Back to Archived Journals » Hypoxia » Volume 7
Reperfusion Microvascular Ischemia After Prolonged Coronary Occlusion: Implications And Treatment With Local Supersaturated Oxygen Delivery
Authors Spears JR ![]()
Received 31 May 2019
Accepted for publication 20 September 2019
Published 21 October 2019 Volume 2019:7 Pages 65—79
DOI https://doi.org/10.2147/HP.S217955
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
James Richard Spears
Cardiovascular Research Laboratory, Department of Medicine, Division of Cardiology, Beaumont Heart & Vascular Center, Dearborn, MI 48124, USA
Correspondence: James Richard Spears
Cardiovascular Research Laboratory, Department of Medicine, Division of Cardiology, Beaumont Heart & Vascular Center, 22060 Beech St, Suite 200, Dearborn, MI 48124, USA
Tel +1 313-228-0505
Fax +1 313-228-0508
Email [email protected]
Abstract: Following a prolonged coronary arterial occlusion, heterogeneously scattered, focal regions of low erythrocyte flow are commonly found throughout the reperfused myocardium. Experimental studies have also demonstrated the presence of widespread, focally patchy regions of microvascular ischemia during reperfusion (RMI). However, the potential contribution of RMI to tissue viability and function has received little attention in the absence of practical clinical methods for its detection. In this review, the anatomic/functional basis of RMI is summarized, along with the evidence for its presence in reperfused myocardium. Advances in microcirculation research related to obstructive responses of vascular endothelial cells and blood elements to the effects of hypoxia and low shear stress are discussed, and a potential cycle of intensification of RMI from such responses and progressive loss of functional capillary density is presented. In capillaries with impaired erythrocyte flow, compensatory increases in the delivery of oxygen, because of its low solubility in plasma, are effective only at high partial pressures. As discussed herein, attenuation of the cycle with oxygen at hyperbaric levels in plasma is, very likely, responsible for improved tissue level perfusion noted experimentally. Observed clinical benefits from intracoronary SuperSaturated oxygen (SSO2) delivery, including infarct size reduction, can be attributed to attenuation of RMI with improvement in microvascular blood flow.
Keywords: oxygen, ischemia, capillaries, reperfusion, plasma
Background
Microvascular flow impairment associated with ST segment myocardial infarction (STEMI) reperfusion may progress over hours to days. Noninvasive imaging methods have provided important insights regarding the prognosis of patients with myocardial regions of no reflow and/or hemorrhage, pathologies within the infarct necrotic core representing the extreme end of the spectrum of this problem.1–4 Reductions in microvascular flow can also progress temporally throughout the reperfused area at risk, but in a focally heterogeneous distribution of occluded and low erythrocyte flow capillaries, as noted by histology and microvascular casts.5 Intermixed focal regions of hyperemia and arteriovenous shunting within the zone of reperfusion render conventional measures of regional flow misleading regarding the severity of focal flow reductions and associated reperfusion microvascular ischemia (RMI). The term denotes persistent/uncorrected and/or progressive microvascular ischemia during reperfusion. Although global microvascular dysfunction within myocardium subtending a reperfused coronary artery can be assessed clinically with a variety of methods,6 the spatial resolution required to detect heterogeneously distributed microscopic regions of low flow is currently not achievable clinically. Moreover, interpretation of any measure of low flow in the absence of an epicardial stenosis is ambiguous without knowledge of metabolic demand, and clinical methods for directly imaging myocardial hypoxia or ischemia are not readily available.7–10 The potential effects of RMI on myocardial viability, function, and healing have therefore received little attention clinically.
In this review of RMI, anatomic/functional heterogeneities in focal myocardial flow, as a substrate for this problem, are discussed. A paradigm consisting of a positive feedback cycle of RMI and obstructive responses of vascular endothelial cells (ECs) and blood elements is presented (Figure 1; Tables 1–3), along with the rationale for treatment of RMI with methods that effectively deliver oxygen in plasma. Relevant literature is then reviewed in support of the concept that treatment of RMI can enhance tissue viability as a result of improvement in microvascular blood flow.
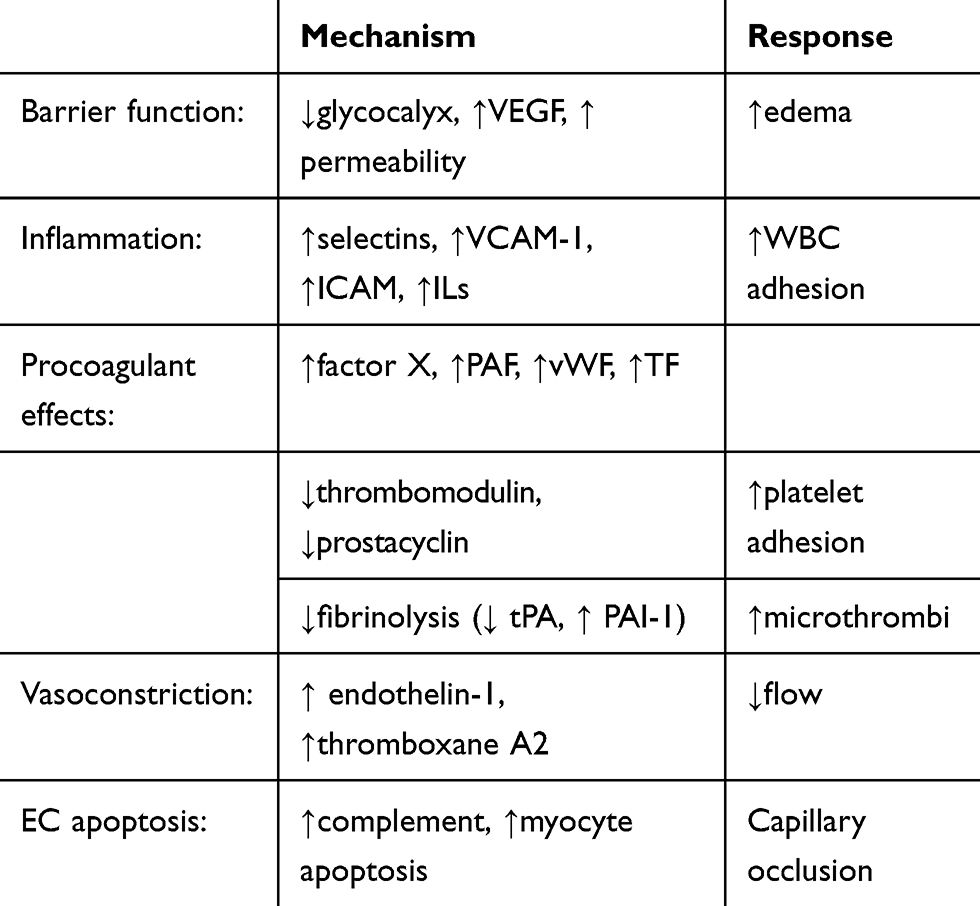 |
Table 1 Endothelial Cell Microvascular Obstructive Responses To Acute Hypoxia |
 |
Table 2 Other Microvascular Obstructive Responses To Acute Hypoxia |
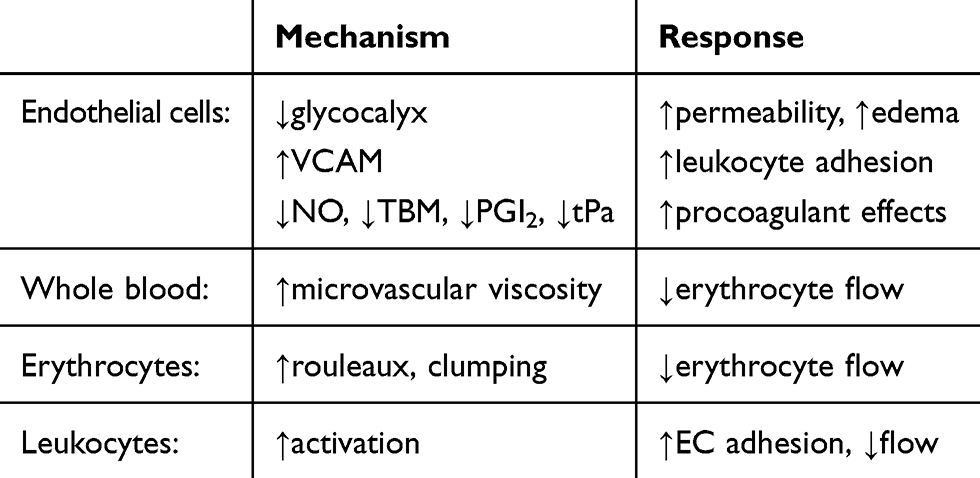 |
Table 3 Microvascular Obstructive Responses To Low Shear Stress |
 |
Figure 1 Cycle of reperfusion microvascular ischemia (RMI) and obstructive responses. Effective increase in O2 delivery attenuates both limbs of the cycle, with improvement of microvascular blood flow, viability, and function. Pathogenetic mechanisms associated with lethal reperfusion injury can be better assessed after RMI has been effectively treated. |
Myocardial Microvascular Heterogeneities
Blood flow in the normal heart is heterogeneous within all transmural layers, reflecting the fractal nature of vascular branching patterns.11 Regional flow variability increases with progressively fine spatial resolutions down to <1 g. Focal ischemia is not normally present because heterogeneity of metabolic demand12–14 matches that of flow. The vulnerability to ischemia is nevertheless heterogeneous, contributing to a patchy distribution of infarction.15 During hypoxic coronary perfusion, a markedly heterogeneous pattern is seen in the severity of regional ischemia, as viewed with NADH fluorescence imaging.16 Recovery from global ischemia or high-flow anoxia similarly is spatially heterogeneous at the capillary level, by both NADH fluorescence imaging and PO2 mapping during normoxic reperfusion.17,18 The size of the regulatory unit governing myocardial heterogeneity during low flow perfusion has been found to vary between tens of microns to about 200 microns.19
Effects Of Microvascular Flow Heterogeneities
Oxygen transport to tissue is dependent on convective supply by erythrocytes to capillaries and diffusion between capillaries. The potential contribution of problems with both types of oxygen transport, as a result of heterogeneities of microvascular flow, to the pathogenesis of organ dysfunction and failure in critical illness has been elucidated clinically with the advent of sidestream dark field and orthogonal polarization spectroscopic (OPS) imaging techniques.20–22 Common findings include capillaries with sluggish or absent erythrocyte flow, focal reductions in functional capillary density (FCD), and presence of arteriovenous shunts. Evidence of tissue ischemia (eg, lactate production) may be found despite globally normal levels of blood flow and oxygen extraction. Ince and coworkers have described such ischemically vulnerable focal regions in tissues, including myocardium, as “microcirculatory weak units”.12,18 In septic shock, persistent heterogeneous sublingual microvascular flow abnormalities are associated with organ failure and are more predictive of mortality than hemodynamic parameters.22 In a relevant experimental model, Ellis et al found a heterogeneous maldistribution of microscopic O2 delivery, with a marked increase in the number of stopped flow capillaries and a three-fold increase in O2 extraction.23 Similar peripheral microcirculatory abnormalities have been found with OPS imaging in association with cardiac surgery, congestive heart failure, and cardiogenic shock.24,25
In reperfused myocardium, experimental findings consistent with these observations include a reduction in FCD26 and an increase in arteriovenous shunting.27 Evidence of myocyte injury upon reperfusion following a relatively brief (20 mins) coronary occlusion was seen only in regions with poor capillary flow within heterogeneous microvascular flow patterns.26
A heterogeneous pattern of microvascular obstruction was reported in an experimental myocardial infarction model.28 A cycle of hypoxia-driven VEGF expression, increased EC permeability, and myocardial edema contributed to progressive infarct expansion.
As another example of potential functional consequences of RMI, Kay et al demonstrated that microscopic sites of ectopic beats and tachyarrhythmias correlated with focal regions of myocardial ischemia during postischemic low flow reperfusion of isolated rat hearts.29
Tissue hypoxia in the perinecrotic border zone of myocardial rat infarcts was quantitated by the use of a nitroimidazole stain by Wang et al.30 Fluorescent and immunohistological stains were also used to quantitate vascular density. Over a 1 to 4 week period post MI, a marked decrease in the density of perfused microvessels was found. Correspondingly greater diffusion distances and more severe tissue hypoxia were found between vessels. In a follow-up analysis of the data with a microvascular transport model,31 it was estimated that, 2 weeks post MI, 29% of the perinecrotic infarct zone was severely hypoxic (PO2 <2 mmHg) from a reduction in microvascular density. The vascular density estimated by the model to prevent critical hypoxia was 75% of normal values.
Although clinical data regarding RMI are extraordinarily difficult to obtain, a unique clinical study by Al-Obaidi et al32 provides some insight. Using microelectrodes in patients undergoing coronary bypass surgery, they found that 74% of the 29 focally interrogated myocardial regions showed delayed recovery of PO2 over a 32-mins period of reperfusion. No recovery was noted in 13% of the regions, compatible with persistent severe ischemia. A poor correlation was observed between changes in local flow, measured with the same microelectrodes, and changes in PO2. The authors surmised that hypoxic regions during reperfusion contribute to deterioration of left ventricular function after bypass surgery. Pathologic microvascular changes noted in a relevant porcine model provide an anatomic basis for these observations.33 Scanning electron microscopy (SEM) of microvascular casts demonstrated large increases in intercapillary distances at 30 mins of reperfusion compared to normal hearts. FCD and luminal dimensions by SEM were significantly reduced, with a resultant reduction in vascular cast density.
Focal reductions in capillary hematocrit may further contribute to impairment of oxygen delivery. The number of capillaries perfused by cell-free plasma may greatly exceed the number perfused by erythrocytes in pathologic microvascular settings.34 In addition, the hematocrit may be reduced in the capillary bed perfused by a vasoconstricted arteriole,35 below levels associated with a physiologic Fahraeus effect. During low flow myocardial ischemia, Eliasen and Amtorp36 found that mean microvascular hematocrit decreased to 23% from a control level of 31%, as a result of 25% and 34% decreases in red cell volume in the subepicardium and subendocardium, respectively. Importantly, Barker et al demonstrated that a moderate reduction in capillary hematocrit significantly reduced mean capillary PO2 and the amplitude of erythrocyte-associated PO2 transients.37
Thus, many factors contribute to the potential presence of heterogeneous, focally severe microvascular ischemia during myocardial reperfusion. As discussed by De Backer et al,21 based on an analysis of capillary oxygen transport data,38 a loss of FCD produces a more severe level of tissue hypoxia compared to a similar, but homogeneous reduction in blood flow through preserved microvessels.
Paradigm: Cycle Of RMI And Obstructive Responses Of The Microcirculation
The severity of microvascular hypoxia associated with RMI ranges across all three thresholds defined by Connett et al,39 with oxygen availability not coping with aerobic ATP requirements. In regions without myocyte viability, the term would apply to the ischemic microvasculature alone.
Because of microvascular injury from ischemia/reperfusion (I/R),40,41 coronary flow reserve is blunted. As demonstrated with microsphere embolization of normal coronaries,42 hyperemic responses may nevertheless occur in non-ischemic segments adjacent to focally ischemic ones, possibly enhanced by diffusion of adenosine. During RMI, the known obstructing responses of capillary endothelial cells (ECs) and blood elements to hypoxia, very likely the most important component of RMI, would be expected to participate in a cycle of progressive RMI and continued obstructive responses to RMI (Tables 1–3), commonly leading to capillary occlusion and/or loss of capillary integrity (Figure 1). By increasing oxygen delivery to the hypoxic microcirculation, RMI is attenuated. The hypoxic responses of RMI are diminished, and the cycle is slowed, if not broken. As microvascular flow improves, potential residual obstructive responses related to low shear stress (Table 3) are reduced, resulting in persistent increases in capillary patency and flow.
RMI: Specialized Methods Required For Its Detection And Imaging
Direct detection of spatially heterogeneous microvascular ischemia presents extreme technical challenges. Balaban and Arai13 suggested that an appropriate spatial resolution for assessment of myocardial flow heterogeneity is a tissue volume (0.3 mm3) perfused by a single 150 micron diameter arteriole. This flow resolution may be achievable experimentally with fast high-resolution MRI experimentally.11 However, the spatial resolution for registration of functional, metabolic (eg, BOLD and TOLD), and flow CMR images is likely to be significantly lower. Moreover, a single arteriole as above normally subtends approximately 450 capillaries.43 If reduced FCD and arteriovenous shunting are present within this region, measures of mean flow and oxygenation may not reflect the severity of even smaller focal regions of microscopic ischemia.
The limitations of conventional clinical methods for diagnosis and/or imaging of microvascular ischemia associated with other myocardial pathologies were well summarized by Pries et al.6 Direct noninvasive imaging of tissue hypoxia is even more challenging.8 Experimentally, molecular imaging techniques,44 such as PET imaging of activated platelets that can detect minimal ischemia45 are promising new approaches. Electron paramagnetic resonance (EPR) imaging of tissue PO2 has also been used only experimentally to study the bioenergetics of ischemia/reperfusion. The latter approach was used to show that hyperoxia normalizes the hypoxic border zone (penumbra) adjacent to the necrotic core of reperfused ischemic brain lesions.46
Obstructive Responses Of The Microcirculation To Hypoxia And Low Shear Stress
ECs can tolerate lower oxygen levels than some other cells,47 but the luminal obstructive responses of ECs and blood elements to either severe hypoxia48–64 or low shear stress65–70 are numerous and profound ((Tables 1–3). While physiologically adaptive in many types of wound repair, eg, walling off an infectious agent or a site of bleeding, such responses are maladaptive (“dysfunctional”) during reperfusion. The changes are similar to those described for Types I and II activation of microvascular ECs associated with generic responses to an altered microenvironment.50 Prior to apoptosis,53–55 earlier responses of ECs to hypoxia include loss of barrier function, cytoplasmic swelling, and interstitial edema. Hypoxia has been shown to disrupt the integrity of the endothelial surface glycocalyx of cardiac capillaries, with an increase in permeability. Retraction of the lateral margins of adjacent ECs results from actin filament contraction associated with a reduction in intracellular cyclic AMP needed for cytoskeleton maintenance; the integrity of intercellular junctions is reduced, resulting in interstitial edema from paracellular leakage and in EC shape change (rounding). Stabilization of hypoxia-inducible factor (HIF-1) by hypoxia and activation of numerous genes produce phenotypic changes in ECs, including increased permeability from VEGF production.28 Potential adverse effects of edema include increased distance for oxygen diffusion and increased interstitial pressure.71 Capillary luminal dimensions may be compromised by intracellular EC edema/shape change and by external compression. Microvascular flow resistance follows Poisouille’s law and is inversely proportional to the fourth power of diameter,34 so that small reductions in capillary diameter would be expected to significantly reduce flow. In addition, small reductions in capillary diameter below 5 microns, similar to the mean value of capillaries in the human left ventricle,72 exponentially increase apparent blood viscosity in an extension of the Fahraeus–Lindqvist effect.73
Hypoxia also induces proinflammatory phenotypic changes in ECs, primarily via HIF-1 and other transcription factors, including nuclear factor-kB (NF-kB).56–62 Expression of adhesion molecules (eg, E-selectin) promotes leukocyte rolling and subsequent binding to leukocyte integrins with ligands such as VCAM-1 and ICAM-1, facilitated by activation of chemoattractant/activator molecules including platelet-activating factor (PAF). Production of proinflammatory cytokines and increased expression of tissue factor by ECs exposed to hypoxia may further contribute to an inflammatory response. Also in response to hypoxia, Weibel-Palade body exocytosis from ECs and shedding of EC microparticles74 may increase neutrophil recruitment and inflammation. As reviewed recently by Eltzschig and Carmeliet, hypoxia is a common trigger of inflammatory responses in many tissues, and such responses may contribute to progression of lesion hypoxia.62
Hypoxia-activated ECs release endothelin-1,63 a potent vasoactive peptide, and endothelial NO levels may decrease from ROS scavenging. The resultant vasoconstricting effects may reduce downstream capillary hematocrit as well as erythrocyte flow.75
Procoagulant pathways in ECs are enhanced49,76,77 and fibrinolytic activity is reduced78 by hypoxia. Hypoxia has also been shown to stimulate platelet aggregation and formation of thromboxane A2.79 The latter may contribute to arteriolar vasoconstriction, superimposed on fibrin and microthrombi deposition.
Recently, ischemia-induced pericyte contraction that segmentally obstructs capillary erythrocyte flow during reperfusion has been noted in a model of brain I/R.80 Pericytes are also known to line capillaries in the mammalian heart.81 It is possible that focal constrictions along the capillary lumen noted by Glyn et al82 in reperfused rat hearts represent the same phenomenon.
Low shear stress represents another important aspect of RMI. Apparent blood viscosity increases at low shear rates, and erythrocyte rouleau formation, aggregation, and clumping may further reduce erythrocyte flow through capillaries. Erythrocyte aggregation from increased local capillary flow resistance, as a result of non-uniform leakage of plasma through ECs, can contribute to flow heterogeneity.83
Low-velocity flow per se can activate leukocytes66 and result in microvascular sequestration.65–67 Leukocyte adhesion to ECs in postcapillary venules during low flow can markedly increase resistance to flow.67 Low shear stress may also promote obstructing responses of ECs, via mechanotransduction originating in the glycocalyx.69 Low flow in the setting of vascular injury has also been shown to enhance platelet activation.70
Sluggish flow of erythrocytes through injured capillaries reduces oxygen delivery to ECs, and the cycle of profound hypoxia and obstructive responses to hypoxia and low shear stress can continue unabated until capillary occlusion occurs. If EC viability is maintained, chronic responses to hypoxia may occur. These include continued HIF-1 stabilization and numerous, complex biochemical pathways that may be associated with either pathologic or adaptive responses.84
How Can Oxygen Delivery Be Effectively Increased To Regions Of RMI?
On room air breathing, hemoglobin O2 saturation approaches 100% in most patients with adequate lung function. Moreover, the solubility of O2 in plasma is low.85 Therefore, oronasal supplementation in such patients is ineffective in increasing oxygen delivery, and studies of such treatment in patients with acute coronary syndromes show no benefit.86–88 The O2 content of plasma at hyperbaric PO2s, however, is sufficient to at least partially compensate for low erythrocyte flow. At HBOT exposures of 2.0 to 3.0 ATA (101 to 202 kPa above atmospheric), sufficient O2 can be dissolved in plasma to meet metabolic demands at extremely low hemoglobin levels.89 Although physiologic autoregulation of blood flow during hyperoxia results in vasoconstriction of precapillary sphincters in non-ischemic tissues,90 median tissue PO2 nevertheless increases in normal myocardium and other tissues.91 In regions of myocardial ischemia, hyperoxia increases blood flow and reduces ischemia.92,93
Liquid infusion of oxygen (aqueous oxygen = AO) is a new, catheter-based approach for achieving regional, site-specific hyperbaric levels of oxygen in blood.94 AO at high dissolved O2 concentrations can be delivered into host liquids at ambient pressure in a bubbleless manner because heterogeneous nucleation is prevented.95 The oxygen level of host liquids such as blood can be predictably increased to >133 kPa (1000 mmHg) without bubble nucleation. Oxygenation occurs quite rapidly from convective liquid mixing of AO with blood (in contrast to the slow process of oxygen diffusion at a gas/liquid interface) under laminar flow conditions and at physiological velocities. Biocompatibility problems related to a large foreign body surface area in contact with blood and low flow shear stress, as with the use of membrane oxygenators, are eliminated with this new approach.
Clinically, the PO2 of the coronary blood perfusate, termed supersaturated oxygen (SSO2), is adjusted to about 120 ± 13 kPa (900 ± 100 mmHg) by addition of AO (typical [AO] = 1.2 mL O2/mL saline) at approximately 50:1 volume ratio of [arterial blood]/[AO], sufficient to increase the O2 content of plasma to approximately 3 vol %.96 The high O2 tension should markedly increase the effective O2 diffusion distance between capillaries. Although O2 diffusion at the level of arterioles at a normal arterial PO2 does not normally contribute significantly to tissue oxygenation,97 hyperbaric oxygen tensions may facilitate such diffusion.98
Studies Of Methods To Attenuate RMI
The lack of readily available methods for clinical detection of RMI has impeded progress in the potential treatment of the problem. Nevertheless, there has been growing impetus to the concept that methods for improving myocardial tissue perfusion, by addressing the obstructive components of the microcirculation, may promote healing and possibly enhance functional recovery of viable myocardium.99–102
Hyperbaric Oxygen Therapy (HBOT) For Acute Myocardial Infarction And Reperfusion
Experimental studies with HBOT, conducted before the thrombolytic era, demonstrated a reduction in ventricular fibrillation and improved survival. In the 1960s, a few anectodal clinical reports of HBOT for acute myocardial infarction suggested benefit. Subsequently, a small, randomized clinical trial of HBOT for acute MI demonstrated a reduction in mortality (approximately 50% of the control group) that was statistically significant when the low-risk minority group of patients (Peel Index) was excluded.103 Of the 12 patients in cardiogenic shock, the only survivors (n = 3) received HBOT.
In the reperfusion era, experimental studies demonstrated that HBOT reduced infarct size when administered during reperfusion. Two subsequent small, randomized clinical trials of HBOT for STEMI104,105 demonstrated reductions in CPK release and improvement in parameters of LV function, with statistically greater values than those of the control group in one study. In a different randomized study of HBOT for either acute MI or unstable angina, a significant reduction in restenosis and clinical events was noted in the treatment group.106
A Cochrane meta-analysis of six small randomized clinical trials of HBOT for AMI demonstrated significant reductions (p < 0.05) in major adverse events, dysrhythmias, heart block, and time to pain relief, along with a non-significant trend in mortality reduction (p = 0.08).107 Despite the encouraging results of HBOT, practical problems with this cumbersome technology,108 including lack of ready access to critically ill patients within pressurized chambers and undesirable side effects, such as middle ear barotrauma and claustrophobia, along with fire hazards, have limited the conduct of studies in larger groups of patients.
However, in three randomized, double-blinded clinical trials of HBOT for treatment of microvascular dysfunction and ischemia of other tissues, significant improvements (p < 0.05) were noted in skin graft survival;109 healing of crush injuries;110 and cognitive sequelae after carbon monoxide poisoning.111
Advances have also been made experimentally in the understanding of potentially beneficial mechanisms of HBOT in the treatment of a wide variety of reperfused tissues.112 Improved FCD and inhibition of neutrophil-EC adherence have been observed, with potential mechanisms including inhibition of neutrophil beta2-integrin and reduction of EC and neutrophil ICAM-1. Other suggested benefits of HBOT include improved oxygen supply to the ischemic microcirculation; inhibition of apoptosis; reduction of lipid peroxidation; increased superoxide dismutase levels; improved endothelial-dependent vasorelaxation; and stimulation of the endothelial fibrinolytic system.
As perhaps the most extreme problem in reperfusion, resuscitation beyond a 15-mins period of cardiac arrest is usually not possible. However, Van Meter et al demonstrated that high-dose HBOT was usually successful for resuscitation of swine subjected to 25 mins of cardiac arrest compared to normoxic and low-dose HBOT groups.113 HBOT has also been shown to reduce mortality in experimental studies of shock, sepsis, and multiorgan failure.114
Preclinical SSO2 Studies
As a catheter-based approach for providing regional arterial hyperoxemia, SSO2 infusion is a potentially more practical approach than HBOT for critically ill patients, and precise control of regional arterial oxygen levels is achievable. In a canine model of regional low flow myocardial ischemia, intracoronary SSO2 infusion prevented the fall in echo LVEF associated with normoxemic low flow perfusion.94 The potential benefits of intracoronary SSO2 infusion for acutely enhancing post MI reperfusion in animal models were described by Spears et al115–117 and Johnson et al,118 and reviewed by Glazier119 and Bartorelli.120 A 90-mins treatment, initiated 15–30 mins after the onset of reperfusion following a 60–90 mins coronary occlusion, was associated with normalization of left ventricular ejection fraction (LVEF) compared to no improvement of this parameter in control groups. The frequency of ventricular extrasystoles was significantly reduced by the treatment. Microvascular blood flow upon completion of SSO2 infusion was doubled compared to that of controls.116 Histologically, microvascular hemorrhage, tissue myeloperoxidase, and infarct size were each markedly reduced (60–80%)(p < 0.05) by SSO2 compared to three control groups at 3 hrs of reperfusion.117 By transmission electron microscopy (TEM), striking differences were noted between SSO2 (n = 3) and autoreperfusion control (n = 3) groups (Figure 2).115,120 In the control group, EC edema and loss of nuclei were noted, along with prominent disruption of myofibrillar structure. In the SSO2 group, EC edema was not observed, and EC nuclei were preserved. Myofibrillar disruption was not noted.115 Evidence of apoptosis in myocardium reperfused for 3 hrs was found in control group, but not in the SSO2 group (Figure 3).115 The scattered pattern of apoptosis is consistent with the concept of RMI.
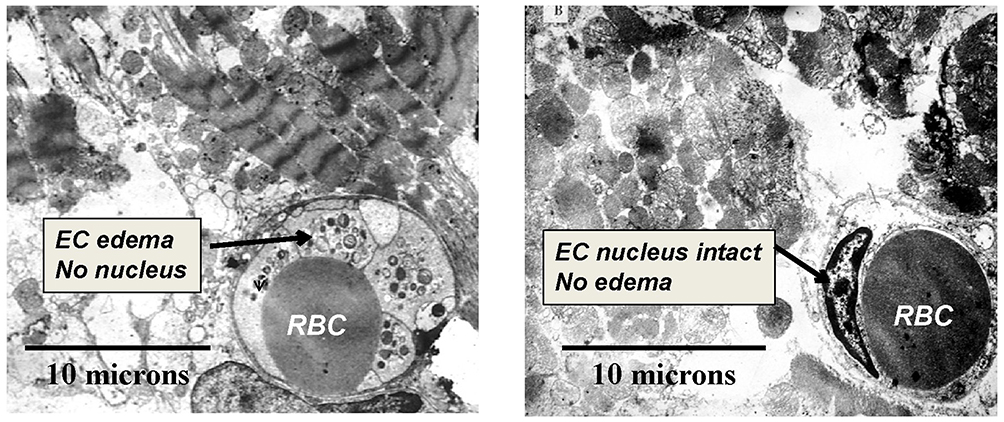 |
Figure 2 Transmission electron microscopy (TEM) of porcine myocardium at 3 hrs of reperfusion following 1 hr of coronary occlusion. Representative images from control (left panel) and SSO2-treated (right panel) groups (masked assignment selection by cardiac pathologist [Vander Heide R]) (data from Spears et al,117). EC = Endothelial Cell. RBC = Red Blood Cell (within lumen of EC). EC edema, loss of EC nuclei, and prominent disruption of myofibrillar structure was evident in the Control group only. Reprinted by permission from Springer Nature (Springer Nature) (Am J Cardiovasc Drugs) (Hyperoxemic perfusion for treatment of reperfusion microvascular ischemia in patients with myocardial infarction) Bartorelli AL., Copyright (2003).120 |
 |
Figure 3 Representative histologic sections of porcine myocardium stained with both propidium iodide (red fluorescence of all nuclei, left column) and terminal deoxynucleotidyl transferase dUTP nick end labeling of apoptotic cells (green fluorescence; DeadEndTM Fluorometric TUNEL System, Promega)(middle column).115 Superimposition of the fluorescent images in right column (Dual). Normal = myocardium not subjected to ischemia (top row). Control AutoRP = 3 hrs of reperfusion (AutoRP = passive reperfusion) after one hour ischemia (LAD occlusion)(middle row). SSO2 = 3 hrs of reperfusion after one hour ischemia (LAD occlusion), with SSO2 perfusion performed for 90 mins, 15 mins after the onset of reperfusion (bottom row). Apoptosis, in a heterogeneously distributed pattern (green dots), was noted in each Control AutoRP animal (n = 3) and in none of the animals in the Normal group (n = 3) and the SSO2 group (n = 3). |
Interestingly, intracoronary SSO2 infusion, when delayed by 24 hrs after the onset of reperfusion following a 1 hr occlusion of the LAD in swine, still resulted in significant improvements in infarct size and LVEF compared to the control group.121 No significant improvement in LVEF was noted in the control group despite 24 hrs of reperfusion. The results suggest that RMI in this model persists for a prolonged period but is nevertheless amenable to treatment. The observations suggest that myocardial “stunning” following 24 hrs of reperfusion in this model simply results from uncorrected microvascular hypoxia that is reversible with SSO2 infusion.
Clinical SSO2 Studies
In a Phase I multicenter clinical trial of SSO2 for treatment of STEMI within 24 hrs of symptom onset, hyperoxemic perfusion was performed for 60 to 90 mins after successful stenting of the infarct coronary artery in 29 patients.122 Over a three-month period after the procedure, progressive improvement in echocardiographic regional wall motion score was observed, primarily as a result of infarct zone improvement. In a similar small trial of SSO2 for patients with anterior STEMI, Trabattoni et al123 found significant improvements in global and regional left ventricular function at 3 months, compared to a matched control group (p < 0.01). The time-to-peak and half-life of creatine kinase were shorter compared to the control group (p < 0.01), and the rate of complete ST segment resolution (78%) compared to the control group (42%) was significantly greater (p < 0.01).
In a randomized trial of SSO2 infusion vs autoreperfusion controls post PCI, STEMI patients were treated within 24 hrs of symptom onset (AMIHOT trial).124 The primary endpoints, SPECT sestamibi infarct size (% LV mass) at 2 weeks (treatment = 9% vs control = 23%), acute ST-segment resolution, and echo wall motion score index at 3 months (0.75 vs 0.54) were significantly improved (p< 0.05) only in the subset of anterior STEMI patients treated within 6 hrs of symptom onset. In the follow-up, AMIHOT II randomized clinical trial, only patients with anterior wall myocardial infarction who were reperfused within 6 hrs of symptom onset were therefore included.125 The trial followed a Bayesian statistical design, wherein the results of the Phase II study were incorporated into those of the follow-up trial, with weighting of the Phase II data according to degree of similarity between trials. A statistically significant, smaller median infarct size, measured at two weeks by SPECT sestamibi imaging, was noted in the treatment group compared to the control group (18.5 vs 25% of left ventricular mass, or a relative infarct size reduction of 26%). In patients with a baseline LVEF <40%, even greater salvage was noted, with an absolute infarct size of 33.5% in controls and 23.5% in SSO2-treated patients. The relatively small infarct size in this STEMI population was recently confirmed by cardiac magnetic resonance imaging at 30 days in 100 anterior STEMI patients treated post-stenting with 60 mins of intracoronary SSO2 (IC-HOT Trial).126 The remarkably low 30-day complication rate (mortality = 0%; new onset CHF = 1%), with no significant change in these complications at 1 year,127 are consistent with a favorable clinical outcome associated with a reduction in infarct size.
Attenuation Of RMI: Potential Mechanisms Of Improved Microvascular Flow
Many of the observed in vivo responses of reperfused tissues to HBOT and SSO2 may be explicable by attenuation of hypoxia-mediated obstructive effects of ECs and blood elements (Table 4). For example, Ali et al demonstrated that the increase in EC monolayer permeability induced by prolonged hypoxia was reversible during a subsequent hour of normoxia.128 A reduction in ROS-mediated cytokine secretion, which was increased by hypoxia, appeared responsible for the effect. In a related study, reoxygenation reduced EC-neutrophil adhesion triggered by hypoxia.129 Since inflammation can trigger increased EC permeability, a reduction in inflammation may improve EC barrier function. Rapid reassembly of EC microfilaments from an increase in ATP levels,130 with restoration of cytoskeleton integrity, may also occur with attenuation of hypoxia.
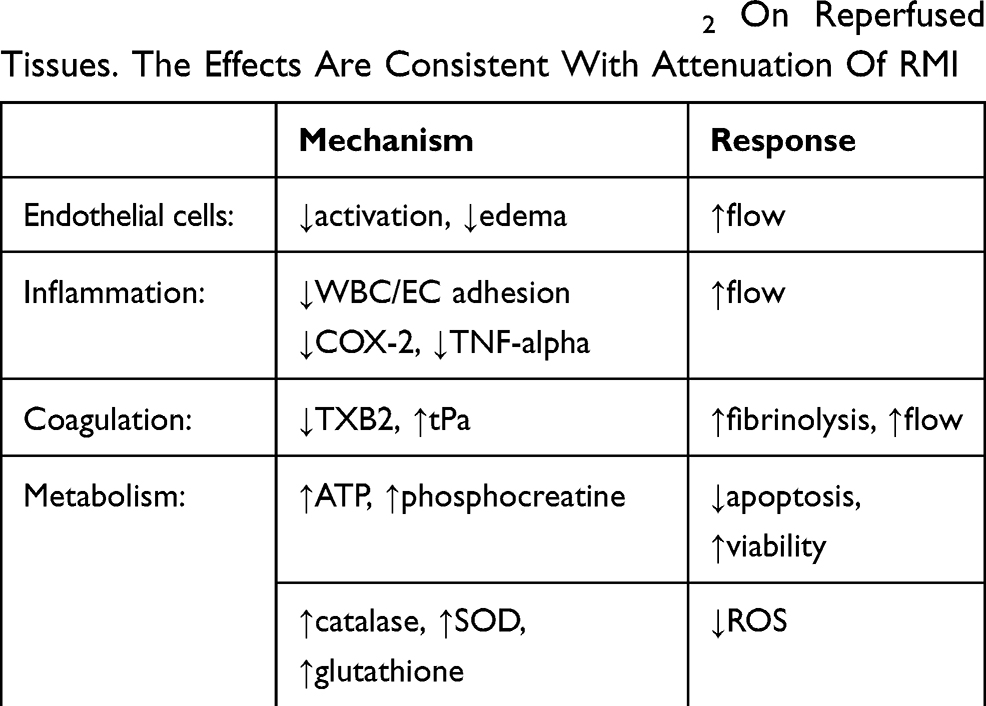 |
Table 4 Observed Effects Of HBOT/SSO2 On Reperfused Tissues. The Effects Are Consistent With Attenuation Of RMI |
Destabilization of HIF-1 upon correction of hypoxia may help attenuate a cycle of hypoxia and inflammatory responses.62 In addition, S-glutathionylation of protein thiols under oxidative stress provides a reversible metabolic switch governing eNOS uncoupling.131 As another potentially reversible pathway, hypoxic repression of eNOS may occur from eviction of histones from the eNOS promoter, while the ATP-dependent chromatin-remodeling enzyme brahma-related gene 1 (BRG1) can help restore eNOS expression following reoxygenation.132 Additional studies will also be required to define reversible biochemical pathways associated with shear stress-triggered mechanotransduction, but S-nitrosylation of EC proteins may represent one example.133
Injured but still viable myocytes may be rescued by improvement in oxygen delivery. The potential contribution of improvement in microvascular integrity on infarct size, function, healing, and survival was described by Weis et al.28 Molecular mechanisms related to improved capillary integrity and myocyte survival from attenuation of RMI may be difficult to define, however, in view of the complex biochemistry of cell death,134,135 including apoptosis, necrosis, and autophagy. Reduction of tissue hypoxia might reduce oxidative stress, with inhibition of mitochondrial permeability pore transition opening.136 Scarabelli et al137 found in a rat heart I/R model that apoptosis of ECs occurs earlier than myocyte apoptosis; moreover, release of soluble pro-apoptotic mediators associated with EC apoptosis appears to induce myocyte apoptosis. Prevention of EC apoptosis may prevent myocyte apoptosis. The findings of Pozzi et al,138 showing that hypoxia alone, without the need for reperfusion, causes apoptosis of myocytes in Langendorff-perfused hearts, are consistent with this concept. Maintainance of myocyte viability may allow time for other beneficial effects, such as resolution of myofibrillar edema with improvement of myocyte function.139
Even in the absence of myocyte viability, the presence of microcirculatory flow through the infarct zone is a critical determinant of complications. Hypoxia may increase the activity of certain metalloproteinases (MPs)140 and decrease that of tissue inhibitors of MPs.141 Enhanced degradation of structural myocardial proteins such as collagen by MPs may contribute to myocardial wall thinning, infarct expansion, and adverse left ventricular remodeling.142 If so, hyperoxic reperfusion may attenuate such pathologic responses.143
Oxygen Radicals
Hypoxia per se is well known to result in an excessive load of ROS that can adversely affect proteins, nucleic acids, and lipids.144 Injury to the distal electron transport chain during ischemia is an important mechanism for the formation of ROS during subsequent reperfusion. Although reintroduction of oxygen necessarily results in the generation of ROS, Lesnefsky et al145 found that additional injury to the distal electron transport chain was not observed during reperfusion in the isolated rabbit heart.
The statistically significant, marked reduction in tissue myeloperoxidase levels, a quantitative measure of neutrophil counts,146 in the reperfused myocardium treated with SSO2 compared to control groups in two independent models of myocardial infarction117,118 suggests that inflammation was reduced experimentally by the treatment. If ROS had been increased by SSO2, one would have expected evidence of more inflammation.147 The findings are consistent with inhibition of neutrophil adhesion by HBOT in reperfused tissues. Although a burst of ROS is generated during early myocardial reperfusion, hypoxic reperfusion has been shown by EPR spectroscopy to greatly increase ROS formation compared to normoxic and hyperoxic reperfusion in an isolated rat heart I/R model.148 ROS formation in this model was subsequently found to be compartment-specific, however; low O2 perfusate promoted extracellular ROS, while high O2 perfusate promoted intracellular ROS.149 Given the relatively brief period of ischemia (20 mins), the high O2 perfusate may have produced supranormal intracellular PO2s, rather than attenuated RMI.
As reviewed by Thom, oxidant stress may provide a beneficial role in the effects of HBOT, similar to the role of ROS in signal transduction cascades related to ischemic preconditioning.150 In addition to the possibility that attenuation of RMI may reduce leukocyte activation, hyperoxia per se has been shown to increase nitrosylation of B-actin, so that filamentous actin distribution within the cell is altered and inhibits B2 integrin clustering at the membrane surface. HBOT has also been shown to inhibit lipid peroxidation via a reaction between hydroperoxyl radicals and organic radicals.150 The reaction, in addition to attenuation of ischemia, may help explain the reduction in lipid peroxidation products associated with HBOT for carbon monoxide poisoning.
Limitations
The remarkably low adverse event rate at 30 days and 1 year in the IC-HOT trial126,127 is consistent with clinical observations regarding the effect of intracoronary hyperoxemia during reperfusion on infarct size.124–126 However, the optimal level and duration of SSO2 reperfusion is unknown. Whether brief intermittent infusions or repeat infusions of SSO2 over several days would be advantageous has also not been studied. Real-time assessment of cellular ischemia or hypoxia during treatment would be helpful but currently possible only in animal models with technologies still in evolution. The potential contribution of osmotic removal of tissue edema (including within endothelial cells) by hyperoxemia151 is unknown but may acutely reduce myocardial edema that compresses capillaries (similar to “compartment syndromes” that respond to HBOT). Unlike HBOT, however, dissolved nitrogen is not removed in association with AO-induced hyperoxemia, so that this mechanism may be enhanced.
In order to mimic the clinical setting, SSO2 infusion has been performed in animal models 15–30 mins after the onset of coronary artery reperfusion. Substantial microvascular and myocyte injury resulting from pathogenetic mechanisms associated with reperfusion per se (eg, Ca2+ overload, ROS, mitochondrial permeability transition pore opening, etc.) may have already occurred prior to treatment. Indeed, Johnson et al118 showed experimentally that immediate perfusion with SSO2 post coronary ischemia via a retrograde coronary venous route was more effective in infarct size reduction compared to similar treatment following a 30-mins period of reperfusion.
It is possible that successful treatment of pathogenetic mechanisms associated with reperfusion would reduce the severity of RMI. Conversely, improvement in microvascular flow by reduction of RMI may enhance the efficacy of therapeutic strategies directed at specific aspects of potentially lethal reperfusion injury.152–154
Conclusions
Physiologic heterogeneities in flow and in vulnerability to ischemia provide a substrate for RMI after a prolonged arterial occlusion. Highly specialized methods have been required for its detection even experimentally, however, so that the problem has received little attention clinically. The recent advances in OPS imaging of the microcirculation in man have provided important insights regarding the implications of a reduction in functional capillary density and heterogeneously low erythrocyte flow on tissue viability. The results of studies of HBOT and SSO2 during postinfarction reperfusion provide compelling evidence that enhanced oxygen delivery in plasma can be used to attenuate a cycle of RMI and obstructive responses of ECs and blood elements, thereby improving tissue level flow and myocardial viability.
Acknowledgment
The author is grateful to Li Li, PhD (Wayne State University, Center for Molecular Medicine and Genetics) for expert review of selected sections of the manuscript, as well as for her contribution to Figure 3.
Disclosure
JRS is the scientific co-founder of Therox, Inc. (1994). He owns no stock in Therox, Inc.; all relevant patents issued to JRS, assigned to Wayne State University, and licensed to Therox, Inc., have expired. The author reports no other conflicts of interest in this work.
References
1. Jeremy RW, Links JM, Becker LC. Progressive failure of coronary flow during reperfusion of myocardial infarction: documentation of the no reflow phenomenon with positron emission tomography. J Am Coll Cardiol. 1990;16:695–704. doi:10.1016/0735-1097(90)90362-s
2. Wu KC, Zerhouni EA, Judd RM, et al. Prognostic significance of microvacular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation. 1998;97:765–772. doi:10.1161/01.cir.97.8.765
3. Rochitte CE, Lima JAC, Bluemke DA, et al. The magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation. 1998;98:1006–1014. doi:10.1161/01.CIR.98.10.1006
4. O’Regan DP, Ariff B, Neuwirth C, Tan Y, Durighel G, Cook SA. Assessment of severe reperfusion injury with T2 cardiac MRI in patients with acute myocardial infarction. Heart. 2010;96:1885–1891. doi:10.1136/hrt.2010.200634
5. Ambrosio G, Weisman HF, Mannisi JA, Becker LC. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation. 1989;80:1846–1861. doi:10.1161/01.cir.80.6.1846
6. Pries AR, Habazettl H, Ambrosio G, et al. A review of methods for assessment of coronary microvascular disease in both clinical and experimental settings. Cardiovasc Res. 2008;80:165–174.
7. Swartz HM, Dunn JF. Measurements of oxygen in tissues: overview and perspectives on methods. Adv Exp Med Biol. 2003;530:1–12. doi:10.1007/978-1-4615-0075-9_1
8. Vikram DS, Zweier JL, Kuppusamy P. Methods for noninvasive imaging of tissue hypoxia. Antioxid Redox Signal. 2007;9:1745–1756. doi:10.1089/ars.2007.1717
9. Jain D, He Z-X. Direct imaging of myocardial ischemia: a potential new paradigm in nuclear cardiovascular imaging. J Nucl Card. 2008;15:617–630. doi:10.1016/j.nuclcard.2008.07.004
10. Handley MG, Medina RA, Nagel E, Blower PJ, Southworth R. PET imaging in cardiac hypoxia: opportunities and challenges. J Mol Cell Cardiol. 2011;51(5):640–650. doi:10.1016/j.yjmcc.2011.07.005
11. Bauer WR, Hiller K-H, Galuppo P, et al. Fast high-resolution magnetic resonance imaging demonstrates fractality of myocardial perfusion in microscopic dimensions. Circ Res. 2001;88:340–346. doi:10.1161/01.res.88.3.340
12. Zuurbier CJ, van Iterson M, Ince C. Functional heterogeneity of oxygen supply-consumption ratio in the heart. Cardiovasc Res. 1999;44:488–497. doi:10.1016/s0008-6363(99)00231-x
13. Balaban RS, Arai A. Function, metabolic, and flow heterogeneity of the heart. The View Is Getting Better. Circ Res. 2001;88:265–267.
14. Pries AR, Secomb TW. Origins of heterogeneity in tissue perfusion and metabolism. Cardiovasc Res. 2009;81:328–335. doi:10.1093/cvr/cvn318
15. Ghaleh B, Shen YT, Vatner SF. Spatial heterogeneity of myocardial blood flow presages salvage versus necrosis with coronary artery reperfusion in conscious baboons. Circulation. 1996;94:2210–2215. doi:10.1161/01.cir.94.9.2210
16. Steenbergen C, Deleeuw G, Barlow C, Chance B, Williamson JR. Heterogeneity of the hypoxic state in perfused rat heart. Circ Res. 1977;41:606–615. doi:10.1161/01.res.41.5.606
17. Ince C, Vink H, Wieringa PA, Giezeman M, Spaan JA. Heterogeneous NADH fluorescence during post-anoxic hyperemia in saline perfused heart. Adv Exp Med Biol. 1990;277:477–482. doi:10.1007/978-1-4684-8181-5_53
18. Ince C, Ashruf JF, Avontuur JA, Wieringa PA, Spaan JA, Bruining HA. Heterogeneity of the hypoxic state in rat heart is determined at the capillary level. Am J Physiol. 1993;264:H294–H301. doi:10.1152/ajpheart.1993.264.2.H294
19. Vetterlein F, Prange M, Lubrich D, Pedina J, Neckel M, Schmidt G. Capillary perfusion pattern and microvascular geometry in heterogeneous hypoxic areas of hypoperfused rat myocardium. Am J Physiol. 1995;268:H2183–H2194. doi:10.1152/ajpheart.1995.268.6.H2183
20. Groner W, Winkelman JW, Harris AG, et al. Orthogonal polarization spectral imaging: a new method for study of the microcirculation. Nat Med. 1999;5:1209–1212. doi:10.1038/13529
21. De Backer D, Ospina-Tascon G, Salgado D, Favory R, Creteur J, Vincent JL. Monitoring the microcirculation in the critically ill patient: current methods and future approaches. Intensive Care Med. 2010;36:1813–1825. doi:10.1007/s00134-010-2005-3
22. Sakr Y, Dubois M-J, De Backer D, Creteur J, Vincent J-L. Persistent microcirculatory alterations are associated with organ failure in patients with septic shock. Crit Care Med. 2004;32:1825–1831. doi:10.1097/01.ccm.0000138558.16257.3f
23. Ellis CG, Bateman RM, Sharpe MD, Sibbald WJ, Gill R. Effect of maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002;2002(282):H156–H164. doi:10.1152/ajpheart.2002.282.1.H156
24. De Backer D, Creteur J, Dubois MJ, Sakr Y, Vincent JL. Microvascular alterations in patients with acute severe heart failure and cardiogenic shock. Am Heart J. 2004;147:91–99. doi:10.1016/j.ahj.2003.07.006
25. De Backer D, Dubois MJ, Schmartz D, et al. Microcirculatory alterations in cardiac surgery: effects of cardiopulmonary bypass and anesthesia. Ann Thorac Surg. 2009;88:1396–1403. doi:10.1016/j.athoracsur.2009.07.002
26. Camilleri JP, Nlom MO, Michel JB, Barres D, Mignot J. Capillary perfusion patterns in reperfused ischemic subendocardial myocardium: experimental study using fluorescent dextran. Exp Mol Pathol. 1983;39:89–99. doi:10.1016/0014-4800(83)90043-6
27. Downey HF, Bashour FA, Jishi B, Parker PE. Arteriovenous shunts in dilated or reperfused canine coronary vasculature. Microvasc Res. 1979;17:22–26. doi:10.1016/0026-2862(79)90004-9
28. Weis S, Shintani S, Weber A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–894. doi:10.1172/JCI20702
29. Kay M, Swift L, Martell B, Aruntunyan A, Sarvazyan N. Locations of ectopic beats coincide with spatial gradients of NADH in a regional model of low-flow reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H2400–H2405. doi:10.1152/ajpheart.01158.2007
30. Wang B, Ansari R, Sun Y, Postlethwaite AE, Weber KT, Kiani MF. The scar neovasculature after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2005;289:H108–H113. doi:10.1152/ajpheart.00001.2005
31. Wang B, Scott RC, Pattillo CB, Prabhakarpandian B, Sundaram S, Kiani MF. Microvascular transport model predicts oxygenation changes in the infarcted heart after treatment. Am J Physiol Heart Circ Physiol. 2007;293:H3732–H3739. doi:10.1152/ajpheart.00735.2007
32. Al-Obaidi MK, Etherington PJ, Barron DJ, Winlove CP, Pepper JR. Myocardial tissue oxygen supply and utilization during coronary artery bypass surgery: evidence of microvascular no-reflow. Clin Sci. 2000;98:321–328. doi:10.1042/cs0980321
33. Pathi VL, McPhaden AR, Morrison J, et al. The effects of cardioplegic arrest and reperfusion on the microvasculature of the heart. Europ J Cardio-Thor Surg. 1997;11:350–357. doi:10.1016/S1010-7940(96)01037-8
34. Lipowski HH. Microvascular rheology and hemodynamics. Microcirculation. 2005;12:5–15. doi:10.1080/10739680590894966
35. Boerma EC, Ince C. The role of vasoactive agents in the resuscitation of microvascular perfusion and tissue oxygenation in critically ill patients. Intensive Card Med. 2010;36:2004–2018. doi:10.1007/s00134-010-1970-x
36. Eliasen P, Amtorp O. Oxygen transport to the canine left ventricle distal to a flow-limiting coronary artery stenosis. Int J Microcirc Clin Exp. 1985;4:329–341.
37. Barker MC, Golub AS, Pittman RN. Erythrocyte-associated transients in capillary PO2: an isovolemic hemodilution study in rat spinotrapezius muscle. Am J Physiol Heart Circ Physiol. 2007;292:H2540–H2549. doi:10.1152/ajpheart.00915.2006
38. Goldman D, Bateman RM, Ellis CG. Effect of sepsis on skeletal muscle oxygen consumption and tissue oxygenation: interpreting capillary oxygen transport data using a mathematical model. Am J Physiol Heart Circ Physiol. 2004;287:H2535–H2544. doi:10.1152/ajpheart.00889.2003
39. Connett RJ, Honig CR, Gayeski TE, Brooks GA. Defining hypoxia: a systems view of VO2, glycolysis, energetics, and intracellular pO2. J Appl Physiol. 1990;68:833–842. doi:10.1152/jappl.1990.68.3.833
40. Sezer M, van Royen N, Umman B, et al. Coronary microvascular injury in reperfused acute myocardial infarction: a view from an integrative perspective. J Am Heart Assoc. 2018;7(21):e009949. doi:10.1161/JAHA.118.008528
41. Kloner RA, Dai W, Hale SL. No-reflow phenomenon. A new target for therapy of acute myocardial infarction independent of myocardial infarct size. J Cardiovasc Pharmacol Ther. 2018;23(3):273–276.
42. Hori M, Inoue M, Kitakaze M, et al. Role of adenosine in hyperemic response of coronary blood flow in microcirculation. Am J Physiol. 1986;250:H509–H518. doi:10.1152/ajpheart.1986.250.3.H509
43. Kassab GS, Rider CA, Tang NJ, Fung YC. Morphometry of pig coronary arterial trees. Am J Physiol. 1993;265:H350–H365. doi:10.1152/ajpheart.1993.265.1.H350
44. Wang X, Peter K. Molecular imaging of atherothrombotic diseases. Seeing Is Believing. Arterioscler Thromb Vasc Biol. 2017;37:1029–1040. doi:10.1161/ATVBAHA.116.306483
45. Ziegler M, Alt K, Paterson BM, et al. Highly sensitive detection of minimal cardiac ischemia using positron emission tomography imaging of activated platelets. Sci Rep. 2016;6:38161. doi:10.1038/srep38161
46. Liu S, Liu W, Ding W, Miyake M, GA R, Liu KJ. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:1274–1284. doi:10.1038/sj.jcbfm.9600277
47. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA. 2006;103:5379–5384. doi:10.1073/pnas.0601026103
48. Ten VS, Pinsky DJ. Endothelial response to hypoxia: physiologic adaptation and pathologic dysfunction. Curr Opin Crit Care. 2002;8:242–250.
49. Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol. 2004;164:1875–1882. doi:10.1016/S0002-9440(10)63747-9
50. Pober JS. Physiology and pathobiology of microvascular endothelium. In: Tuma RE, Duran WN, Ley K, editors. Handbook of Physiology: Microcirculation.
51. Pober JS. Physiology and Pathobiology of Microvascular Endothelium. Compr Physiol. 2011; Supplement 9:37–55. Handbook of Physiology, The Cardiovascular System, Microcirculation. First published in print 2008. doi: 10.1002/cphy.cp020402.
52. Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemostasis. 2005;3:1392–1406. doi:10.1111/j.1538-7836.2005.01328.x
53. Matsushita H, Morishita R, Nata T, et al. Hypoxia-induced endothelial apoptosis through nuclear factor-kB (NF-kB)-mediated bcl-2 suppression. In vivo evidence of the importance of NF-kB in endothelial cell regulation. Circ Res. 2000;86:974–981. doi:10.1161/01.res.86.9.974
54. Walford GA, Moussignac R-L, Scribner AW, Loscalzo J, Leopold JA. Hypoxia potentiates nitric oxide-mediated apoptosis in endothelial cells via peroxynitrite-induced activation of mitochondria-dependent and -independent pathways. JBC. 2004;279:4425–4432. doi:10.1074/jbc.M310582200
55. Ohta T, Eguchi R, Suzuki A, Miyakaze S, Ayuzawa R, Kaji K. Hypoxia-induced apoptosis and tube breakdown are regulated by p38 MAPK but not by caspase cascade in an in vitro capillary model composed of human endothelial cells. J Cell Physiol. 2007;211:673. doi:10.1002/jcp.20975
56. Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi:10.1182/blood-2004-07-2958
57. Winning S, Splettstoesser F, Fandrey J, Frede S. Acute hypoxia induces HIF-independent monocyte adhesion to endothelial cells through increased intercellular adhesion molecule-1 expression: the role of hypoxic inhibition of prolyl hydroxylase activity for the induction of NF-kappa B. J Immunol. 2010;185:1786–1793. doi:10.4049/jimmunol.0903244
58. Tian F, Zhou AX, Smits AM, et al. Endothelial cells are activated during hypoxia via endoglin/ALK-1/SMAD1/5 signaling in vivo and in vitro. Biochem Biophys Res Commun. 2010;392:283–288. doi:10.1016/j.bbrc.2009.12.170
59. Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–247. doi:10.1161/CIRCRESAHA.107.151860b
60. He P. Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled? Cardiovasc Res. 2010;87:281–290. doi:10.1093/cvr/cvq140
61. Bartels K, Grenz A, and Elzschig HK. Hypoxia and inflammation are two sides of the same coin. PNAS. 2013;110(46):18351–18352. doi:10.1073/pnas.1318345110
62. Eltzchig HK, Carmeliet P. Hypoxia and inflammation. New Engl J Med. 2011;364:656–665. doi:10.1056/NEJMra0910283
63. Aversa CR, Oparil S, Caro J, et al. Hypoxia stimulates human proendothelin-1 promoter activity in transgenic mice. Am J Physiol. 1997;273:L848–L855. doi:10.1152/ajplung.1997.273.4.L848
64. Parthasarathi K, Lipowski HH. Capillary recruitment in response to tissue hypoxia and its dependence on red cell deformability. Amer J Physiol Heart Circ Physiol. 1999;46:H2145–H2157. doi:10.1152/ajpheart.1999.277.6.H2145
65. Ritter LS, McDonagh PF. Low-flow reperfusion after myocardial ischemia enhances leukocyte accumulation in coronary microcirculation. Am J Physiol. 1997;273:H1154–H1165.
66. Kubes P. The role of shear forces in ischemia/reperfusion-induced neutrophil rolling and adhesion. J Leukoc Biol. 1997;62:458–464. doi:10.1002/jlb.62.2.268
67. Eppihimer MJ, Lipowski HH. Leukocyte sequestration in the microvasculature in normal and low flow states. Am J Physiol. 1994;267:H1122–H1134. doi:10.1152/ajpheart.1994.267.3.H1122
68. Sriramarao P, Languino LR, Altieri DC. Fibrinogen mediates leukocyte-endothelium bridging in vivo at low shear forces. Blood. 1996;88:3416–3423.
69. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. doi:10.1038/nrm2596
70. Bassiouny HS, Song RH, Kocharyan H, Kins E, Glagov S. Low flow enhances platelet activation after acute experimental arterial injury. J Vasc Surg. 1997;27:910–918. doi:10.1016/S0741-5214(98)70272-1
71. Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res. 2010;87:331–339. doi:10.1093/cvr/cvq145
72. Tang Y, Nyengaard JR, Andersen JB, Baandrup U, Gundersen HJG. The application of stereological methods for estimating structural parameters in the human heart. Anat Rec. 2009;292:1630–1647. doi:10.1002/ar.20952
73. Pries AR, Neuhaus D, Gaehtgens P. Blood viscosity in tube flow: dependence on diameter and hematocrit. Am J Physiol. 1992;263:H1770–H1778. doi:10.1152/ajpheart.1992.263.6.H1770
74. Vince RV, Chrismas B, Midgley AW, McNaughton LR, Madden LA. Hypoxia mediated release of endothelial microparticles and increased association of S100A12 with circulating neutrophils. Oxid Med Cell Longev. 2009;2:2–6.
75. Boerma EC, Ince C. The role of vasoactive agents in the resuscitation of microvacular perfusion and tissue oxygenation in critically ill patients. Intensive Care Med. 2010;36:2004–2018. doi:10.1007/s00134-010-1970-x
76. Ogawa S, Shreeniwas R, Brett J, Clauss M, Furie M, Stern DM. The effect of hypoxia on capillary endothelial cell function: modulation of barrier and coagulant function. Br J Haematol. 1990;75(4):517–524. doi:10.1111/j.1365-2141.1990.tb07792.x
77. Gupta N, Zhao -Y-Y, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. 2019;181:77–83. doi:10.1016/j.thromres.2019.07.013
78. Gertler JP, Perry L, L’Italien G, et al. Ambient oxygen tension modulates endothelial fibrinolysis. J Vasc Surg. 1993;18(6):939–946.
79. Ponicke K, Sternitsky R, Mest HJ. Stimulation of aggregation and thromboxane A2 formation of human platelets by hypoxia. Prostaglandins Leukot Med. 1987;29:49–59.
80. Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi:10.1038/nm.2022
81. Schulze C, Firth JA. Junctions between pericytes and the endothelium in rat myocardial capillaries: a morphometric and immunogold study. Cell Tissue Res. 1993;271:145–154. doi:10.1007/bf00297552
82. Glyn MC, Lawrenson JG, Ward BJ. Change in endothelial cell shape in response to ischaemia in the cardiac and retinal capillary beds. J Anat. 2002;200:526–534. doi:10.1046/j.1469-7580.2002.00047_11.x
83. Chenghai S, Jain RK, Munn LL. Non-uniform plasma leakage affects local hematocrit and blood flow: implications for inflammation and tumor perfusion. Ann Biomed Eng. 2007;35:2121–2129. doi:10.1007/s10439-007-9377-8
84. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–1480. doi:10.1152/jappl.2000.88.4.1474
85. Battino R, ed, Oxygen Data Series, Vol 7: Oxygen and Ozone. New York, NY: Pergamon Press; 1981:1–40.
86. Hofmann R, James SK, Jernberg T, et al. Oxygen therapy in suspected acute myocardial infarction. N Engl J Med. 2017;377:1240–1249. doi:10.1056/NEJMoa1706222
87. Khoshnood A, Carlsson M, Akbarzadeh M, et al. Effect of oxygen therapy on myocardial salvage in ST elevation myocardial infarction: the randomized SOOCER trial. Eur J Emerg Med. 2018;25(2):78–84. doi:10.1097/MEJ.0000000000000431
88. Sepehrvand N, James SK, Stub D, Khosnood A, Ezekowitz JA, Hofmann R. Effect of supplemental oxygen therapy in patients with suspected acute myocardial infarction: a meta-analysis of randomized clinical trials. Heart. 2018;104(20):1691–1698. doi:10.1136/heartjnl-2018-313089
89. Van Meter KW. A systematic review of the application of hyperbaric oxygen in the treatment of severe anemia: an evidence-based approach. Undersea Hyperb Med. 2005;32:61–83.
90. Duling BR. Microvascular responses to alterations in oxygen tension. Circ Res. 1972;31:481–489. doi:10.1161/01.res.31.4.481
91. Piantadosi CA. Pulmonary gas exchange, oxygen transport, and tissue oxygenation. In: Neuman TS, Thom SR, editors. Physiology and Medicine of Hyperbaric Oxygen Therapy. Philadelphia: Saunders; 2008:133–158.
92. Cason BA, Wisneski JA, Neese RA, et al. Effects of high arterial oxygen tension on function, blood flow distribution, and metabolism in ischemic myocardium. Circulation. 1992;85:828–838. doi:10.1161/01.cir.85.2.828
93. Ribeiro LGT, Louie EK, Davis MA, Maroko PR. Augmentation of collateral blood flow to the ischemia myocardium by oxygen inhalation following experimental coronary artery occlusion. Cardiovasc Res. 1979;13:160–166. doi:10.1093/cvr/13.3.160
94. Spears JR, Wang B, Wu X, et al. Aqueous oxygen: a highly O2-supersaturated infusate for regional correction of hypoxemia and production of hyperoxemia. Circulation. 1997;96:4385–4391. doi:10.1161/01.cir.96.12.4385
95. Brereton GJ, Crilly RJ, Spears JR. Nucleation in small capillary tubes. Chem Phys. 1998;230:253–265. doi:10.1016/S0301-0104(98)00052-4
96. Dixon SR, O’Neill WW, Bartorelli AL, et al. Early recovery of infarct zone function with intracoronary hyperoxemia reperfusion after primary angioplasty for acute myocardial infarction. JACC. 2002;39:387–392. doi:10.1016/s0735-1097(01)01771-5
97. Propel AS, Gross JF. Analysis of oxygen diffusion from arteriolar networks. Amer J Physiol. 1979;237:H681–H689. doi:10.1152/ajpheart.1979.237.6.H681
98. Sharan M, Singh MP, Singh B. An analytical model for oxygen transport in tissue capillaries in a hyperbaric environment with first order metabolic consumption. Math Comput Modelling. 1995;22:99–111. doi:10.1016/0895-7177(95)00171-W
99. Reffelmann T, Kloner RA. The “no-reflow” phenomenon: basic science and clinical correlates. Heart. 2002;87:162–168. doi:10.1136/heart.87.2.162
100. Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection. The “dark side” of reperfusion. Circulation. 2009;120:2105–2112. doi:10.1161/CIRCULATIONAHA.108.814640
101. Rodrigues SF, Granger DN. Role of blood cells in ischemia-reperfusion induced endothelial barrier failure. Cardiovasc Res. 2010;87:291–299. doi:10.1093/cvr/cvq090
102. Bekkers SCAM, Yazdani SK, Virmani R, Waltenberger J. Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J Am Coll Cardiol. 2010;55:1649–1660. doi:10.1016/j.jacc.2009.12.037
103. Thurston JGB, Greenwood TW, Bending MR, Connor H, Curwen MP. A controlled investigation into the effects of hyperbaric oxygen on mortality following acute myocardial infarction. Q J Med. 1973;42:751–770.
104. Stavitsky Y, Shandling AH, Ellestadt MH, et al. Hyperbaric oxygen and thrombolysis in myocardial infarction: the “HOT MI” randomized mulitcenter study. Cardiology. 1998;90:131–136. doi:10.1159/000006832
105. Dekleva M, Neskovic A, Vlahovic A, Putnikovic B, Beleslin B, Ostojic M. Adjunctive effect of hyperbaric oxygen treatment after thrombolysis on left ventricular function in patients with acute myocardial infarction. Am Heart J. 2004;148:E14. doi:10.1016/j.ahj.2004.05.032
106. Sharifi M, Fares W, Abdel-Karim I, Koch JM, Sopko J, Adler D. Usefulness of hyperbaric oxygen therapy to inhibit restenosis after percutaneous coronary intervention for acute myocardial infarction or unstable angina pectoris. Am J Cardiol. 2004;93:1533–1535. doi:10.1016/j.amjcard.2004.03.009
107. Bennett MH, Lehm JP, Jepson N. Hyperbaric oxygen therapy for acute coronary syndrome. Cochrane Database Syst Rev. 2015;(7): CD004818.
108. Ambiru S, Furuyama N, Aono M, Otsuka H, Suzuki T, Miyazaki M. Analysis of risk factors associated with complications of hyperbaric oxygen therapy. J Crit Care. 2008;23:295–300. doi:10.1016/j.jcrc.2007.08.002
109. Perrins DJD, Cantab MB. Influence of hyperbaric oxygen on the survival of split skin grafts. Lancet. 1967;II:868–871. doi:10.1016/S0140-6736(67)91428-6
110. Bouachour G, Cronier P, Gouello JP, Toulemonde JL, Talha A, Alquier P. Hyperbaric oxygen therapy in the management of crush injuries: a randomized double-blinded placebo-controlled clinical trial. J Trauma. 1996;41:333–339. doi:10.1097/00005373-199608000-00023
111. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347:1057–1067. doi:10.1056/NEJMoa013121
112. Buras JA, Garcia-Covarrubias L. Ischemia-reperfusion injury: hyperbaric oxygen and basic studies. In: Neuman TS, Thom SR, editors. Physiology and Medicine of Hyperbaric Oxygen Therapy. Philadelphia: Saunders Elsevier; 2008:160–184.
113. Van Meter K, Sheps S, Kriedt F, et al. Hyperbaric oxygen improves rate of return of spontaneous circulation after prolonged normothermic porcine cardiopulmonary arrest. Resuscitation. 2008;78:200–214. doi:10.1016/j.resuscitation.2008.02.026
114. Oter S, Radermacher S, Matejovic M. Can (hyperbaric) oxygen turn off the motor of multiorgan dysfunction? Intensive Care Med. 2006;32:1694–1696.
115. Spears JR, Henney C, Prcevski P, et al.Reperfusion microvascular ischemia attenuated with aqueous oxygen infusion in a porcine coronary occlusion model. Circulation. 1999;102(Suppl. I):I–512. abstract, presented by Spears JR at the 72nd Scientific Sessions of the AHA, November 1999.
116. Spears JR, Prcevski P, Xu R, et al. Aqueous oxygen attenuation of reperfusion microvascular ischemia in a canine model of myocardial infarction. Asaio J. 2003;49:716–720. doi:10.1097/01.mat.0000094665.72503.3c
117. Spears JR, Henney C, Prcevski P, et al. Aqueous oxygen hyperbaric reperfusion in a porcine model of myocardial infarction. J Invas Cardiol. 2002;14:160–166.
118. Johnson LL, Schofield L, Bouchard M, et al. Hyperbaric oxygen solution infused into the anterior interventricular vein at reperfusion reduces infarct size in swine. Am J Physiol Heart Circ Physiol. 2004;287:H2234–H2240. doi:10.1152/ajpheart.00333.2003
119. Glazier JJ. Attenuation of reperfusion microvascular ischemia by aqueous oxygen: experimental and clinical observations. Am Heart J. 2005;149:580–584. doi:10.1016/j.ahj.2004.12.009
120. Bartorelli AL. Hyperoxemic perfusion for treatment of reperfusion microvascular ischemia in patients with myocardial infarction. Am J Cardiovasc Drugs. 2003;3:253–263. doi:10.2165/00129784-200303040-00004
121. Spears JR, Prcevski P, Jiang A, Brereton GJ, Vander Heide R. Intracoronary aqueous oxygen perfusion, performed 24 hrs after the onset of postinfarction reperfusion, experimentally reduces infarct size and improves left ventricular function. Int J Cardiol. 2006;113:371–5.
122. Dixon SR, Bartorelli AL, Marcovitz PA, et al. Initial experience with hyperoxemic reperfusion after primary angioplasty for acute myocardial infarction: results of a pilot study utilizing intracoronary aqueous oxygen therapy. J Am Coll Cardiol. 2002;39:387–392. doi:10.1016/s0735-1097(01)01771-5
123. Trabbatoni D, Bartorelli AL, Fabbiocchi F, et al. Hyperoxemic perfusion of the left anterior descending coronary artery after primary angioplasty in anterior ST-elevation myocardial infarction. Catheter Cardiovasc Interv. 2006;67:859–865. doi:10.1002/ccd.20704
124. O’Neill WW, Martin JL, Dixon SR, et al.; AMIHOT Investigators. Acute myocardial infarction with hyperoxemic therapy (AMIHOT): a prospective, randomized trial of intracoronary hyperoxemic reperfusion after percutaneous coronary intervention. J Am Coll Cardiol. 2007;50:397–405. doi:10.1016/j.jacc.2007.01.099
125. Stone GW, Martin JL, de Boer M-J, et al., for the AMIHOT-II Trial Investigators. Effect of supersaturated oxygen delivery on infarct size after percutaneous coronary intervention in acute myocardial infarction. Circulation. 2009;2:366–375.
126. David SW, Khan ZA, Patel NC, et al. Evaluation of intracoronary hyperoxemic oxygen therapy in acute anterior myocardial infarction: the IC-HOT study. Catheter Cardiovasc Interv. 2019;93(5):882–890.
127. Stone GW. Symposium Chairman, SuperSaturated Oxygen in STEMI to Reduce Infarct Size: A New FDA-Approved Therapy. Oral presentation: SSO2 Therapy: Clinical Evidence and Outcomes. TCT Conference, San Francisco, CA, September 26, 2019
128. Ali MH, Schlidt SA, Chandel NS, Hynes KL, Schumacker PT, Gewertz BL. Endothelial permeability and IL-9 production during hypoxia: role of ROS in signal transduction. Am J Physiol. 1999;277:L1057–L1065. doi:10.1152/ajplung.1999.277.5.L1057
129. Schmitz K, Jennewein M, Pohlemann T, Seekamp A, Oberringer M. Reoxygenation attenuates the adhesion of neutrophils to microvascular endothelial cells. Angiology. 2011;62:155–162. doi:10.1177/0003319710375943
130. Hinshaw DB, Armstrong BC, Beals TF, Hyslop PA. A cellular model of endothelial cell ischemia. J Surg Res. 1988;44:527–537. doi:10.1016/0022-4804(88)90158-8
131. Chen C-A, Wang T-Y, Varadharaj S, et al. S-gluthionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi:10.1038/nature09599
132. Fish JE, Yan MS, Matouk CC, et al. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010;285:810–826. doi:10.1074/jbc.M109.067868
133. Huang B, Chen SC, Wang DL. Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc Res. 2009;83:536–546. doi:10.1093/cvr/cvp154
134. Stamenkovic A, Pierce GN, Ravandi A. Review: Many avenues to cardiac cell death. Phospholipid oxidation products in ferroptotic myocardial cell death. Am J Physiol Heart Circ Physiol. 2019;317:H156–H163. doi:10.1152/ajpheart.00076.2019
135. Tang D, Kang R, Vanden Berghe T, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Research. 2019;29:347–364. doi:10.1038/s41422-019-0164-5
136. Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res. 2008;102:1082–1090. doi:10.1161/CIRCRESAHA.107.167072
137. Scarabelli T, Stephanou A, Rayment N, et al. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation. 2001;104:253. doi:10.1161/01.CIR.104.3.253
138. Pozzi S, Malferrari G, Biunno I, Samaja M. Low-flow ischemia and hypoxia stimulate apoptosis in perfused hearts independently of reperfusion. Cell Physiol Biochem. 2002;12:39–46. doi:10.1159/000047825
139. Bragadeesh T, Jayaweera AR, Pascotto M, et al. Post-ischemic myocardial dysfunction (stunning) results from myofibrillar oedema. Heart. 2008;94:166–171. doi:10.1136/hrt.2006.102434
140. O’Toole EA, van Koningsveld R, Chen M, Woodley DT. Hypoxia induces epidermal keratinocyte matrix metalloproteinase-9 secretion via the protein kinase C pathway. J Cell Physiol. 2007;214:47–55. doi:10.1002/jcp.21160
141. Norman JT, Clark IM, Garcia PL. Regulation of TIMP-1 expression by hypoxia in kidney fibroblasts. Ann NY Acad Sci. 2006;878:503–505. doi:10.1111/j.1749-6632.1999.tb07709.x
142. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi:10.1152/physrev.00012.2007
143. Liu W, Sood R, Chen Q, et al. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107:1196–1205. doi:10.1111/j.1471-4159.2008.05664.x
144. Behn C, Araneda OF, Llanos AJ, Celedon G, Gonzalez G. Hypoxia-related lipid peroxidation: evidences, implications and approaches. Respir Physiol Neurobiol. 2007;158:143–150. doi:10.1016/j.resp.2007.06.001
145. Lesnefsky EJ, Chen Q, Slabe TJ, et al. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Amer J Physiol Heart Circ Physiol. 2004;287:H258–H267. doi:10.1152/ajpheart.00348.2003
146. Trush MA, Egner PA, Kensler TW. Myeloperoxidase as a biomarker of skin irritation and inflammation. Food Chem Toxicol. 1994;32:143–147. doi:10.1016/0278-6915(94)90175-9
147. McCord JM. Oxygen-derived radicals: a link between reperfusion and inflammation. Fed Proc. 1987;46:2402–2406.
148. Angelos MG, Kutala VK, Torres CA, et al. Hypoxic reperfusion of the ischemic heart and oxygen radical generation. Am J Physiol Heart Circ Physiol. 2006;290:H341–H347. doi:10.1152/ajpheart.00223.2005
149. Stoner JD, Clanton TL, Aune SE, Angelos MG. O2 delivery and redox state are determinants of compartment-specific reactive O2 species in myocardial reperfusion. Am J Physiol Heart Circ Physiol. 2007;292:H109–H116. doi:10.1152/ajpheart.00925.2006
150. Thom SR. Oxidative stress is fundamental to hyperbaric oxygen therapy. J Appl Physiol. 2009;106:988–995. doi:10.1152/japplphysiol.91004.2008
151. Babchin A, Levich E, Melamed Y, Sivashinsky G. Osmotic phenomena in application for hyperbaric oxygen treatment. Colloids Surf B Biointerfaces. 2011;83:128–132. doi:10.1016/j.colsurfb.2010.11.019
152. Ziegler M, Wang X, Peter K. Platelets in cardiac ischaemia/reperfusion injury: a promising therapeutic target. Cardiovasc Res. 2019;115(7):1178–1188. doi:10.1093/cvr/cvz070
153. Hausenloy DJ, Botker HE, Engstrom T, et al. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J. 2017;38(13):935–941. doi:10.1093/eurheartj/ehw145
154. Davidson SM, Ferdinandy P, Andreadou I, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2019;73(1):89–99. doi:10.1016/j.jacc.2018.09.086
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.