")
Back to Journals » International Medical Case Reports Journal » Volume 17
Report of Two Contrasting Cases of Epstein–Barr Virus–Associated Hemophagocytic Lymphohistiocytosis: Comparison to Infectious Mononucleosis and Flow Cytometric Analysis of Bone Marrow
Authors Ono S , Yoshimoto K , Matsubara M , Nishimura N, Kawashima H , Yoneima R, Yada N, Nishio K
Received 6 November 2023
Accepted for publication 24 December 2023
Published 20 January 2024 Volume 2024:17 Pages 43—49
DOI https://doi.org/10.2147/IMCRJ.S443996
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Shiro Ono, Kiyomi Yoshimoto, Masaki Matsubara, Nobushiro Nishimura, Hiromasa Kawashima, Ryo Yoneima, Noritaka Yada, Kenji Nishio
Department of General Medicine, Nara Medical University, Kashihara, Nara, Japan
Correspondence: Shiro Ono, Department of General Medicine, Nara Medical University, 840 Shijo-cho, Kashihara, Nara, 634-8522, Japan, Tel +81-744-29-8905, Fax +81-744-24-5739, Email [email protected]
Purpose: This study aims to investigate the characteristics of Epstein–Barr virus associated-hemophagocytic lymphohistiocytosis (EBV-HLH) and HLH caused by a severe form of infectious mononucleosis (IM-HLH) compared to IM by EBV, and thus also to assist in early diagnosis and providing appropriate treatment.
Methods: Data for this analysis were collected from patients at the Department of General Medicine, Nara Medical University, between April 1, 2012, and August 1, 2020. EBV infection was diagnosed using clinical presentation and laboratory tests. HLH diagnosis followed the HLH-2004 protocol, supplemented by plasma EBV DNA detection. A range of clinical and laboratory parameters were collected, including age, sex, clinical outcomes, blood cell counts, hemoglobin, platelets, and various serum values. Plasma EBV DNA levels and flow cytometric analysis (FCM) of bone marrow were performed for HLH cases.
Results: Among 1850 hospitalized patients, 14 cases were identified, including 2 HLH cases and 12 IM cases. Comparative analysis revealed distinctive features of HLH, including lower lymphocyte and platelet counts and higher levels of ferritin, soluble interleukin 2 receptor (sIL-2R), and D dimer compared to IM. Notably, one HLH case responded well to corticosteroid monotherapy, while the other case did not, resulting in a fatal outcome. Detection of a cluster of CD5-CD7 lymphocytes in bone marrow is a hallmark of EBV-HLH and useful to distinguish from IM-HLH.
Conclusion: This study underscores the importance of early differentiation among EBV-HLH, IM-HLH, and IM in adults to guide appropriate treatment strategies. While specific laboratory markers help distinguish HLH from IM, a more detailed analysis of FCM is crucial for precise diagnosis of HLH cases and tailored therapeutic interventions.
Keywords: hemophagocytic lymphohistiocytosis, Epstein–Barr virus infection, Epstein–Barr virus–associated hemophagocytic lymphohistiocytosis, diagnosis of EBV-HLH, flow cytometric analysis of bone marrow
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening, hyperinflammatory clinical syndrome induced by uncontrolled immune activation.1 HLH is classified into primary (familial) HLH, caused by genetic mutations affecting immune regulation, and secondary (acquired) HLH, triggered by underlying inflammatory disease, including infections, malignancies, autoimmune/autoinflammatory disorders, and drugs.2–4 Early recognition and treatment of HLH are crucial for a favorable outcome. The diagnosis of the underlying disease is also important, as the optimal treatment varies according to etiology.
Infection-related HLH is the most common form of secondary HLH,5 and Epstein–Barr virus–associated HLH (EBV-HLH) has particularly high severity and mortality. Treatment of EBV-HLH requires aggressive chemotherapy, including bone marrow transplantation, and delay in treatment can be fatal. Most of the findings of EBV-HLH are from pediatric cases; however, it can occur in adults.6
Primary EBV infections in adults usually present as infectious mononucleosis (IM) and resolve spontaneously. Both IM and EBV-HLH present similar acute inflammatory symptoms, though EBV-HLH should be identified in the early stage of the clinical course. Complicating matters are the fact that severe form of IM also causes secondary HLH in the hyper-cytokine state (described as IM-HLH in this report).7,8 Although they are in the same HLH state, EBV-HLH and IM-HLH have different prognoses and treatment options. Therefore, a clinician should distinguish between the two conditions appropriately and not make a diagnostic and therapeutic decision solely based on HLH status.
In this study, we report adult cases of IM-HLH and EBV-HLH and investigate the clinical features compared with those of IM due to EBV. This study aims to assist in the early differentiation of IM, IM-HLH, and EBV-HLH to help determine appropriate treatment at an early stage.
Methods
Patients
We analyzed data from patients with confirmed EBV infection in the Department of General Medicine of Nara Medical University from April 1, 2012, through August 1, 2020. EBV infection was diagnosed by typical clinical presentation such as fever, lymph node swelling, hepatosplenomegaly, detection of atypical lymphocytes, plasma anti-EBV-capsid antigen, and/or EBV DNA. Diagnosis of HLH was based on the HLH-2004 protocol and plasma EBV DNA. This study was approved by the Ethical Committee of Nara Medical University (approval number: 2695).
Clinical and Laboratory Features
Age, sex, clinical outcome, white blood cell counts, neutrophil counts, lymphocyte counts, hemoglobin, platelet counts, and serum values of aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), triglyceride (TG), ferritin, soluble interleukin 2 receptor (sIL-2R), and D dimer were obtained. Plasma EBV DNA levels and flow cytometric analysis (FCM) of bone marrow were obtained for HLH cases.
Statistical Analysis
The Shapiro–Wilk normality test was used for the IM group. We did not perform a statistical comparison between groups because of the small number of HLH patients. Analyses were conducted with R software version 4.0.0.
Results
Patient
A total of 14 patients, two cases of HLH and 12 cases of IM, were identified from 1850 hospitalized patients. HLH Case 1 was a 21-year-old female and met six items of the eight items in the HLH 2004 diagnostic criteria; fever, splenomegaly, hypertriglyceridemia (299 mg/dL), pancytopenia (white-blood cell; 1700/μL, hemoglobin; 10.0 g/dL, and, platelet; 3.5 × 104 /μL) and hemophagocytosis in bone marrow, the elevation of serum ferritin (6539 ng/mL), and elevation of soluble IL-2 receptor (14,709 IU/L). HLH Case 2 was a 35-year-old female and met five items: fever, splenomegaly, cytopenia (white-blood cell; 2600/μL, hemoglobin; 11.6 g/dL, and, platelet; 4.4 × 104 /μL) and hemophagocytosis in bone marrow, elevation of serum ferritin (9.992 ng/mL), and elevation of soluble IL-2 receptor (18,938 IU/L).
Clinical and Laboratory Features
Clinical and laboratory features are noted in Table 1. Data from the HLH cases were obtained on the day when corticosteroids were initiated, with the exception of plasma EBV-DNA and sIL-2R, which were obtained after two doses of methylprednisolone (1 g/day) were administered. Data were obtained from the IM group on the day when LDH was elevated the most in the clinical course (mean: 8.9 days after development of fever). In the IM group, TG, ferritin, sIL-2R, and D dimer were frequently missing, so no statistical analysis was performed on these results.
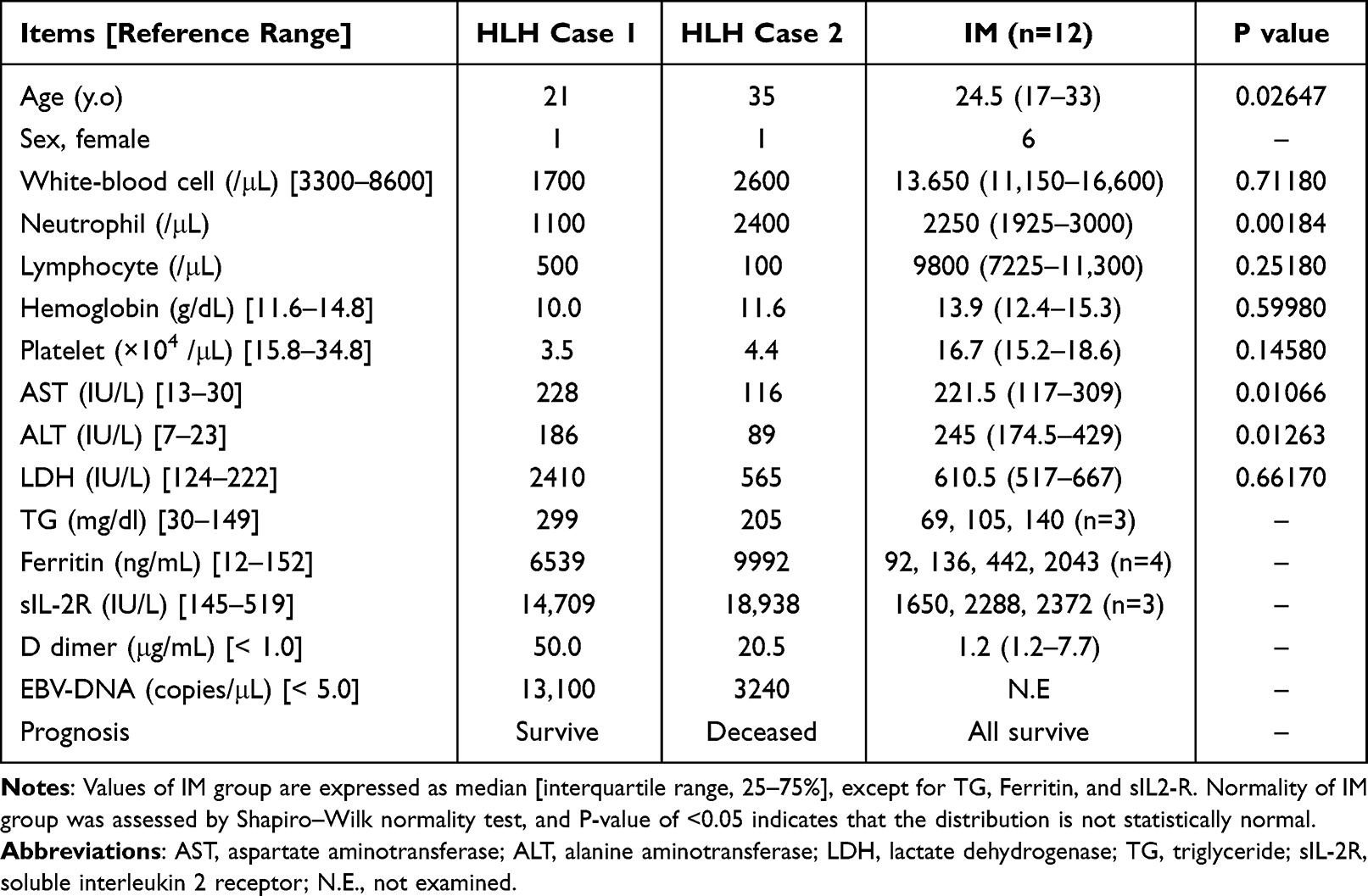 |
Table 1 Clinical and Laboratory Features |
Compared with the IM group, lymphocyte and platelet counts were lower and ferritin, sIL-2R, and D dimer were higher in the HLH patients. LDH was markedly elevated in HLH patient 1, but not in HLH patient 2. Among the IM group, white blood cell, lymphocyte, and platelet counts; hemoglobin; and LDH were statistically normal.
HLH Case 1 received only corticosteroid treatment and improved within a week (Figure 1). HLH Case 2 was treated with corticosteroid, cyclosporine, etoposide, and several antimicrobial agents including ceftriaxone, meropenem, sitafloxacin, and acyclovir; however, she died 18 days after presentation (Figure 2). The IM group was treated with supportive care, such as fluid administration and antipyretics, and all patients survived.
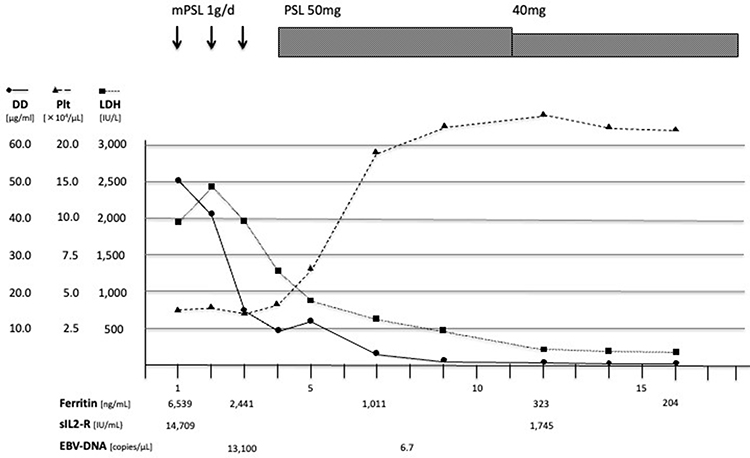 |
Figure 1 The clinical course of HLH case 1: A 21-year-old woman was admitted for persistent high fever and diagnosed with HLH based on the HLH-2004 guidelines. Soon after corticosteroid (CS) therapy treatment was initiated, her fever resolved and laboratory abnormalities improved. CS was reduced and the treatment was completed in two months. Abbreviations: mPSL, methylprednisolone; PSL, prednisolone; DD, D dimer; Plt, platelet; LDH, lactate dehydrogenase; sIL-2R, soluble interleukin-2 receptor. |
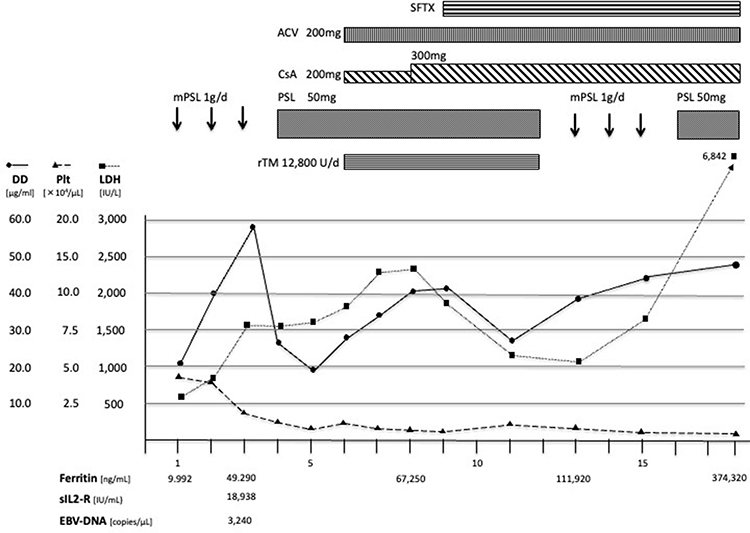 |
Figure 2 The clinical course of HLH case 2: A 40-year-old woman was admitted for persistent high fever and diagnosed with HLH based on the HLH-2004 guidelines. Corticosteroid therapy resulted in only partial fever relief and no improvement of laboratory findings. Cyclosporine, Acyclovir, recombinant thrombomodulin, antimicrobial agents, and repeated mPSL pulse therapy were unsuccessful. The patient died from a cerebral hemorrhage on day 18. Abbreviations: mPSL, methylprednisolone; PSL, prednisolone; SFTX, sitafloxacin; ACV, acyclovir; CsA, cyclosporine; rTM, recombinant thrombomodulin; DD, D dimer; Plt, platelet; LDH, lactate dehydrogenase; sIL-2R, soluble interleukin-2 receptor. |
The CD38+ lymphocytes in the bone marrow of the HLH patients were analyzed with FCM (CD45-SSC gating; Figure 3). In HLH case 1, both CD4+ and CD8+ cells were observed (Case 1 C), and CD5+CD7+ cells were predominant (Case 1 D). However, in HLH case 2, a cluster of CD8+ cells was observed, and only a few CD4+cells were detected (Case 2 C). CD5–CD7– cells, not CD5+CD7+ cells, were predominant (Case 2 D).
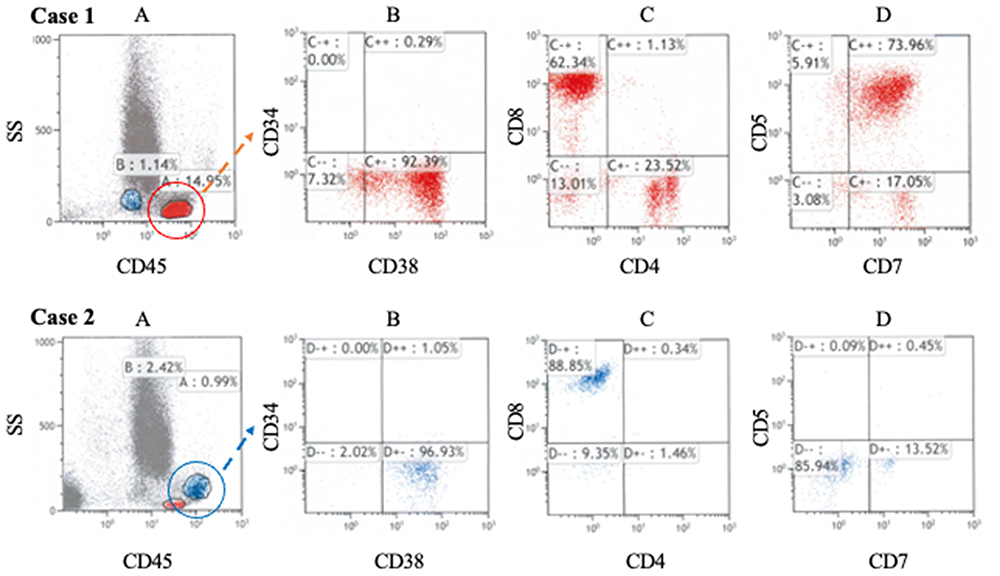 |
Figure 3 Flow cytometric analysis of lymphocytes in bone marrow (A) CD45 gating. The population with the highest CD45 fluorescence intensity corresponds to lymphocytes. (B) CD38+ cells were dominant in both cases. (C) Both CD4+ cells and CD8+ cells existed in case 1; however, CD8+ cells were dominant in case 2. (D) CD5 expression was reduced significantly in case 2, but not in case 1. These findings suggest clonal proliferation of CD5–CD8+ T lymphocytes in case 2. |
Discussion
In this study, we investigated the clinical and laboratory characteristics of EBV-HLH, IM-HLH, and IM to assist in their early differentiation and providing appropriate treatment for each condition. First, we discuss the comparison of HLH (including both EBV-HLH and IM-HLH) and IM. In HLH cases, we observed elevation of ferritin and sIL-2R and decreases of lymphocyte and platelet counts. Elevation of ferritin and sIL-2R are signs of hypercytokinemia due to aberrant activation of the immune system, and the decrease in lymphocytes and platelets is the result of hemophagocytosis that is triggered by hypercytokinemia. Elevation of D dimer, which indicates coagulopathy following hypercytokinemia, is also a distinctive feature of HLH, and this result is consistent with those of a previous study.9 These results suggest that hypercytokinemia-induced hemophagocytosis and coagulopathy are the hallmarks of HLH. AST and ALT were elevated in both groups and are therefore not useful to distinguish HLH from IM. This result suggests that severe liver injury is not associated with the development of HLH. LDH was highly elevated in HLH case 1. However, it was elevated to the same degree in HLH case 2 and IM group. Thus, LDH does not help distinguish between HLH and IM.
We identified two HLH cases, each showing a different clinical course. HLH case 1 improved rapidly with corticosteroid monotherapy, while HLH case 2 did not respond to repeated corticosteroid pulse therapy and cyclosporine and died in a few weeks. Regarding laboratory tests performed at the presentation, only the LDH level on day 1 was clearly different between HLH case 1 and HLH case 2 (Table 1). However, we considered LDH was not a prognostic factor, since the LDH level in HLH case 2 gradually increased during the following course (Figure 1). Plasma EBV-DNA, measured after two doses of methylprednisolone, was also elevated to a similar degree in both cases. In consequence, a general blood test and plasma EBV-DNA are not enough to predict treatment response and prognosis of HLH cases.
HLH can develop from several forms of EBV infection.10,11 EBV-HLH and chronic active EBV infection (CAEBV) are well-known forms of HLH triggered by EBV infection,6 and severe form of IM can also potentially lead to HLH by reactive hypercytokinemia (IM-HLH). In EBV-HLH and CAEBV, virus-infected T cells or Natural killer cells proliferate mono- or oligoclonally and exhibit lymphoma-associated HLH-like properties. In IM, infected B cells proliferate polyclonally and cause a reactive inflammatory response with no neoplastic component.8
Several previous studies analyzed the immunophenotypes of bone marrow and peripheral blood lymphocytes in EBV-HLH.12 They reported that the presence of CD5–CD8+ T cells was a remarkable feature of EBV-HLH and that these proliferating T cells played a critical role in the pathogenesis of EBV-HLH.12 In our HLH case 1, both CD4+ T cells and CD8+ T cells were detected, which was consistent with polyclonal proliferation of T cells (Figure 3 Case 1C); however, only a small number of CD5– T cells were confirmed (Figure 3 Case 1D). In contrast, in case 2, CD5–CD8+ T cells were dominant in the bone marrow (Figure 3 Case 2C and D), suggesting monoclonal proliferation of infected T cells. According to previous studies, HLH case 1 corresponds to IM-HLH, and HLH case 2 corresponds to EBV-HLH. We assume that this diagnostic distinction can explain the difference of therapeutic response in each case. Based on previous reports of FCM of EBV-HLH and our two cases, we recommend analyzing the FCM of bone marrow cells in addition to plasma or serum EBV-DNA measurements in diagnosing EBV-related HLH (Figure 4). In cases where clonal proliferation is suggested, as with our HLH case 2, prompt and vigorous treatment is warranted. On the other hand, if polyclonal proliferation is confirmed, as with our HLH case 1, corticosteroid monotherapy can be sufficient treatment, and potentially harmful treatments should be avoided.
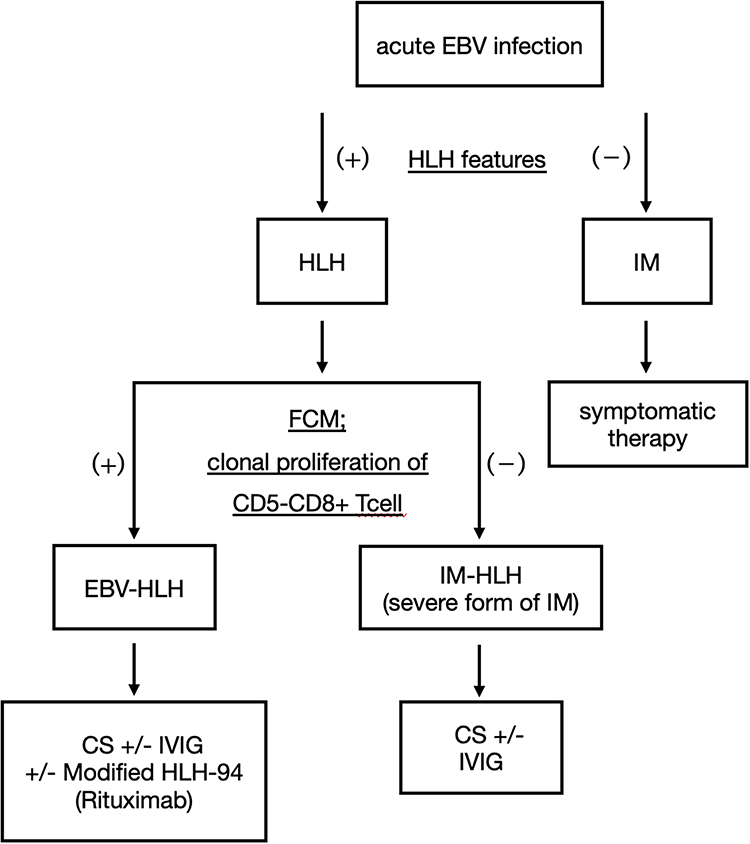 |
Figure 4 Diagnostic algorithm for EBV-HLH and IM-HLH In patients with a diagnosis of acute EBV infection, investigate for features of HLH. Once the diagnosis of HLH is made, we recommend performing a flow cytometric analysis of bone marrow cells to confirm the presence of a clonal proliferation of CD5-CD8+ T cells. If positive, EBV-HLH is suggested and early, strong treatment should be considered. If negative, IM-HLH is suggested, and corticosteroid therapy may be effective. Abbreviations: EBV-HLH, Epstein–Barr virus–associated hemophagocytic lymphohistiocytosis. IM-HLH, infectious mononucleosis-induced hemophagocytic lymphohistiocytosis. FCM, flow cytometric analysis. CS, corticosteroid. IVIG, Intravenous Immunoglobulin. |
Several limitations of this study should be acknowledged. First, we only analyzed a limited number of cases and did not conduct statistical analyses between groups. More cases are needed to confirm the reliability of our recommendation. Second, EBV-DNA was measured after the initiation of corticosteroid therapy and may not reflect the natural course of the disease. The number of missing values in the IM group was another factor that made comparison between the groups difficult. Third, we have not analyzed any HLH cases other than those caused by EBV infection. We therefore cannot say whether the features we found in this study are also valid for other types of HLH. Various pathogens have been reported to cause HLH. As for viruses, herpes simplex virus, varicella-zoster virus, entero virus, and cytomegalovirus are well-known causative pathogens. In addition, HLH caused by viruses that cause hemorrhagic fever, such as Hantaan, Seoul, and Puumala viruses, have also been reported.13 To the best of our knowledge, it is unrevealed that activated and/or proliferating cell types depend on virus species. We hope that these limitations will be overcome in future studies.
In conclusion, this study indicates that cytopenia, coagulopathy, and elevation of ferritin and sIL-2R are the hallmarks of HLH that can be used to differentiate it from IM. However, it is not enough to diagnose HLH to provide appropriate treatment. We recommend that the type of underlying EBV infection should be differentiated in more detail. For this purpose, analyzing the immunophenotype of lymphocytes in the bone marrow enables clinicians to differentiate EBV-HLH from IM-HLH.
Patient Consent for Publication
We obtained written informed consent from two patients of HLH case for the publication of this report. This study was also approved by the Ethical Committee of Nara Medical University (approval number: 2695). The signed consent forms are retained by the corresponding author.
Disclosure
All authors report no conflict of interest for this study.
References
1. Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi:10.1002/pbc.21039
2. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi:10.1016/S0140-6736(13)61048-X
3. Riviere S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127(11):1118–1125. doi:10.1016/j.amjmed.2014.04.034
4. Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine. 2014;93(2):100–105. doi:10.1097/MD.0000000000000022
5. Kim WY, Montes-Mojarro IA, Fend F, Quintanilla-Martinez L. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71. doi:10.3389/fped.2019.00071
6. Zhao Y, Li Z, Zhang L, et al. Clinical features and outcomes of patients with hemophagocytic lymphohistiocytosis at onset of systemic autoinflammatory disorder and compare with Epstein-Barr virus (EBV)-related hemophagocytic lymphohistiocytosis. Medicine. 2020;99(1):e18503. doi:10.1097/MD.0000000000018503
7. Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010;20(2):93–105. doi:10.1002/rmv.638
8. Kofteridis D, Valachis A. Mononucleosis and Epstein–Barr virus infection: treatment and medication. Virus Adapt Treat. 2012;23. doi:10.2147/VAAT.S17837
9. Valade S, Joly BS, Veyradier A, et al. Coagulation disorders in patients with severe hemophagocytic lymphohistiocytosis. PLoS One. 2021;16(8):e0251216. doi:10.1371/journal.pone.0251216
10. Park S, Ko YH. Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disorders. J Dermatol. 2014;41(1):29–39. doi:10.1111/1346-8138.12322
11. Kimura H, Fujiwara S. Overview of EBV-associated T/NK-cell lymphoproliferative diseases. Front Pediatr. 2018;6:417. doi:10.3389/fped.2018.00417
12. Toga A, Wada T, Sakakibara Y, et al. Clinical significance of cloned expansion and CD5 down-regulation in Epstein-Barr Virus (EBV)-infected CD8+ T lymphocytes in EBV-associated hemophagocytic lymphohistiocytosis. J Infect Dis. 2010;201(12):1923–1932. doi:10.1086/652752
13. De Smet MAJ, Bogaert S, Schauwvlieghe A, Dendooven A, Depuydt P, Druwe P. Case report: hemorrhagic fever with renal syndrome presenting as hemophagocytic lymphohistiocytosis. Front Med Lausanne. 2022;9:1096900. doi:10.3389/fmed.2022.1096900
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.