl.2020.04.013
41. Chan CS, Lin FZ, Chen YC, Higa S, Chen SA, Chen YJ. Nicotine exacerbates arrhythmogenesis in rabbit right ventricular outflow tract triggered by chronic obstructive pulmonary disease. J Cell Mol Med. 2025;29(12):e70664. doi:10.1111/jcmm.70664
42. Tonon J, Cecchini AL, Brunnquell CR, Bernardes SS, Cecchini R, Guarnier FA. Lung injury-dependent oxidative status and chymotrypsin-like activity of skeletal muscles in hamsters with experimental emphysema. BMC Musculoskelet Disord. 2013;14(1):39–49. doi:10.1186/1471-2474-14-39
43. Abraham WM. Modeling of asthma, COPD and cystic fibrosis in sheep. Pulm Pharmacol Ther. 2008;21(5):743–754. doi:10.1016/j.pupt.2008.01.010
44. Sabater JR, Lee TA, Abraham WM. Comparative effects of salmeterol, albuterol, and ipratropium on normal and impaired mucociliary function in sheep. Chest. 2005;128(5):3743–3749. doi:10.1378/chest.128.5.3743
45. Zhi YANG, Bing FU, Chunping LI, et al. Evaluation of the changes of right heart secondary to early COPD with 3.0T MRI: an experimental study. Radiol Pract. 2016;31(2):145–150. DOI:10.13609/j.cnki.1000-0313.2016.02.012
46. Borel F, Sun H, Zieger M, et al. Editing out five Serpina1 paralogs to create a mouse model of genetic emphysema. Proc Natl Acad Sci U S A. 2018;115(11):2788–2793. doi:10.1073/pnas.1713689115
47. Richmond BW, Brucker RM, Han W, et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat Commun. 2016;7(1):11240. doi:10.1038/ncomms11240
48. Li J, Wang Y, Yin P, et al. The burden of sulfur dioxide pollution on years of life lost from chronic obstructive pulmonary disease: a nationwide analysis in China. Environ Res. 2021;194:110503. doi:10.1016/j.envres.2020.110503
49. Wagner U, Staats P, Fehmann HC, Fischer A, Welte T, Groneberg DA. Analysis of airway secretions in a model of sulfur dioxide induced chronic obstructive pulmonary disease (COPD). J Occup Med Toxicol. 2006;1(1):12. doi:10.1186/1745-6673-1-12
50. Tran HM, Lin YC, Tsai FJ, et al. Short-term mediating effects of PM2.5 on climate-associated COPD severity. Sci Total Environ. 2023;903:166523. doi:10.1016/j.scitotenv.2023.166523
51. Zhao J, Li M, Wang Z, et al. Role of PM2.5 in the development and progression of COPD and its mechanisms. Respir Res. 2019;20(1):120. doi:10.1186/s12931-019-1081-3
52. He F, Liao B, Pu J, et al. Exposure to ambient particulate matter induced COPD in a rat model and a description of the underlying mechanism. Sci Rep. 2017;7(1):45666. doi:10.1038/srep45666
53. Kotlyarov S. High-Density Lipoproteins: a Role in Inflammation in COPD. Int J Mol Sci. 2022;23(15):8128. doi:10.3390/ijms23158128
54. Reiter BFE, Bordag N, Schnoegl D, et al. The acrolein–lipopolysaccharide mouse model for frequent exacerbations in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2025;73(3):343–352. doi:10.1165/rcmb.2024-0507MA
55. Shanshan WANG, Dianlei WANG, Xiuhua TAO, et al. Functional analysis of MRP1 in lung bronchial epithelium of rats with chronic obstructive pulmonary disease induced by lipopolysaccharide. Acta Laboratorium Animalis Scientia Sinica. 2014;22(3):30–34,5.
56. Junbo LIU, Mengshan HUANG, Chenxi YU, Yan WANG, Fadi TANG. Establishment of a rat model of chronic obstructive pulmonary disease by repetitive intratracheal instillation of lipopolysaccharide. Acta Laboratorium Animalis Scientia Sinica. 2011;19(2):129–133,191.
57. Pera T, Zuidhof A, Valadas J, et al. Tiotropium inhibits pulmonary inflammation and remodelling in a Guinea pig model of COPD. Eur Respir J. 2011;38(4):789–796. doi:10.1183/09031936.00146610
58. yu GX, Chu X, Zeng XL, Bao HR, Liu XJ. Effects of PM2.5 exposure on the notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ Pollut. 2017;226:163–173. doi:10.1016/j.envpol.2017.03.070
59. Hisata S, Racanelli AC, Kermani P, et al. Reversal of emphysema by restoration of pulmonary endothelial cells. J Exp Med. 2021;218(8):e20200938. doi:10.1084/jem.20200938
60. Sabit R, Thomas P, Shale DJ, Collins P, Linnane SJ. The effects of hypoxia on markers of coagulation and systemic inflammation in patients with COPD. Chest. 2010;138(1):47–51. doi:10.1378/chest.09-2764
61. Luan X, Zhu D, Hao Y, et al. Qibai pingfei capsule ameliorated inflammation in chronic obstructive pulmonary disease (COPD) via HIF-1 α/glycolysis pathway mediated of BMAL1. Int Immunopharmacol. 2025;144:113636. doi:10.1016/j.intimp.2024.113636
62. Xie J, Liu M, Gao Y, et al. Integration of metabolomics and network pharmacology to reveal the protective mechanism underlying qibai pingfei capsule on chronic obstructive pulmonary disease. Front Pharmacol. 2023;14:1258138. doi:10.3389/fphar.2023.1258138
63. Rabelo MAE, Lucinda LMF, Reboredo MM, et al. Acute lung injury in response to intratracheal instillation of lipopolysaccharide in an animal model of emphysema induced by elastase. Inflammation. 2017;41(1):174–182. doi:10.1007/s10753-017-0675-5
64. Collie DDS, McLean N, Sallenave JM, et al. Local lung responses following endobronchial elastase and lipopolysaccharide instillation in sheep. Int J COPD. 2006;1(2):189–199. doi:10.2147/copd.2006.1.2.189
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.