Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Rescuing compound bioactivity in a secondary cell-based screening by using γ-cyclodextrin as a molecular carrier
Authors Claveria-Gimeno R, Vega S, Grazu V, Martinez de la Fuente J, Lanas A, Velazquez-Campoy A, Abian O
Received 17 December 2014
Accepted for publication 22 January 2015
Published 19 March 2015 Volume 2015:10(1) Pages 2249—2259
DOI https://doi.org/10.2147/IJN.S79480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Rafael Claveria-Gimeno,1–3 Sonia Vega,3 Valeria Grazu,4 Jesús M de la Fuente,4–6 Angel Lanas,2,8–10 Adrian Velazquez-Campoy,2,3,7 Olga Abian1–3,8
1Instituto Aragonés de Ciencias de la Salud (IACS), Zaragoza, Spain; 2IIS Aragón, Zaragoza, Spain; 3Institute of Biocomputation and Physics of Complex Systems (BIFI), Joint Unit IQFR-CSIC-BIFI, Universidad de Zaragoza, Zaragoza, Spain; 4Instituto de Nanociencia de Aragon (INA), Universidad de Zaragoza, Zaragoza, Spain; 5Instituto de Ciencia de Materiales de Aragón (ICMA), CSIC-Universidad de Zaragoza, Zaragoza, Spain; 6Institute NanoBiomedicine and Engineering, Shanghai Jiao Tong University, Shanghai, People’s Republic of China; 7Fundacion ARAID, Government of Aragon, Spain; 8Centro de Investigación Biomédica en Red en el Área Temática de Enfermedades Hepáticas y Digestivas (CIBERehd), Barcelona, Spain; 9Servicio de Aparato Digestivo, Hospital Clínico Universitario Lozano Blesa, Zaragoza, Spain; 10Department of Medicine, University of Zaragoza, Zaragoza, Spain
Abstract: In vitro primary screening for identifying bioactive compounds (inhibitors, activators or pharmacological chaperones) against a protein target results in the discovery of lead compounds that must be tested in cell-based efficacy secondary screenings. Very often lead compounds do not succeed because of an apparent low potency in cell assays, despite an excellent performance in primary screening. Primary and secondary screenings differ significantly according to the conditions and challenges the compounds must overcome in order to interact with their intended target. Cellular internalization and intracellular metabolism are some of the difficulties the compounds must confront and different strategies can be envisaged for minimizing that problem. Using a novel screening procedure we have identified 15 compounds inhibiting the hepatitis C NS3 protease in an allosteric fashion. After characterizing biophysically the interaction with the target, some of the compounds were not able to inhibit viral replication in cell assays. In order to overcome this obstacle and potentially improve cellular internalization three of these compounds were complexed with γ-cyclodextrin. Two of them showed a five- and 16-fold activity increase, compared to their activity when delivered as free compounds in solution (while γ-cyclodextrin did not show antiviral activity by itself). The most remarkable result came from a third compound that showed no antiviral activity in cell assays when delivered free in solution, but its γ-cyclodextrin complex exhibited a 50% effective concentration of 5 µM. Thus, the antiviral activity of these compounds can be significantly improved, even completely rescued, using γ-cyclodextrin as carrier molecule.
Keywords: primary and secondary screenings, drug activity, antiviral compounds, cyclodextrins, vehiculization, drug delivery, hepatitis C, NS3 protease, virus replicon system
Introduction
There is a permanent need for identifying new specific bioactive compounds. Regardless of their effect (inhibitors or activators), they must interact with a biomolecular target at a certain location within a given organism. High-throughput screening procedures allow for acceleration of the identification of lead compounds as potential candidates for drug development.1,2 Although high-throughput screening for identifying bioactive molecules can be performed by implementing activity assays at different levels and contexts of biological complexity (molecular target, cell, organism), in vitro primary screening is often based on probing the interaction of large collections of compounds with the isolated, purified biomolecular target. At this stage, biophysical studies are instrumental for confirming and providing direct evidence for the interaction of the selected compounds with the molecular target. Identified lead compounds are then assessed in cell-based activity assays as part of secondary screenings (regarding eg, activity, toxicity, side-effects etc). Very often lead compounds do not succeed and are discarded as false positives, because of an apparent low activity in cell-based assays, despite an excellent performance in primary screenings. However, primary and secondary screenings differ significantly according to the experimental conditions and the challenges the compounds must overcome in order to interact with their intended target at the appropriate intracellular location. Among others, cellular internalization and intracellular metabolism are some of the difficulties the compounds must confront in cell-based assays.
There are two main approaches to rescue the bioactivity level observed in primary screenings: 1) structural-functional modifications can be introduced in the compound in order to improve membrane transport or reducing cellular metabolic degradation, yet maintaining the bioactivity level; 2) a suitable non-toxic molecular carrier able to interact with the compound can be employed for storing, protecting and releasing the compound at the required intracellular location. Of course, if cellular metabolic degradation is the main cause for the failure of the lead compound, combination therapy with a second drug interfering with CYP-dependent metabolism can boost the pharmacodynamic profile of certain drugs (eg, pharmacological enhancement of the HIV-1 protease inhibitors by adding a second antiretroviral, usually ritonavir, at a concentration lower than that associated with antiretroviral activity).
Hepatitis C infection is a worldwide health problem of epidemic proportions affecting about 200 million people in the world.3,4 Recently, two NS3-4A protease inhibitors, telaprevir and boceprevir, were approved, adding to the traditional combination therapy based on IFN-α and ribavirin. Despite their remarkable antiviral potency, drug resistance may develop at early stages during clinical therapy; therefore, potential long-term efficacy problems related to drug-resistance associated complications and viral breakthrough can be foreseen.5 The current triple therapy approach (pegylated IFN-α/ribavirin plus one of the protease inhibitors) represents a considerable improvement in comparison with the previous dual therapy.6–8 However, the efficacy of triple therapy still has some limitations in specific groups of patients; in particular, more than 85% of cirrhotic patients who are null-responders to the traditional therapy would not achieve sustained virologic response rates with the triple therapy.9 More recently, new specific direct-acting antivirals have been approved: sofosbuvir (against NS5B polymerase)10 and simeprevir (against NS3 protease).11 In addition, IFN-free regimens are being developed and tested with promising results, thus, overcoming the severe side-effects and low patient adherence associated with IFN treatment.12 Nevertheless, new antivirals with high efficacy and high barrier to resistance development are needed.
Recently, an alternative, physiologically relevant conformational state for NS3 protease has been described and the complex conformational landscape of the NS3 protease has been exploited for identifying new allosteric inhibitors.13,14 Considering this alternative conformational state as a new drug target, chemical compounds binding to it will stabilize and trap the protein into that inactive partially-folded state and, therefore, will block the viral life cycle. This new allosteric mechanism is the basis for a novel screening strategy for identifying potential antivirals that has been recently developed.15 Using this technique, 15 compounds were identified and all of them exhibited good in vitro activity. After the evaluation of their activity and cytotoxicity in cell assays, eight of them showed promising characteristics: low molecular weight, moderate-to-high potency (from micromolar to nanomolar 50% effective concentration [EC50]) and moderate-to-low cytotoxicity.15 However, three of these compounds exhibited low activity in cell assays, which could be related to their incapability to enter into the cells. After exploring different strategies to improve cell internalization for these compounds, we have assessed cyclodextrins (CDs) as molecular carriers.
The chemical structure of CDs, cyclic oligosaccharides composed of α-1,4-glycosidic-linked glycosyl residues, provides them structural and physico-chemical properties that allow their use as molecular carriers.16–21 In their hydrophobic cavity a wide range of compounds ranging from ions to proteins22–25 can be trapped. In addition, CDs exhibit low toxicity and low immunogenicity,26 and they have been used in the pharmaceutical field by promoting CD-drug complexes in order to improve the absorption, distribution, metabolism, excretion, and toxicity (ADMET)-related properties of a drug (eg, solubility, stability, delivery and release, membrane permeability and absorption, toxicity). Currently, more than 30 products can be found in the market based on CD complexes.27–38
The determination of the interaction parameters of CDs and the guest compounds is essential to understand the molecular process of the complex formation. The information derived from that analysis will allow preparation of the most suitable complex to be used with each particular drug in a particular application. Isothermal titration calorimetry (ITC) provides direct thermodynamic information on intermolecular binding interactions. From ITC, the equilibrium association constant (Ka) and the binding enthalpy (ΔH) can be directly and simultaneously determined, from which the Gibbs free energy (ΔG) and the entropy (ΔS) of the interaction can be estimated.39–42
In this work we have evaluated the antiviral activity of three compounds, identified using experimental molecular screening and inhibiting NS3 protease in vitro, but exhibiting poor inhibition activity on viral replication in cellular assays. We provide strong evidence for CD-dependent cellular internalization and delivery as an appropriate strategy to improve or even rescue the anti-hepatitis C virus (anti-HCV) potency of NS3 protease inhibitors with limited activity in cell-based assays, which would be otherwise discarded as failed lead compounds.
Materials and methods
Chemical library
A collection of 1,200 compounds, supplied dissolved in dimethyl sulfoxide (DMSO) 100% at a concentration of 4 mM, was commercially available (Prestwick Chemical, Illkirch-Graffenstaden, France). According to the manufacturer, the compounds are US Food and Drug Administration (FDA)-approved drugs selected for their high chemical and pharmacological diversity. In addition, information on their bioavailability, as well as toxicity and safety, in humans is available.
NS3 protease
The N-terminal domain from full-length NS3 protein corresponding to NS3 protease was expressed and purified as described elsewhere.13,14 The isolated protease domain exhibits similar properties (enzymatic activity, inhibition constants, and allosteric activation mechanism) to those of the full-length protein.43
Identification of NS3 protease inhibitors
A screening procedure based on targeting a non-native inactive partially-folded conformation of the NS3 protease populated in the absence of Zn+2 was implemented. Although this conformational state is highly unstructured, it maintains a significant amount of residual structure and may be stabilized by ligands. Ligands binding to and stabilizing this inactive conformation will act as allosteric inhibitors trapping the NS3 protease into that inactive conformation. The molecular target, the Zn+2-free non-native inactive partially-folded conformation of the NS3 protease, is physiologically relevant given that intracellular zinc is tightly regulated at rather low concentration and the viral polyprotein has limited access to zinc as it is synthesized at the ribosome.
Ligands for NS3 protease have been identified by an experimental screening procedure based on a thermal-shift assay similar to that employed previously for identifying small-molecule compounds acting as Helicobacter pylori flavodoxin inhibitors and human phenylalanine hydroxylase chaperones.44,45 Ligands targeting the inactive partially-folded NS3 protease state have been identified as those compounds inducing a stabilizing effect on NS3 protease against thermal denaturation in the absence of Zn+2.35 Ligand-induced NS3 stabilization was assessed by monitoring the thermal denaturation of recombinant pure NS3 protease in a FluoDia T70 High Temperature Fluorescence Microplate Reader (Photon Technology International, Stanmore, UK). Protein-ligand solutions (100 μL) were dispensed into 96-well microplates (ThermoFast 96 skirted plates; Thermo Fisher Scientific, Waltham, MA, USA) and overlaid with 20 μL mineral oil to prevent evaporation. Protein solutions contained 2 μM NS3 protease in 100 mM sodium acetate, 2 mM ethylenediaminetetraacetic acid (EDTA), pH 5, and 100 μM 8-anilinonaphthalene-1-sulfonic acid (ANS). Ligands dissolved in DMSO were added at 100 μM (with a final 2.5% residual concentration of DMSO) to microplates containing the protein solutions and incubated at 25°C for 30 minutes before loading into the microplate reader. Control experiments with NS3 protease samples with/without DMSO and/or Zn+2 were routinely performed in each microplate. Thermal denaturation was monitored by following the increase in ANS fluorescence intensity associated with protein unfolding (λexc = 395 and λem = 500 nm) where λexc is the excitation wavelength and λem is the emission wavelength. Unfolding curves were registered from 25°C to 75°C in 1°C steps. The system was allowed to equilibrate at each temperature for 1 minute before each fluorescence acquisition. In practice, this represents an operational heating rate of 0.25°C/min approximately.
Although in the absence of Zn+2 NS3 retains some structure, it shows very low stability against thermal denaturation.23 Approximately 40% of the molecules are completely unfolded at 25°C. Therefore, the native baseline in the pre-unfolding region is absent in the thermal denaturation assays. The mid-transition temperature can be operationally defined as the temperature for maximal slope in the unfolding curve or, alternatively, the temperature at which half of the maximal change in the signal is achieved. The absence of the native pre-unfolding baseline makes somewhat difficult the evaluation of the mid-transition temperature following the second criterion. Therefore, hits were identified as those compounds shifting the temperature for maximal slope toward higher temperatures, compared to the internal controls in each microplate.
NS3 protease activity is dependent on its interaction with NS4A accessory viral protein. In vitro biochemical studies are usually conducted in the presence of pNS4A, a peptide mimicking the action of NS4A. However, because the molecular target is the Zn+2-free partially unfolded conformational state of the enzyme, not able to bind NS4A, pNS4A was not considered.
The selected compounds were further tested: direct interaction with NS3 protease by titration calorimetry and enzymatic inhibition assays, antiviral potency in cell-based HCV replicon assays, and cytotoxicity in cell-based assays. Three compounds (compounds 1, 2 and 3) were further selected because they showed low (1 and 2) or negligible (3) antiviral potency in cell-based assays, while exhibiting significant in vitro inhibition effect.
ITC assay
Binding of compounds 1–3 to NS3 protease and γ-cyclodextrin (γ-CD) was determined with a high-sensitivity isothermal titration VP-ITC microcalorimeter (MicroCal, Malvern, Worcestershire, UK). Experiments with NS3 protease were performed at 25°C in 100 mM sodium acetate, 2 mM EDTA, pH 5. NS3 protease 20 μM solution in the calorimetric cell was titrated with 300 μM compound solution. Control experiments were performed under the same experimental conditions. Experiments with γ-CD were performed at 25°C in 100 mM phosphate-buffered saline (PBS), pH 7. Compound solution (120 μM) in the calorimetric cell was titrated with γ-CD 1.8 mM solution. Control experiments were performed under the same experimental conditions.
The heat evolved after each ligand injection was obtained from the integral of the calorimetric signal. The heat due to the binding reaction was obtained as the difference between the reaction heat and the corresponding heat of dilution, the latter estimated as a constant heat throughout the experiment, and included as an adjustable parameter in the analysis. The Ka and the ΔH were obtained through non-linear regression of experimental data to a model considering one class of ligand binding sites. Data were analyzed using software developed in our laboratory implemented in Origin 7 (OriginLab, Northampton, MA, USA).
In vitro enzymatic inhibition assay
The inhibition effect of the selected compounds on NS3 protease was assessed employing the fluorescence resonance energy transfer (FRET) substrate Ac-Asp-Glu-Asp(EDANS)-Glu-Glu-Abu-L-lactoyl-Ser-Lys(DABCYL)-NH2 (Bachem AG, Bubendorf, Switzerland).44–46 A solution of purified NS3 protease at 1 μM final concentration (buffer sodium acetate pH 5) was treated sequentially adding: 1) EDTA at 100 μM final concentration; 2) compound at 0 or 25 μM final concentration (compound stock solutions are 10 mM in 100% DMSO; therefore, the same volume of DMSO was added to the control samples without compounds); 3) Zn+2 at either 200, 100, or 50 μM final concentration. Each addition of EDTA, compound and Zn+2 was followed by 30-minute incubation at room temperature. Finally, substrate was added at 4 μM final concentration for initiating the hydrolysis reaction. Fluorescence intensity was measured in triplicate using a Synergy HT Multimode Reader (BioTek Instruments Inc., Winooski, VT, USA) using 380 nm and 500 nm for excitation and emission wavelengths, respectively. NS3 protease activity was determined as the initial slope of the curve. The quotient between the activity in the presence (25 μM) and the absence of a given compound provides the percentage of inhibition.
Preparation of γ-CD/compound complexes
Selected compounds were incubated 24 hours at room temperature with continuous stirring in a tube roller mixer. Different concentrations of γ-CD (from Carbosynth Company, Berkshire, UK) were added (50, 100, and 200 mM) to a fixed concentration of 20 mM compound in PBS (ie, 1:2.5, 1:5, and 1:10 molar ratios) and the complex formation was followed by spectroscopy. Main absorbance peak wavelengths for compounds 1–3 were shifted toward higher values after 24 hours as an indication of complexation process. The concentration of compound bound to γ-CD was determined using the Ka obtained by ITC. Serial dilutions of the complexes from the stock solutions were prepared to final concentrations of 120, 60, 30, 15, 7.5, 3.75 and 1.85 μM for activity and toxicity assays in cell cultures.
Docking prediction
Binding pose predictions for the γ-CD/compound complexes were performed by computational docking using CycloPredict, available online at the website http://interactions.cyclodextrin.net/web. Compound information was extracted from PubChem database. Dreiding force field was used for energy minimization using built-in Chemaxon tools. PM6 semi-empirical charges calculated by MOPAC2007 from James JP Stewart were added to the ligand atoms. Non-polar hydrogen atoms were merged, and rotatable bonds were defined. Docking calculations were carried out on beta-cyclodextrin (BCD) model. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools.47 Affinity (grid) maps of 20×20×20 Å grid points and 0.375 Å spacing were generated using the Autogrid program.47 AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. Docking simulations were performed using the Lamarckian genetic algorithm and the Solis and Wets local search method.48 Initial position, orientation, and torsions of the ligand molecules were set randomly. Each docking experiment was derived from ten different runs that were set to terminate after a maximum of 250,000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of five were applied.
Cells and replicon system
The highly permissive cell clone Huh 7-Lunet, as well as Huh 7 cells containing subgenomic HCV replicons I389luc-ubi-neo/NS3-3′/5.1 (Huh 5-2), I377NS3-3′/wt (Huh 9-13) or I389/hygro-ubi-NS3-3/5.1 (a kind gift from Dr V Lohmann and Dr R Bartenschlager) have been described recently.49–52 Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (PAN-Biotech GmbH, Aidenbach, Germany), 1× non-essential amino acids (Thermo Fisher Scientific), 100 IU/mL penicillin (Thermo Fisher Scientific), 100 μg/mL streptomycin (Thermo Fisher Scientific), and 250 μg/mL Geneticin (G418; Thermo Fisher Scientific).
Antiviral assay with Huh 5-2 cells
Antiviral assays for assessing the activity of the selected compounds were performed as described in literature.49–53 Briefly, Huh 5-2 cells were seeded at a density of 5·103 cells per well in a tissue culture-treated white 96-well view plate (Techno Plastic Products AG, Trasadingen, Switzerland) in complete DMEM supplemented with 250 μg/mL G418. After incubation for 24 hours at 37°C medium was removed and two-fold serial dilutions in complete DMEM (without G418) of the test compounds were added in a total volume of 100 μL. After 3 days of incubation at 37°C cell culture medium was removed and luciferase activity was determined using the Bright-Glo™ Luciferase Assay System (Promega Corporation, Fitchburg, WI, USA). The luciferase signal was measured using a Synergy HT Multimode Reader (BioTek Instruments Inc.). The EC50 was defined as the concentration of compound that reduced the luciferase signal by 50%.
Cytostatic assay
Cytostatic assays for assessing the cell viability of the selected compounds were performed as described in literature.49–54 Briefly, Huh 5-2 cells were seeded at a density of 5·103 cells per well in a 96-well plate in complete DMEM with the appropriate concentrations of G418. Serial dilutions of the test compounds in complete DMEM (without G418) were added 24 hours after seeding. Cells were allowed to proliferate for 3 days at 37°C, after which the cell number was determined by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega Corporation). The 50% cytostatic concentration was defined as the concentration that inhibited the proliferation of exponentially growing cells by 50%.
Results
Identification of NS3 protease ligands by experimental screening
Lead compounds were identified as those compounds shifting the temperature for maximal slope to higher temperatures, compared to the internal controls (Figure 1). The extent of the stabilization effect against thermal denaturation does not necessarily correlate with the binding affinity of the different compounds, as discussed below. Therefore, the increase stability is a valid index for selecting hits, but not a valid index for establishing an affinity ranking. Compounds 1–3 were selected from this screening procedure (Figure 2) for further characterization. The selected compounds do not show high affinity; their moderate affinity toward the molecular target is typical from primary hits that must progress through optimization.
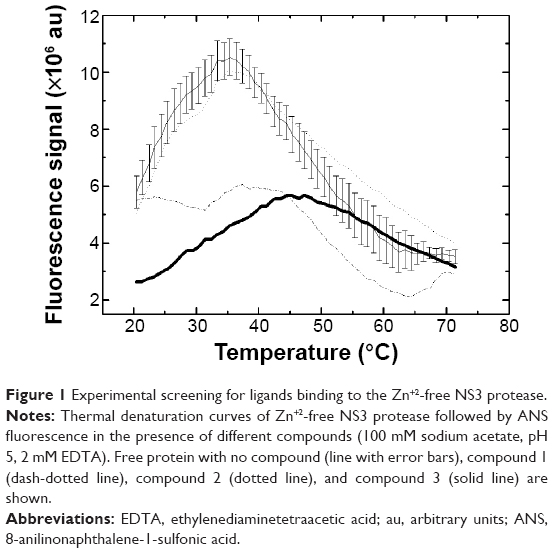 | Figure 1 Experimental screening for ligands binding to the Zn+2-free NS3 protease. |
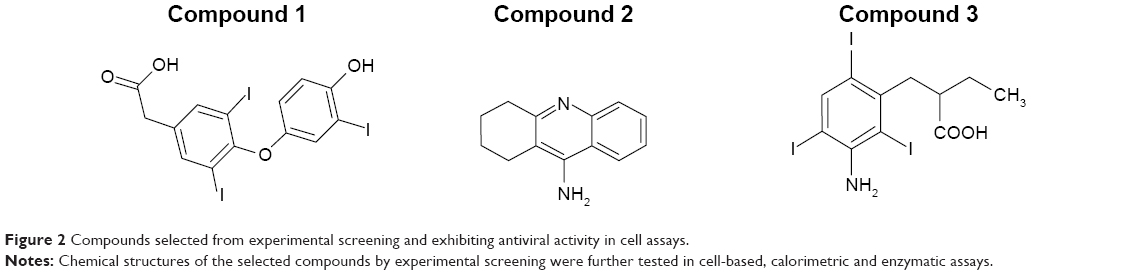 | Figure 2 Compounds selected from experimental screening and exhibiting antiviral activity in cell assays. |
Binding of selected compounds to NS3 protease by ITC
The dissociation constants (Kd) for the interaction between NS3 protease with the selected compounds 1–3 were determined by ITC (Table 1). The Kd for these compounds are in the micromolar range. The binding affinity of these compounds is moderate, but it is important to point out that these are lead compounds obtained directly from molecular screening, with no further optimization.
 | Table 1 Interaction of NS3 protease with selected compounds |
Inhibition of NS3 protease in vitro by selected compounds
Direct inhibition of NS3 protease was tested with the selected compounds. The protocol is based on the Zn+2-free non-native inactive partially-folded conformation of the NS3 protease, a physiologically relevant conformation given that intracellular zinc is tightly regulated at rather low concentration and the viral polyprotein has limited access to zinc as it is synthesized at the ribosome. Selected compounds will bind and stabilize that inactive alternative conformational state acting as allosteric inhibitors. However, because Zn+2 and these compounds bind to different conformational states of the enzyme, they behave as competitive ligands according to a standard Michaelis-Menten mechanism.
The protocol consisted of: 1) removing Zn+2 from the enzyme by adding EDTA (to obtain Zn+2-free NS3 protease); 2) adding compound (to the Zn+2-free NS3 protease); 3) adding Zn+2 to the enzyme; 4) and initiating the catalytic reaction by adding the substrate. According to the Kd for the EDTA-Zn+2 at pH 5 (Kd of 0.8 nM),11 and the total concentrations of Zn+2 (50, 100 and 200 μM) and EDTA (100 μM) employed in the inhibition assay, the concentration of free Zn+2 for each case is 0.8 nM, 0.9 μM, and 0.1 mM, covering five orders of magnitude.
Figure 3 shows the reduction in NS3 protease activity in the presence of the selected compounds. Zn+2 concentration modulates the extent of the inhibitory effect: at typical intracellular Zn+2 concentrations (in the low nanomolar range or below) the inhibition of the enzyme is much larger than at higher concentration of Zn+2. This was expected, since zinc promotes proper folding of the enzyme.
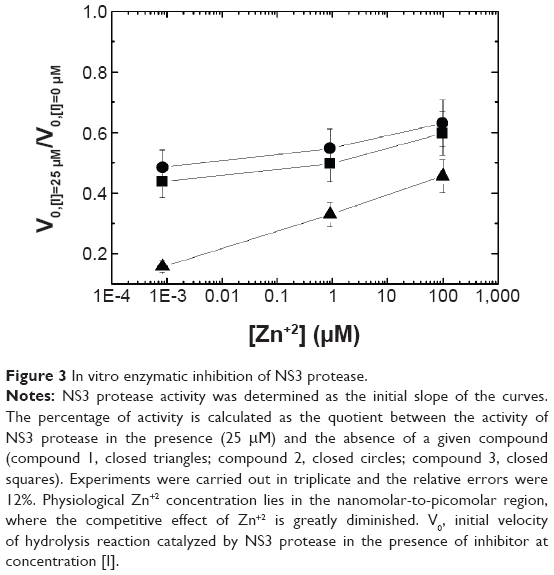 | Figure 3 In vitro enzymatic inhibition of NS3 protease. |
Binding of selected compounds to γ-CD
The thermodynamic parameters of γ-CD interaction with the selected compounds have been determined by ITC (Table 2) allowing the determination of the amount of compound (1–3) complexed to γ-CD by applying chemical equilibrium and mass conservation equations. In order to visually describe the interaction, an algorithm has been applied (from MOPAC2007 computer program) and a possible interaction pose can be visualized (Figure 4). Prediction results from computational docking in terms of interaction energy are in agreement with the experimental results (Table 2).
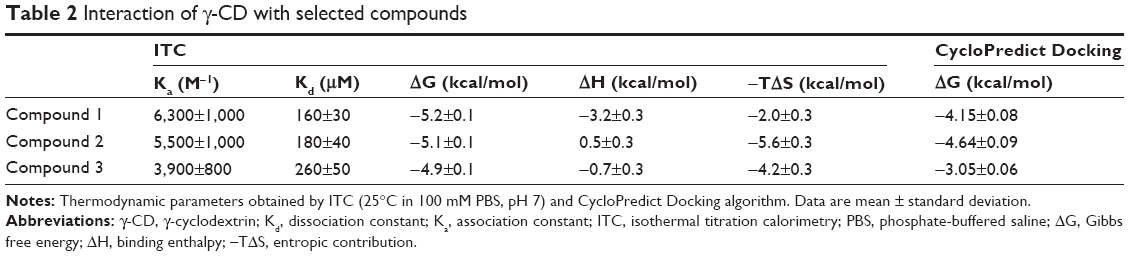 | Table 2 Interaction of γ-CD with selected compounds |
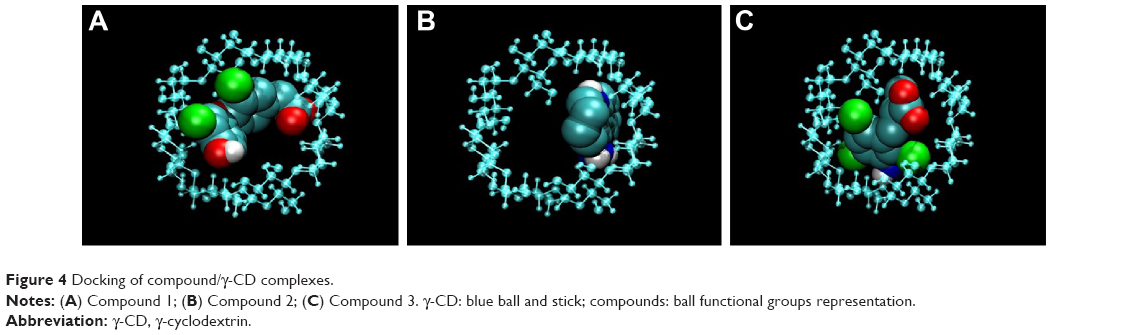 | Figure 4 Docking of compound/γ-CD complexes. |
Antiviral activity of selected compounds and γ-CD complexes in HCV sub-genomic replicon cells
Our novel screening method is based on the identification of compounds that act as allosteric inhibitors by interacting with the inactive partially-folded Zn+2-free NS3 protease conformation. The next logical step consisted of testing their potency in inhibiting viral replication in cell-based assays. In vitro HCV replication assays were performed with genotype 1b Con1 HCV sub-genomic replicon (Huh 5-2 cells). Viral replication levels can be easily monitored by quantifying the reporter (luciferase).
Despite their interaction with the protease with a Kd of 10 μM (Table 1), the selected compounds exhibited poor EC50 values in cell culture: 25, 40, and >120 μM for compound 1, compound 2, and compound 3 respectively (Table 3). Compounds 1 and 2 probably would be discarded from further consideration based on the low activity in cell assays, and compound 3 would be considered as a false positive (EC50 could not even be determined at the experimental conditions), if the primary screening has not been previously performed (Table 3). The antiviral activity of the three compounds was determined, either as a free species or in complex with γ-CD, in order to test the improvement brought about by the protecting role of the molecular carrier.
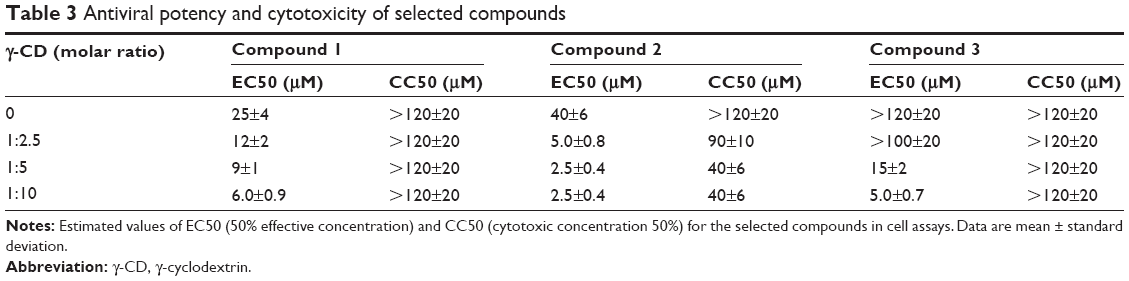 | Table 3 Antiviral potency and cytotoxicity of selected compounds |
All three γ-CD complexes inhibited HCV replicon replication in a dose-dependent manner with EC50 lower than 100 μM (Figure 5A and Table 3). The anti-HCV activity was not the result of a cytostatic effect, since the 50% cytostatic concentration values for these compounds were significantly higher than the EC50 values. In addition, a correlation can be observed between the percentage of the viral replication inhibition and the concentration of the γ-CD used (Figure 5B), indicating a dose-dependent specific effect from the molecular carrier interacting with each compound and providing a suitable vehicle for internalization and intracellular delivery. All experiments included appropriate control tests with compound-free γ-CD, where the luminescence and viability data remained at 100% (data not shown). Therefore, the observed effect for the γ-CD-compound complexes is due to the presence of the compounds.
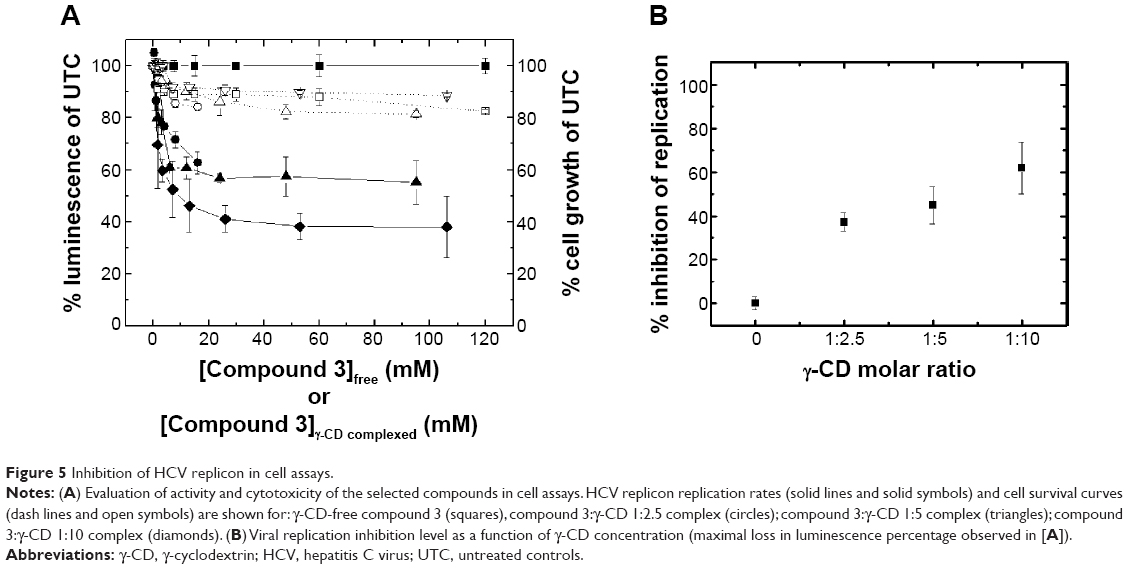 | Figure 5 Inhibition of HCV replicon in cell assays. |
Discussion
Primary and secondary screenings are designed and implemented on the basis of different biological complexity levels (molecular target, cell, organism), different reporter probes, and under different experimental conditions. Selected compounds from primary screenings are directed to further optimization (eg, biophysical characterization, designing of functional derivatives) and parallel cell-based secondary screenings, where, in case their performance is poor, they may either be rejected as compounds exhibiting insufficient activity or be considered as false positives. Taking into consideration the primary screening data, together with further pieces of evidence from a biophysical characterization of the selected compounds with the molecular target, it is possible to devise strategies for rescuing the activity of lead compounds in cell-based secondary screenings.
In this work we present three compounds identified from a primary screening as inhibitors of the hepatitis C NS3 protease. Despite substantive evidence for a direct interaction with the target, they show low or negligible activity in cell-based viral replication assays. A strategy based on γ-CD as a molecular carrier (potentially beneficial for storing, transporting, protecting, and delivering the compounds) has been employed for rescuing their activity for further optimization.
The thermal-shift assay is a fast and easy methodology for identifying compounds binding to a given protein target. However, there are some important caveats that must be stressed. The extent of the stabilizing effect of a ligand on a given protein (ie, increase in the mid-transition temperature) not only depends on the binding affinity and the concentration of compound, but also on the ΔH and, to a lesser extent, the binding heat capacity. For example, two compounds binding with similar affinities will promote mid-transition temperature increases very differently if their binding enthalpies are very different. Similarly, compounds binding with very different affinities may promote similar mid-transition temperature increases if their binding enthalpies are very different. In addition, compounds binding to both the folded and the unfolded states of the protein with similar binding affinities may induce little or no stabilization at all (leading to false negatives). Therefore, the extent of the mid-transition temperature increase in thermal shift assays is not valid for establishing an affinity ranking for the ligands, but just a criterion for selecting ligands binding to the protein target. Additional biophysical (eg, calorimetry, spectroscopy) or enzymatic assays are always further required for providing more direct information on the protein-ligand interaction.
Using this screening method, three compounds were identified. They were able to bind to the inactive protease and increase its stability (Figures 1 and 2), and their Kd for the interaction with NS3 protease, as determined by ITC, were in the μM range (Table 1). In addition, they could also inhibit the NS3 protease activity in vitro (Figure 3). Additional assays with human pancreatic α-chymotrypsin showed a small or negligible inhibition effect (results not shown), indicating they are specific for NS3 protease. However, when their antiviral activity in cell culture was evaluated, diminished HCV replicon inhibition was observed; in particular, compound 3 showed no antiviral effect when administered free (Figure 5), and compounds 1 and 2 showed significantly reduced antiviral potency (Table 3).
It is interesting to point out that, overall, the primary screening hit rate was 1.25% (15 positive hits from a collection of 1,200 compounds), and all 15 hits (100%) exhibited significant inhibition activity against NS3 protease. However, less than 50% of the compounds (seven out of 15) showed significant antiviral activity in cell assays (EC50 lower than 50 μM).
The discrepancy between the activity assays and the cell-based assays could be explained considering an insufficient cellular internalization for the three compounds, leading to an insufficient intracellular concentration for HCV replicon inhibition. In order to overcome this difficulty and recover the antiviral activity of these compounds, we exploited the possibility of using CDs as compound carriers for cellular uptake and distribution.
We studied the interaction of these three compounds with three types of CDs (α-, β-, and γ-CD) by ITC. γ-CD was finally chosen for its higher affinity in all cases, with Ka very similar for the three compounds (Ka from 4,000 to 6,000 M−1). In the case of compound 1, the interaction seemed to be more specific because the enthalpic contribution to the binding Gibbs energy was larger than the entropic one. And for compound 3, the situation was reversed, with a larger entropic contribution. For drug administration and delivery a substantial affinity of γ-CD for the compound in the extracellular environment coupled to a fast release in the intracellular medium would be desirable.
In order to substantiate the ITC binding data, we performed a computational simulation of the interaction using a specific tool for CD docking, and the results obtained were consistent with those determined experimentally (Figure 4). In compound 1, two aromatic rings are twisted through its linker and the interaction with γ-CD covers both sides of its internal cavity; this compound was the one that exhibited the greatest enthalpy term and, probably, the most specific binding. In the case of compound 2, with three rings without linker, the interaction occurs in the same side of the γ-CD cavity. In compound 3, the interaction also covers both sides of the γ-CD cavity (as in compound 1), but the presence of only one aromatic ring could reduce the strength and specificity of binding, which is in agreement with the experimental thermodynamic data.
In all three compounds their complexation with γ-CD increased their potency as antivirals in cell-based assays, compared to the free compounds. Compound 1 showed a five-fold activity increase, and its cytotoxic effect in cells did not change. Compound 2 showed a 16-fold activity increase, but its cytotoxicity effect was also increased (at least three-fold). Compound 3 constitutes the most successful improvement, since the free compound exhibited no antiviral effect at all and its complex with γ-CD shows an EC50 value of 5 μM, with no increase in cell cytotoxicity. In addition, the effect of γ-CD is dependent on the γ-CD:compound molar ratio, indicating a specific effect derived from the formation of the complexes.
We have been able to enhance the antiviral potency of in vitro selected compounds by complexation with γ-CD (in the case of compounds 1 and 2). More importantly, the antiviral activity in compound 3 was completely recovered. It is important to point out that compound 3 would have been discarded if only a cell-based inhibition assay had been carried out. Therefore, a combination of activity and cell-based assays provide invaluable information for screening and selecting lead compounds for further optimization. For a compound that interacts and inhibits the protein target, but shows no antiviral potency in cellular assays, insufficient cellular internalization or substantial efflux transport would be two plausible causes for that outcome. Chemical modification of the compound for improving cellular uptake or minimizing efflux transport, yet maintaining antiviral potency, would be a possible strategy for overcoming that problem. Alternatively, the use of carrier molecules facilitating the cellular internalization and evading the efflux machinery constitutes an alternative advantageous approach, because no chemical modification in the compound is required. We have provided evidence for the improvement in the antiviral activity of three compounds in cell culture by using γ-CD as a carrier molecule. Therefore, if compound vehiculization studies are performed in parallel with compound screening and optimization procedures, it will be possible to decrease the concentration of a given compound required for obtaining a certain pharmacological effect within the cell (dose reduction), and to recover the activity of those compounds that could have been initially discarded because of their low (or even lack of) potency in cell assays, thus reducing false negatives.
The specific features of the interaction of the carrier molecule with a given cargo (eg, binding affinity, interaction enthalpy and entropy, kinetic association and dissociation rate constants) will be key factors for designing and optimizing an efficient molecular delivery system, and for that purpose we are currently performing a thorough study with a collection of antiviral compounds targeting the NS3 protease.
Conclusion
Compounds exhibiting in vitro bioactivity may show reduced activity in cell culture assays because of insufficient cellular internalization or significant cellular efflux. The employment of carrier/delivery molecules represents a potential strategy for recovering those compounds for further optimization. We have proven successful this approach for three inhibitors targeting the NS3 protease from the HCV by complexation with γ-CD. Thus, it is possible not only to decrease the concentration of a given compound required for obtaining a certain pharmacological effect within the cell (reduction of dose with the added benefit of reducing potential side-effects), but also to recover the activity of those compounds that could have been initially discarded because of their low (or even lack of) potency in cell assays, thus reducing false negatives. Future work will involve: 1) optimization of binding affinity and toxicity of the compounds; 2) characterization of the activity profile regarding different viral replicons and the emergence of resistance-associated mutations; 3) assessment of alternative carrier molecules; 4) optimization of the compound/carrier molar ratio; 5) evaluation of in vivo efficacy in animal models.
Acknowledgments
This work was supported by Spanish Ministerio de Ciencia e Innovación (BFU2010-19451 to AVC, PTA2009-2341-I to SV), Spanish Ministerio de Economía y Competitividad (BFU2013-47064-P to AVC), Spanish Ministerio de Educación, Cultura y Deporte (Grant FPU13/3870 to RCG), Miguel Servet Program from Instituto de Salud Carlos III (CP07/00289 to OA), Fondo de Investigaciones Sanitarias (PI10/00186 to OA, PI11/02578 to AL), grant ERC-Starting Grant (239931-NANOPUZZLE project to JML), Diputación General de Aragón (Grant B136/13 to RCG, Protein Targets Group B89 to AVC, Digestive Pathology Group B01 to OA, RCG, and AL, and Nanotherapy and Nanobiosensors Group E93 to JMF), Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBERehd) to AL and OA, and Fondo Social Europeo to JMF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors declare no conflicts of interest in this work.
References
Inglese J, Johnson RL, Simeonov A, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. | ||
Bleicher KH, Böhm HJ, Müller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov. 2003;2(5):369–378. | ||
Nature Outlook: Hepatitis C. Nature. 2011;474:S1–S21. | ||
Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. | ||
Lange CM, Zeuzem S. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol. 2013;58(3):583–592. | ||
Bacon BR, Gordon, SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1207–1217. | ||
Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364(25):2405–2416. | ||
Poordad F, McCone J Jr, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195–1206. | ||
Zeuzem S, Andreone P, Pol S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364(25):2417–2428. | ||
Gane EJ, Stedman CA, Hyland RH, et al. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014;146(3):736–743. | ||
Rosenquist Å, Samuelsson B, Johansson PO, et al. Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J Med Chem. 2014;57(5):1673–1693. | ||
Sarkar S, Lim JK. Advances in interferon-free hepatitis C therapy: 2014 and beyond. Hepatology. 2014;59(4):1641–1644. | ||
Abian O, Neira JL, Velazquez-Campoy A. Thermodynamics of zinc binding to hepatitis C virus NS3 protease: A folding by binding event. Proteins. 2009;77(3):624–636. | ||
Abian O, Vega S, Neira JL, Velazquez-Campoy A. Conformational stability of hepatitis C virus NS3 protease. Biophys J. 2010;99(11):3811–3820. | ||
Abian O, Vega S, Sancho J, Velazquez-Campoy A. Allosteric Inhibitors of the NS3 Protease From the Hepatitis C Virus. PLoS One. 2013;8(7):e69773. | ||
Li S, Purdy WC. Cyclodextrins and their applications in analytical chemistry. Chem Rev. 1992;92(6):1457–1470. | ||
Villiers A. Sur la fermentation de la fecule par l’action du ferment butyrique [The fermentation of starch by the action of butyric]. C R Acad Sci. 1981;112:536–538. French. | ||
Saenger W. Cyclodextrin inclusion compounds in research and industry. Angew Chem Int Ed Engl. 1980;19(5):344–362. | ||
Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998;98(5):2045–2076. | ||
Singh M, Sharma R, Banerjee UC. Biotechnological applications of cyclodextrins. Biotechnol Adv. 2002;20(5–6):341–359. | ||
Villalonga R, Cao R, Fragoso A. Supramolecular chemistry of cyclodextrins in enzyme technology. Chem Rev. 2007;107(7):3088–3116. | ||
Rekharsky MV, Inoue Y. Complexation thermodynamics of cyclodextrins. Chem Rev. 1998;98(5):875–1917. | ||
Fuchs R, Habermann N, Klufers P. Multinuclear sandwich-type complexes of deprotonated β-cyclodextrin and copper(II) ions. Angew Chem Int Ed Eng. 1993;32(6):852–854. | ||
Irie T, Uekama K. Cyclodextrins in peptide and protein delivery. Adv Drug Deliv Rev. 1999;36(1):101–123. | ||
Lysik MA, Wu-Pong S. Innovations in oligonucleotide drug delivery. J Pharm Sci. 2003;92(8):1559–1573. | ||
Irie T, Uekama K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J Pharm Sci. 1997;86(2):147–162. | ||
Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59(7):645–666. | ||
Carrier RL, Miller LA, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. J Control Release. 2007;123(2):78–99. | ||
Hirayama F, Uekama K. Cyclodextrin-based controlled drug release system. Adv Drug Deliv Rev. 1999;36(1):125–141. | ||
Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present, and future. Nat Rev Drug Discov. 2004;3(12):1023–1035. | ||
Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: effects on drug permeation through biological membranes. J Pharm Pharmacol. 2011;63(9):1119–1135. | ||
Matsuda H, Arima H. Cyclodextrins in transdermal and rectal delivery. Adv Drug Deliv Rev. 1999;36(1):80–99. | ||
Loftsson T, Jarvinen T. Cyclodextrins in ophthalmic drug delivery. Adv Drug Deliv Rev. 1999;36(1):59–79. | ||
Merkus FW, Verhoef JC, Marttin E, et al. Cyclodextrins in nasal drug delivery. Adv Drug Deliv Rev. 1999;36(1):41–57. | ||
Szente L, Szejtli J. Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv Drug Deliv Rev. 1999;36(1):17–28. | ||
Duchene D, Ponchel G, Wouessidjewe D. Cyclodextrins in targeting application to nanoparticles. Adv Drug Deliv Rev. 1999;36(1):29–40. | ||
Sinha VR, Nanda A, Kumria R. Cyclodextrins as sustained-release carriers. Pharmaceutical Technology. 2002;44:36–44. | ||
Loftsson T, Duchene D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329(1–2):1–11. | ||
Fini P, Castagnolo M, Catucci L, Cosma P, Agostiano A. Inclusion complexes of Rose Bengal and cyclodextrins. Thermochim Acta. 2004;418:33–38. | ||
Sun DZ, Qiu XM, Li L, Wei XL, Yin BL. A study of a-cyclodextrin with a group of cationic Gemini surfactants utilizing isothermal titration calorimetry and NMR. J Chem Thermodyn. 2006;38(6):773–777. | ||
Liu Y, Cao R, Chen Y, He JY. Effect of b-cyclodextrin charge type on the molecular recognition thermodynamics of reactions with (ferrocenylmethyl)dimethylaminium derivatives. J Phys Chem B. 2008;112(5):1445–1450. | ||
Denadai AM, Teixeira KI, Santoro MM, Pimenta AM, Cortes ME, Sinisterra RD. Supramolecular self-assembly of b-cyclodextrin: an effective carrier of the antimicrobial agent chlorhexidine. Carbohydr Res. 2007;342(15):2286–2296. | ||
He Y, King MS, Kempf DJ, et al. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother. 2008;52(3):1101–1110. | ||
Cremades N, Velazquez-Campoy A, Martinez-Julvez M, et al. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem Biol. 2009;4(11):928–938. | ||
Pey AL, Ying M, Cremades N, et al. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest. 2008;118(8):2858–2867. | ||
Taliani M, Bianchi E, Narjes F, et al. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal Biochem. 1996;240(1):60–67. | ||
Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. | ||
Solis FJ, Wets RJ. Minimization by Random Search Techniques. Mathematics of Operations Research. 1981;6:19–30. | ||
Courcambeck J, Bouzidi M, Perbost R, et al. Resistance of hepatitis C virus to NS3-4A protease inhibitors: Mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antivir Ther. 2004;11(7):847–855. | ||
Susser S, Vermehren J, Forestier N, et al. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J Clin Virol. 2011;52(4):321–327. | ||
Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76(24):13001–13014. | ||
Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285(5424):110–113. | ||
Urbani A, Bazzo R, Nardi MC, et al. The metal binding site of the hepatitis C virus NS3 protease. A spectroscopic investigation. J Biol Chem. 1998;273(30):18760–18769. | ||
Vega S, Neira JL, Marcuello C, et al. NS3 protease from hepatitis C virus: Biophysical studies on an intrinsically disordered protein domain. Int J Mol Sci. 2013;14(7):13282–13306. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.