Back to Journals » International Journal of Nanomedicine » Volume 19
Repaglinide–Solid Lipid Nanoparticles in Chitosan Patches for Transdermal Application: Box–Behnken Design, Characterization, and In Vivo Evaluation
Authors Ali HSM ![]() , Namazi N
, Namazi N ![]() , Elbadawy HM
, Elbadawy HM ![]() , El-Sayed AAA, Ahmed SA, Bafail R, Almikhlafi MA, Alahmadi YM
, El-Sayed AAA, Ahmed SA, Bafail R, Almikhlafi MA, Alahmadi YM ![]()
Received 3 October 2023
Accepted for publication 12 December 2023
Published 10 January 2024 Volume 2024:19 Pages 209—230
DOI https://doi.org/10.2147/IJN.S438564
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Eng San Thian
Hany SM Ali,1,2 Nader Namazi,1 Hossein M Elbadawy,3 Abdelaziz AA El-Sayed,4,5 Sameh A Ahmed,6,7 Rawan Bafail,1 Mohannad A Almikhlafi,3 Yaser M Alahmadi8
1Department of Pharmaceutics and Pharmaceutical Industries, College of Pharmacy, Taibah University, Madinah, Al-Madinah Al-Munawwarah, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut, Egypt; 3Department of Pharmacology and Toxicology, College of Pharmacy, Taibah University, Madinah, Al-Madinah Al-Munawarah, Saudi Arabia; 4Biology Department, Faculty of Science, Islamic University of Madinah, Madinah, Al-Madinah Al-Munawarah, Saudi Arabia; 5Zoology Department, Faculty of Science, Zagazig University, Zagazig, Al-Sharqiya, Egypt; 6Department of Pharmacognosy and Pharmaceutical Chemistry, College of Pharmacy, Taibah University, Madinah, Al-Madinah Al-Munawarah, Saudi Arabia; 7Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt; 8Department of Pharmacy Practice, College of Pharmacy, Taibah University, Madinah, Al-Madinah Al-Munawarah, 30001 Saudi Arabia
Correspondence: Hany SM Ali, Tel +966 50 286 4018, Fax +966 4 847 5027, Email [email protected]; [email protected]
Background: Repaglinide (REP) is an antidiabetic drug with limited oral bioavailability attributable to its low solubility and considerable first-pass hepatic breakdown. This study aimed to develop a biodegradable chitosan-based system loaded with REP–solid lipid nanoparticles (REP-SLNs) for controlled release and bioavailability enhancement via transdermal delivery.
Methods: REP-SLNs were fabricated by ultrasonic hot-melt emulsification. A Box–Behnken design (BBD) was employed to explore and optimize the impacts of processing variables (lipid content, surfactant concentration, and sonication amplitude) on particle size (PS), and entrapment efficiency (EE). The optimized REP-SLN formulation was then incorporated within a chitosan solution to develop a transdermal delivery system (REP-SLN-TDDS) and evaluated for physicochemical properties, drug release, and ex vivo permeation profiles. Pharmacokinetic and pharmacodynamic characteristics were assessed using experimental rats.
Results: The optimized REP-SLNs had a PS of 249± 9.8 nm and EE of 78%± 2.3%. The developed REP-SLN-TDDS demonstrated acceptable characteristics without significant aggregation of REP-SLNs throughout the casting and drying processes. The REP-SLN-TDDS exhibited a biphasic release pattern, where around 36% of the drug load was released during the first 2 h, then the drug release was sustained at around 80% at 24 h. The computed flux across rat skin for the REP-SLN-TDDS was 2.481± 0.22 μg/cm2/h in comparison to 0.696± 0.07 μg/cm2/h for the unprocessed REP, with an enhancement ratio of 3.56. The REP-SLN-TDDS was capable of sustaining greater REP plasma levels over a 24 h period (p< 0.05). The REP-SLN-TDDS also reduced blood glucose levels compared to unprocessed REP and commercial tablets (p< 0.05) in experimental rats.
Conclusion: Our REP-SLN-TDDS can be considered an efficient therapeutic option for REP administration.
Keywords: repaglinide, chitosan, solid-lipid nanoparticles, Box–Behnken design, transdermal, bioavailability
Introduction
For decades, lipid-based systems have been utilized to enhance qualities of a wide range of medications.1,2 In particular, lipid-based nanoparticles have shown remarkable benefits in pharmaceutical formulations, including excellent biocompatibility, simplicity of fabrication, tissue specificity, avoidance of reticuloendothelial systems, and nontoxicity.3 Among lipid nanocarriers, solid-lipid nanoparticles (SLNs) have attracted much attention from academia and industry recently due to their fascinating properties. SLNs are colloidal carriers (50–1000 nm) constructed of physiologically tolerable solid lipids and distributed in an aqueous surfactant solution.4 Due to their safety profile, SLNs are recommended as adaptable nanocarriers, an alternative to polymeric nanoparticles, for delivering lipophilic and hydrophilic medicaments. Advantages of SLNs include increased drug-loading capability, improved stability with minimal drug leakage, controlled-release applications, and adaptability for industrial manufacturing.5 SLNs are also effective in penetrating physiological barriers due to their lipophilic nature and small particles.6 Other advantages include enhanced solubility and bioavailability of weakly water-soluble actives and better stability and protection of degraded actives.
Transdermal medication is a method of administering pharmaceutically active substances to healthy skin for systemic treatments. Transdermal drug delivery systems (TDDSs) are successful, noninvasive ways to prevent first-pass metabolism, maintaining plasma levels within the therapeutic window for extended periods.7 When oral delivery is impossible or may result in unpredictable bioavailability, a TDDS provides a welcome alternative. Transdermal patches are simple to administer and widely accepted for transporting medications straight into the circulation at a predetermined rate, with the added benefit of allowing for the easy and fast termination of medication delivery by patch removal.8 Achieving these goals would reduce variability in treatment reactions and enhance patient compliance significantly. Nevertheless, the transdermal route remains a challenge for medication administration due to the the skin’s impermeable epithelium. The physicochemical features of the drug and its delivery system, anatomy, physiology, and biochemical skin structure influence drug absorption and penetration.9 Several polymers have been successfully utilized to fabricate TDDSs. Of these polymers, chitosan (a polycationic polysaccharide), has attracted attention due to its advantageous qualities, such as biodegradability, bioadhesion, permeability enhancement, and intriguing physicochemical properties.10,11
Repaglinide (REP), a meglitinide member, is therapeutically utilized for the management of type 2 diabetes.12 REP acts by reducing early-phase insulin release and decreasing postprandial glucose. This is believed to be crucial in lowering long-term cardiovascular problems associated with diabetes.13 REP is a practically insoluble medication with a reported aqueous solubility of 34 µg.mL−1 at 37°C.14 The bioavailability of REP (~50%) is noticeably variable.15 There is a substantial interindividual variability in plasma concentrations, owing to limited and unpredictable REP solubility in GI contents, as well as a well-documented hepatic first-pass effect.16 REP also has a high affinity for the permeability glycoproteinpresent in intestinal absorptive cells.17 REP is a short-acting antidiabetic with a short half-life of 1 h.18 Treatment with REP necessitates increased frequency of dosing of immediate-release formulations before meals to maintain effective levels of the drug in the blood, and this may cause such side effects as headaches, discomfort in skeletal muscles, and gastrointestinal effects.19 Other drawbacks, particularly with the long-term use of the drug, include its impact on patient compliance and health outcomes. Accordingly, the development of novel formulations that can extend the effect of REP and improve patient compliance is extremely desirable.20,21 To achieve this target, several approaches have been reported, including matrix pellets,22 an osmotic pump system,23 niosomal encapsulation,15 biodegradable microspheres,24 and transdermal microemulsion gels.25
Box–Behnken design (BBD) is a very efficient response surface design offering information on the influence of testing variables and total experimental error with the fewest number of necessary repetitions.26 BBD efficiency outperforms Doehlert design, central composite design, and the three levels of full factorial design.27 BBD also necessitates fewer experimental efforts than three-level complete factorial design and central composite design, making it more cost-effective.28 BBD has been used effectively in pharmaceutical formulations.29,30 In nanopharmaceutical lipid formulations, most BBD-based SLN-optimization studies have focused on oral delivery.31–33 Based on our literature survey, no study has been conducted on optimizing REP-loaded sono-processed SLNs for transdermal applications using BBD with a comprehensive assessment of its performance. Here, we investigated the efficacy of presenting REP as SLNs in biodegradable chitosan-based patches on its bioavailability. To accomplish that, REP-loaded SLNs were fabricated from Compritol 888 ATO and stabilized by a covering of poloxamer 188 surfactant. A combined procedure of hot homogenization and ultrasonication was employed for nanoparticle production. Chitosan-based transdermal systems (patches) were fabricated using REP-SLNs and subjected to in vitro, pharmacokinetic, and pharmacological evaluation.
Methods
Materials
REP was obtained from Henan Coreychem (Henan, China). Glyceryl dibehenate (Compritol 888 ATO) was gifted by Gattefossē SAS (Saint-Priest Cedex, France). Poloxamer 188 was purchased from Spectrum Chemicals (New Brunswick, NJ, USA). Chitosan (medium molecular weight), cetirizine (CTZ), and stearic acid were obtained from Sigma-Aldrich (Darmstadt, Germany). Acetic acid was obtained from Fisher Scientific (Tokyo, Japan). Polyoxyethylene (20) sorbitan monooleate (Tween 80), polyoxyethylene sorbitan monolaurate (Tween 20), macrogolglycerol hydroxystearate (Cremophor RH 40), and castor oil polyoxyethylene ether (Cremophor EL) were obtained from Sigma-Aldrich Chemie (Steinheim, Germany). Sorbitan oleate (Span 80) was purchased from Loba Chemie (Mumbai, India). Streptozotocin (STZ) was purchased from Merck Millipore (Billerica, MA, USA). REP tablets (Novonorm) were produced by Novo Nordisk, Denmark. Ultrapure water was provided using the Millipore water filtration system (Bedford, MA, USA). Other materials were of pharmaceutical grade and utilized as received.
Assay of Repaglinide
REP was assayed according to a validated HPLC method.34 Procedures involved the application of a Hypersil C18 column for drug quantification. The mobile phase consisted of ethanol–acetonitrile–phosphate buffer (pH 3.4; 25.6 mM) prepared at 2:30:68 v/v/v and delivered at a flow rate of 1.5 mL. min−1. The UV detector was set to 240 nm.
Drug Lipid Solubility
REP solubility in solid lipids was initially assessed applying a reported method.35,36 Briefly, in a water bath (Isotemp 110; Fisher Scientific, Bloomington, MN, USA), a particular amount of the hard lipid was put into glass vials and melted at a temperature exceeding its melting point by 5°C. To the melted solutions of lipids, 10 mg REP was added to the vials and instantaneously vortexed. Then, increments of the lipids were added until complete solubilization of REP and the formation of transparent homogeneous mixtures.
Surfactant Screening
A specified quantity of the molten solid lipid (4% w/w), combined with an aqueous surfactant solution (3% w/w) at 70°C in a 10 mL container was utilized. The mixture was vortexed for 5 minutes, then subjected to 2 minutes of sonication. Once the mixture was quickly chilled in an ice bath, the dispersion it produced was measured for particle size (PS) and polydispersity index (PDI) value. Precipitation was visually inspected under suitable lighting after preparation and overnight storage at room temperature.6 Findings were checked and contrasted.
Preparation of SLNs
REP-SLN batches were prepared based on hot emulsification–ultrasonication (or acoustic cavitation–assisted hot emulsification) procedures. A calculated amount of REP was added to the molten lipid at 80°C to get a transparent phase. An aqueous solution of the surfactant was heated similarly to the selected temperature of the lipid phase. The surfactant solution was introduced to the lipid phase and homogenized for 5 minutes at 8000 rpm at the same temperature using an Ultra Turrax T 25 basic homogenizer (IKA, Staufen, Germany). The resultant hot o/w emulsion was then sonicated by a probe sonicator (Sonics and Materials, Newtown, CT, USA) for 5 min at different amplitude levels (20%–40%). The mixture was then put into an ice-water bath for 15 minutes to accelerate lipid crystallization and synthesis of SLNs.
Box–Behnken Design
BBD (three-level, three-factor) was adopted in the planned study to derive the optimal conditions for REP-SLN preparation. The trial experiments (15 batches, including three center points) were based on high, medium, and low levels of CMP, PLX, and sonication amplitude (Tables 1 and 2). These factors were categorized as independent variables. The dependent responses were the size of the generated SLNs and entrapment efficiency (EE). The gathered response values were fitted to linear, linear two-factor interaction (2FI), and quadratic models to obtain various polynomial equations. The model with the highest determination coefficients and significance value at the specified probability level was recommended by the Design-Expert 13 software (Stat–Ease, Minneapolis, MN, USA). All statistical studies were performed using ANOVA, lack-of-fit, and multiple correlation coefficient (R2) tests to determine the significance of the fitted equations. Optimization was achieved with the criteria of smallest size and highest EE. The process variables and their coded experimental values are listed in Tables 1 and 2. For further investigations, 3D surface and contour plots were generated.
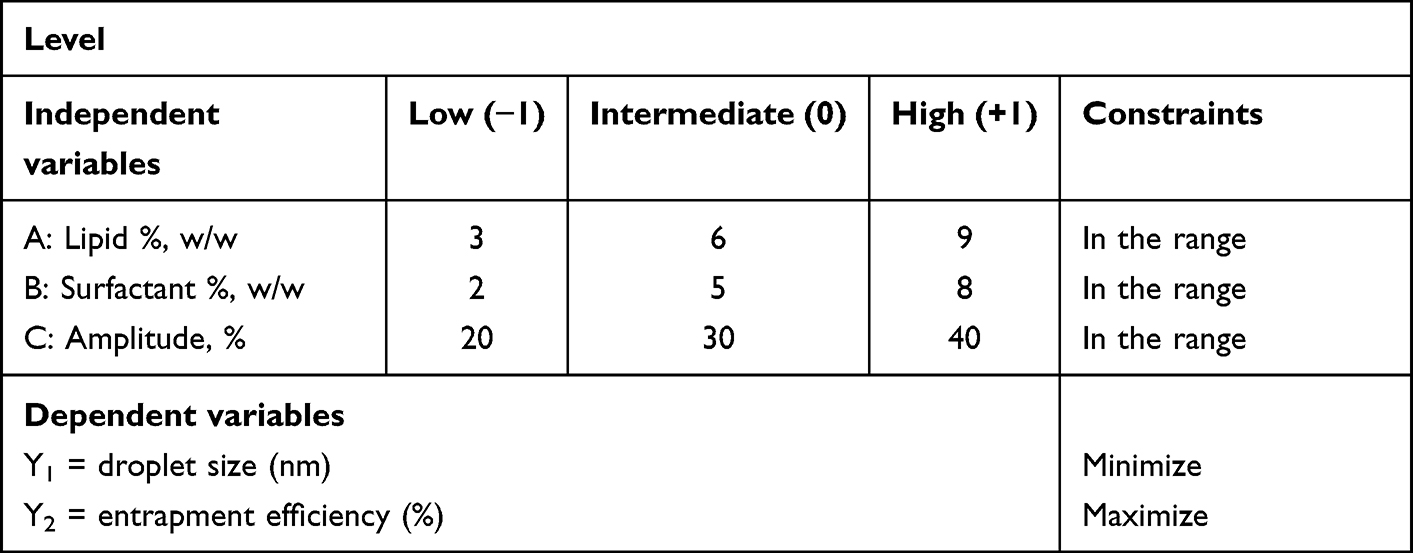 |
Table 1 Variables and levels of the Box–Behnken design |
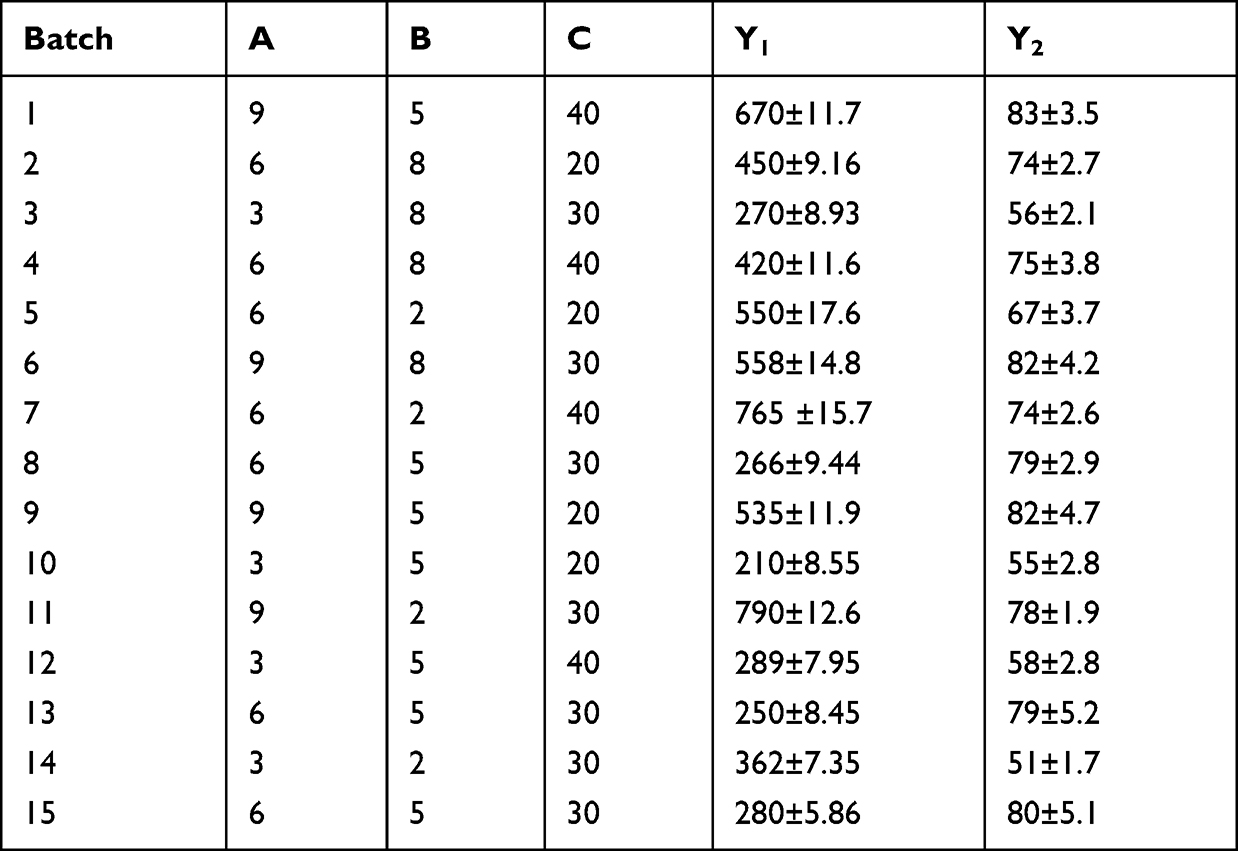 |
Table 2 Randomized experiments of the Box–Behnken design |
Size Characterization
PS and PDI values of the prepared REP-SLNs were evaluated by dynamic light scattering (DLS) using a Microtrac S3500 (Microtrac, Montgomeryville, PA, USA) following suitable dilutions with double-distilled water. Size was measured three times for 120 seconds at 25°C. Evaluation of zeta potential (ZP) was also performed using the same equipment (Microtrac S3500) after proper dilution (100-fold) with de-mineralized water. Transmission electron microscopy (TEM) was employed for further assessment of size and morphologic characterization. Drops from an appropriately diluted REP-SLN representative sample (batch 10) were placed on a copper grid with application of aqueous uranyl acetate solution (2%) for 2 min. The sample was then dried for 5 minutes at ambient temperature to leave a thin film on the grid. Scanning was performed using TEM (JEOL JEM-HR-2100, JEOL, Ltd., Tokyo, Japan).
Entrapment Efficiency
REP-SLN dispersions were centrifuged (Centurion Scientific K3 series, West Sussex, UK) for 30 minutes at 25,000 rpm37 to separate the unentrapped REP fraction. The supernatant was then filtered through a nylon syringe filter 0.45 µm and properly diluted with distilled water/methanol. Supernatants were assayed for REP content using a validated HPLC procedure, as described in section 2.2. The EE in the SLNs was estimated using equation (1):
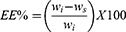
where Wi is the initial weight of REP and Ws is the quantified REP quantity in the supernatant.
Validation of BBD Models
After specifying the desired goals of the responses (Table 1), the numerical predictability software was probed to calculate the highest desirability value. A value between 0 and 1 represents the closeness of the response to its goal value, from least to highly suggested.38 The optimized formulation (OPT-REP-SLNs) was prepared based on the constraints given in Table 1 and the calculated desirability then examined for responses. The observed and anticipated values were compared, and the prediction error percentage was computed.
Lyophilization of Repaglinide–Solid Lipid Nanoparticles
Lyophilization was carried out to extend the shelf life of the SLNs and to facilitate additional characterization and assessment.39 After careful separation, OPT-REP-SLNs were redispersed in an appropriate volume of 5% w/v mannitol (as a cryoprotectant), deep-frozen at −20°C for 24 h, and lyophilized at 7×10−2 mbar and –50°C for 48 h (iLShineBioBase freeze-dryer, Gyeonggi-do, South Korea). The lyophilized powder was harvested and kept in the refrigerator till further use.31
Calorimetric Analysis
A Netzsch thermal analyzer (Netzsch F3 Maia, Germany) was used to obtain DSC thermograms of unprocessed REP (raw REP), CMP, PLX, mannitol and REP-SLNs. Samples (~4 mg) were kept in aluminum pans and scanned over a range of 30°C–300°C at a heating rate of 10°C.min−1. DSC analysis was carried out under a dynamic nitrogen atmosphere.
Transdermal Delivery Systems
Transdermal patches containing REP-SLNs were fabricated using solvent casting procedures.11 Chitosan (1.5% w/v) was dissolved in 1.5% glacial acetic acid, then PEG 400 was incorporated as a plasticizer. Calculated amounts of REP-SLNs were added to the solution and stirred with a propeller (IKA Werke, Staufen, Germany) for 10 minutes. The mixture was then placed into petri plates and allowed to dry at 40°C for 48 h (Binder oven, Tuttlingen, Germany). The backing film was made of aluminum foil. The final patches were isolated and stored in closed containers.
Characterization of the the REP-SLN-TDDS
The thickness of the developed TDDS was measured with a digital Vernier caliper (Mitutoyo, Kanagawa, Japan). Thickness was evaluated at three distinct locations, and the mean value and standard deviation were computed.40 To determine drug content, each medicated patch was cut into smaller fragments and dissolved in a phosphate buffer (pH 7.4, containing 1.5% w/v acetic acid). Solutions were then sonicated (Ultrasons-HD, Barcelona, Spain), filtered, properly diluted, and assayed for REP content, as described in section 2.2.41 For folding-endurance assessment, patch strips (2×1 cm) were divided uniformly and folded repeatedly at the same place till the strip broke. The value of folding endurance was determined by how many times the strip could be folded at the same location without breaking or cracking.42 A halogen moisture analyzer (HB43-S Mettler Toledo, Greifensee, Switzerland) was employed for gravimetric determination of moisture content at 105°C.11 The moisture uptake capacity was determined in accordance with a previously reported method.42 Briefly, potassium chloride was added to the desiccator to keep the humidity at 84%. Strips of known weights were kept in the desiccator at room temperature until recording constant weights. The percentage of moisture uptake was estimated by subtracting the final weight from the initial weight with respect to initial weights of the strips.
Dispersibility
The REP-SLN-TDDS was placed into various volumes of deionized water containing 1.5% w/v glacial acetic acid to dissolve the chitosan, with gentle stirring for 10 min. PS was measured as previously explained (section 2.7).
Drug-Release Characterization
A USP basket-type dissolution apparatus was employed to characterize REP-release profiles from the REP-SLN-TDDS in comparison to raw REP and commercial REP tablets.43 Tested formulations were placed in individual baskets and rotated at 50 rpm in 500 mL pH 7.4 phosphate buffer at 32°C±0.5°C. At predetermined intervals, samples of 5 mL were taken, filtered, and assayed for REP content after appropriate dilution (section 2.2). Equivalent volumes from the release medium were used to replace the withdrawn samples. Findings were processed using zero-order, first-order, Higuchi, and Korsmeyer–Peppas equations to characterize release kinetics of REP from SLN-based formulations.44
Skin Permeation
Based on an approval of an institutional ethics committee (COPTU-REC-66-20230503), rat skin samples were utilized to characterize REP patterns from TDDS. Skin samples were prepared according to reported procedures.45 The cleansed skin was then rinsed with distilled water and kept at –21°C until further usage. Skin samples were kept at room temperature for 1 h prior to hydration with pH 7.4 methanolic phosphate-buffered saline (PBS; 20:80% v/v), which was used in the receiver compartment in Franz diffusion cells.46 Skin sections (3.14 cm2) were mounted between the two compartments of the diffusion cells (PermeGear, Hellertown, PA, USA) with the stratum corneum facing the donor compartment and the dermal side of the skin towards the receiver compartment.25 The REP-SLN-TDDS as placed over the skin in such a way that the release surface of the TDDS was kept in intimate contact with the stratum corneum side of the skin and the backing membrane upward. To assure sink conditions, methanol/PBS of pH 7.4 (20:80% v/v) was used in the receiver compartment. The receptor medium was kept warmed (32°C±0.5°C) with continuous magnetic stirring at 200 rpm.47 The tested formulation was the REP-SLN-TDDS (2 mg REP) compared to an equipotent dose of the control formulation of unprocessed REP suspension. The designated formulations were introduced into the donor chamber. At preselected times, samples were collected from the receiver compartment and replenished with equal volumes of fresh samples. After filtration, REP contents of the collected samples were quantified (section 2.2). A graph representing the cumulative amount of REP permeated (µg/cm2) against time was constructed. The slope of the linear part of the curve was utilized to compute the in vitro skin permeation rate or flux — Jss (μg/cm2/h).48 In order to determine the permeability coefficient (Kp), the flux (Jss) was divided by the initial drug concentration in the donor cell (C0), as in Equation 2:
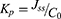
To determine the enhancement ratio (Er), the Jss of the relevant test formulation (the REP-SLN-TDDS) was divided by the Jss of the control REP suspension formulation (Equation 3):
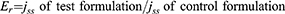
In Vivo Assessment
Induction of Diabetes
All animal experiments were approved by the institutional review board and were implemented in strict accordance with approved guidelines. Ethics approval (COPTU-REC-66-20230503) was obtained before starting the experiments. Adult male albino rats (180–200 g) were obtained from the animal house of the College of Pharmacy at Taibah University. Rats were group-housed in a temperature-controlled room (24°C±1°C) and maintained on a 12-hour light/dark cycle. A standard rodent pelleted diet was supplemented by ad libitum access. For induction of type 2 diabetes mellitus, rats were fasted overnight then injected intraperitoneally with low-dose (20 mg/kg) STZ once per day for 5 days.49 Citrate buffer (0.01 M at PH 4.5) was used to dissolve STZ. Diabetic rats with blood glucose level of 300±20 mg/dL were included in the in vivo experiments.
Pharmacokinetic Profiling
Three groups (n=5) of overnight-fasted rats were treated as follows. Group I received the unprocessed REP at 1 mg/kg and group II received a similar dose of commercial REP tablets (Novonorm, Novo Nordisk, Denmark). For group III, the REP-SLN-TDDS (2 cm2 containing 1 mg REP) was administered to previously shaved dorsal skin areas. At specified times, blood samples were taken from the retro-orbital area using a capillary tube. Rats were anesthetized in desiccators with diethyl ether to reduce discomfort during sampling. Each time, ~0.5 mL blood was collected from each animal in the relevant groups. Blood was collected in precoated EDTA tubes and shaken to eliminate the possibility of clotting during storage. Plasma samples were centrifuged (Biofuge- 13, Heraeus Instruments, Hanau, Germany) and kept in vials at −70° C till being analyzed. Quantification of REP from plasma samples was conducted using a completely validated liquid chromatography–electrospray ionization–tandem mass spectrometric (LC-ESI-MS/MS) method using CTZ as the internal standard.50 An Agilent UPLC system was used consisting of a 1260 Infinity II quaternary pump with an integrated degassing unit and a column thermostat, a 1260 Infinity II autosampler (Agilent), and an NG Castore XS iQ nitrogen generator from LNI Swiss Gas (Versoix, Switzerland). Analysis was carried out using reverse-phase chromatographic separation on a C18 Eclipse Plus RRHD column (50×2.1 mm, 1.8 μm, Agilent, CA, USA). A mobile phase comprising water–methanol–acetonitrile (62.5:20:17.5, v/v/v) and 0.2% formic acid was set to 0.4 mL /min flow rate while the column was maintained at 40°C. For both the standards and the samples, the injection volume was 5 µL. The UPLC system was connected to an Ultivo triple-quadrupole mass spectrometer that operated in positive ESI mode to measure RPG in MRM mode. Mass Hunter quantitative data analysis software was used to process data (Agilent Technologies, Santa Clara, CA, USA). The quantifier MRM transitions were selected based on the most intense fragment ion, and the measurements were made using MRM mode with the transition’s m/z 453.2→230.1 for RPG and 389.1→201.0 for CTZ(IS). The dwell time was set to 25 ms for each MRM transition. The ESI Jet Stream source parameters were adjusted to provide the strongest MRM signals, including gas temperature at 300°C, gas flow at 7 L/min, nebulizer gas at 15 psi, capillary voltage at 4000 V, fragmentor voltage at 70 V and 45 V for RPG and CTZ, respectively, and collision energy (CE) at 27 V and 29 V for RPG and CTZ, respectively. A noncompartmental analysis method was applied to estimate pharmacokinetic parameters from plasma concentration vs time profiles of experimental rats. The maximum REP plasma concentration (Cmax) and the corresponding time (tmax) were read from the graphs. The linear trapezoidal method was applied to assess the area under the curves (AUC0–24·h and AUC0–∞). The elimination rate constant (Kel) was calculated from the slope of the terminal phase of the ln-normal plasma concentration–time data, and then calculating the half-life as t½=0.693/Kel.12
Hypoglycemic Activity
For pharmacodynamic evaluation, rats were randomly allocated into three groups, with five rats in each group. Group I were given the commercial REP tablets in aqueous suspension (1 mg/kg) by oral gavage. Group II were treated with the REP-SLN-TDDS, which was attached to the shaved back, whereas group III were assigned a blank (REP-free) SLN-TDDS. Blood withdrawal from the retro-orbital plexus was performed at the beginning of the experiment (0 hour) and after 1, 3, 6, and 24 h. Blood samples were immediately used for blood glucose determinations (average of three per rat) using a conventional Accu-Check glucometer (Roche Diagnostics, IN, USA).
Statistical Analysis
Results were statistically processed applying Student‘s’ t-test and ANOVA. P<0.05 was considered significant. All values are reported as means ± SD.
Results and Discussion
Selection of SLN Components
Solubility of the drug in the lipid is crucial in SLN preparation. Low drug solubility in the lipid could result in reduced levels of EE and drug loading. Furthermore, binding of the drug molecules to the lipid could be retarded by the little drug lipid and more drug molecules remain unentrapped with failure of sustained-release characteristics.51 The conventional equilibrium solubility approach is not fully appropriate to solid lipids. Accordingly, a modified assessment was applied to study REP solubility.36 Here, the quantity of solid lipid needed to dissolve 10 mg of REP was estimated. CMP and stearic acid were reported as lipids for SLN fabrication. In the current assessment, both CMP and stearic acid showed reasonable solubilization potential towards REP, with respective values of 315±18 and 290±21 mg. CMP was ultimately selected as the lipid core for SLNs because of its popularity in developing SLN formulations and capaity for greater EE. Other features, such as superior flowability, nontoxicity, regulatory approval, and low cost, further favor the use of CMP as a solid lipid.52–54 The glyceride content of CMP permits easier accommodation of the drug within its highly porous structur.55 Regarding surfactant screening, SLN fabrication was explored using a set of commonly used surfactants and examined for PS, ZP, PDI value, and precipitation. As shown in Table 3, the PLX-based SLN preparation possessed the smallest PS (242±11 nm) and acceptable values of ZP and PDI (25.6±0.9 mV and 0.24±0.02, respectively), with no sign of precipitation. The hydrophilic–lipophilic balance (HLB) of the used surfactant is crucial for PS in SLN synthesis, where greater HLB values usually correlate with smaller particles.56 PLX with a high HLB (29), high ratio of hydrophilic blocks and, low molecular weight is effective in sterically stabilizing the generated nanosized lipid particles.6,57,58 These findings supported selection of PLX for further REP-SLN optimization.
 |
Table 3 Results of surfactant screening |
Fitting the Response Surface Models
In comparison with other equivalent designs, the BBD is a straightforward experimental design for analyzing interactive factorial effects and obtaining polynomial equations utilizing fewer experiments. The 15 preparations with three center points and the consequent responses are displayed in Table 1. The data were statistically analyzed to determine the best-fitting model from linear, second-order, cubic, and quadratic models. As confirmed by the software, the quadratic model was found to fit for all responses best (Table 4). ANOVA findings indicated significant model F-values (p<0.0001 for Y1 and Y2), representing appropriate signals with a limited noise impact. Each of the derived polynomial equations shows the influence of each variable on the generated responses, as well as the p-values indicating the significance of the differences caused by each variable. The coefficient of each variable represents the factor’s contribution to the produced responses, and the plus or minus sign denotes its boosting or castrating effect, respectively.59
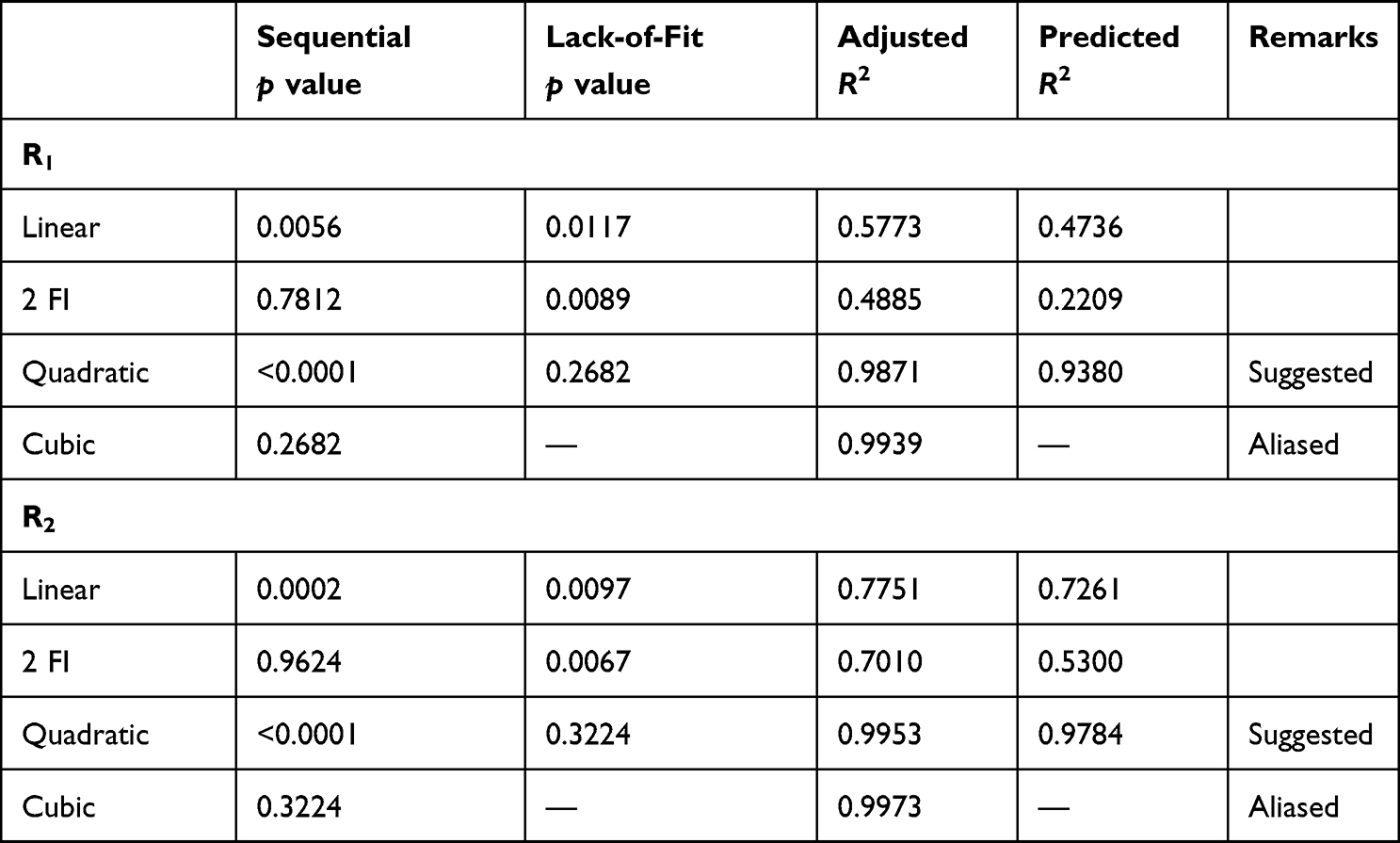 |
Table 4 Regression analysis and ANOVA results of the response surface models |
Impacts on Size of SLNs
PS is important for cellular uptake and ascertaining the physical stability and intestinal lymphatic targeting of nanocarrier systems.60 According to size measurement using DLS, the PS of the developed batches ranged from 210±8.5 to 790±12.6 nm (Table 2, Figure 1). This finding revealed a meaningful influence of the chosen variables on PS. In addition, size characteristics were assessed by TEM scanning. Figure 1B depicts the TEM image of a representative sample (batch 10). The SLNs were found to be more or less spherical with distinct peripheries. The size of the SLNs was found to concur with the size distribution determined by DLS (Figure 1A). In Table 5, the model’s F=132.93 value demonstrates its significance. Values of “Prob > F” <0.05 imply model terms that are significant. The lack-of-fit p=2.52 indicates that the lack of fit is not significant. With the exception of AC, other terms of the model were found significant. The predicted R2 of 0.945 is consistent with the adjusted R2 of 0.989. The ratio of signal to noise demonstrates adequate precision, where a value >4 is desired. The model ratio of 33.08 indicates an adequate signal in this case. The model’s polynomial equation (4) for PS% is shown as:
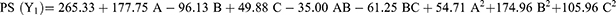
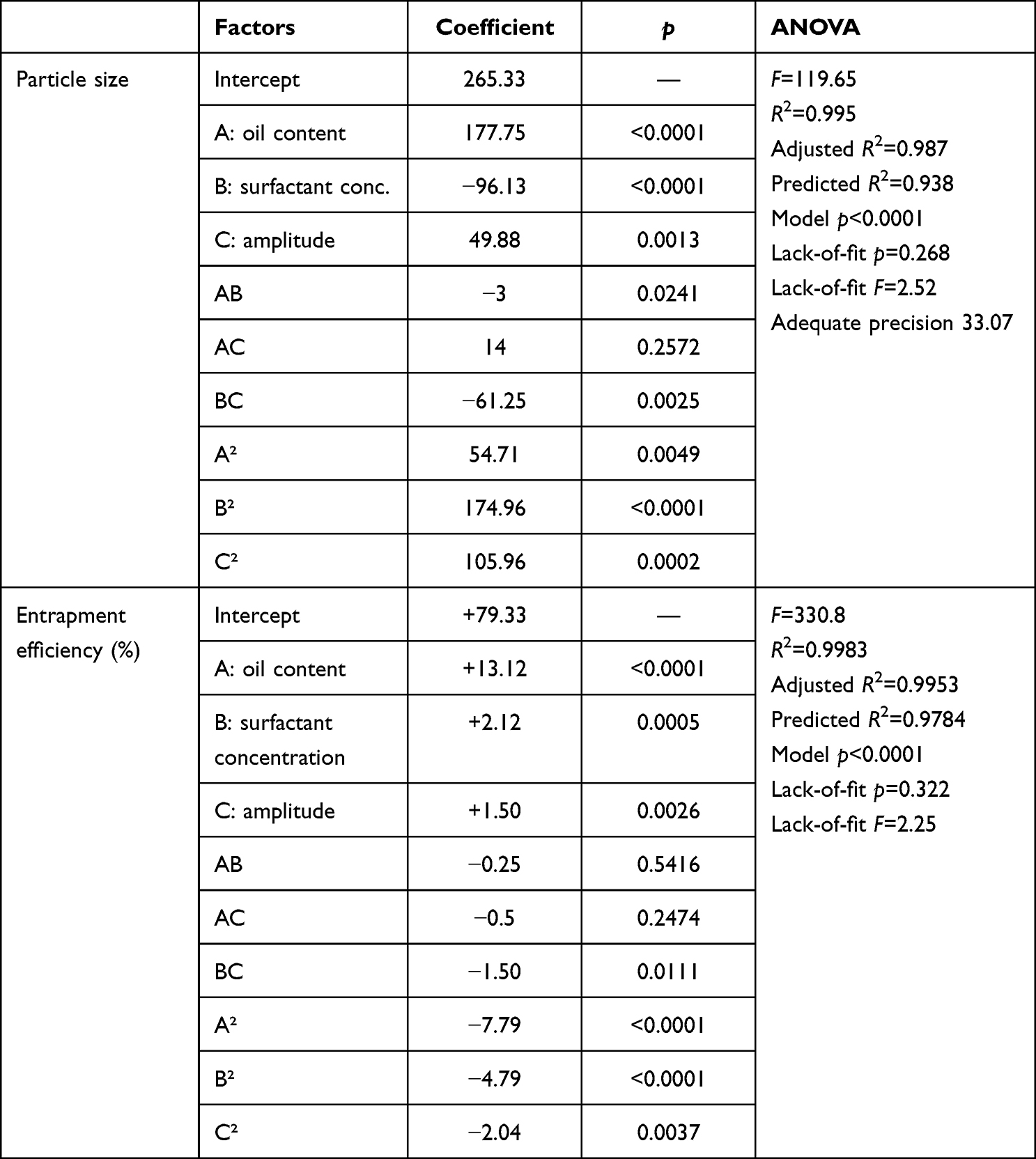 |
Table 5 Statistical analysis of the model generated by Box–Behnken design |
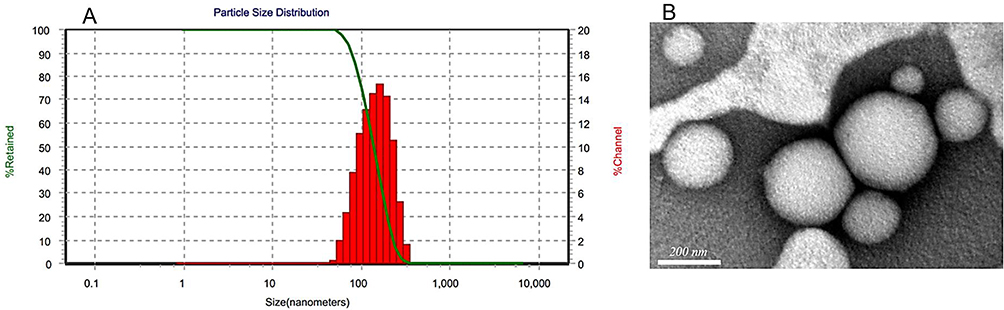 |
Figure 1 Size distribution of representative repaglinide–solid lipid nanoparticles (A) and TEM micrograph (B). |
According to equation 4, it was obvious that PS (Y1) was directly impacted by the lipid content (A) and amplitude level (C), while inversely related to the surfactant concentration (B). When coefficient values of significant model terms of equation 4 are compared, variable A (the lipid content) showed the greatest influence on size of the generated SLNs, whereas the minimum significant impact was shown by the combined variable AB. The influence of lipid content, surfactant concentration, and amplitude level on PS is displayed in their related contour and 3D surface response plots (Figure 1). As implied by Figure 1A and B, with increasing the lipid content of the SLN preparations, a noticeable enlargement in PS was observed. At low lipid levels, sonication energy propagates better in the dilute dispersions than concentrated dispersions, causing more effective diminution of SLNs.35 By increasing the lipid content, the consistency and surface tension of the preparations increase, with consequent enlargement of SLNs.61 It is also clear from Figure 1A and C that PS of SLNs generally decreased with increasing the surfactant concentration. The new surfaces generated during sonication can be stabilized by steric hindrance of surfactant molecules on SLNs. Nonetheless, very high concentrations of surfactant ultimately increase the apparent viscosity of the system, which weakens propagation of the ultrasonic waves and subsequently diminishes the shear stress of the acoustic cavitation. Reduction of the overall threshold and efficiency of cavitation result in formation of larger droplets.62,63 Nevertheless, because of cytotoxicity concerns regarding high stabilizer concentrations, the inclusion of a minimum concentration of stabilizers is recommended in drug-delivery formulations in ultrasound-assisted cavitations within the preparation medium. According to Figures 2b and c, low-to-moderate levels of amplitude proportion were efficient in inducing cavitations that were efficient for particle breakup.64 The excessive energy supply at higher amplitudes creates severe turbulence and encourages collisions between growing particles, coalescence, and size enlargement.62 Overall analysis of the contour plots in Figure 2; the lowest PS of REP-SLNs can be obtained by adjusting lipid content, surfactant concentration, and amplitude level: 3–6, 4%–7%, and 22%–36% respectively.
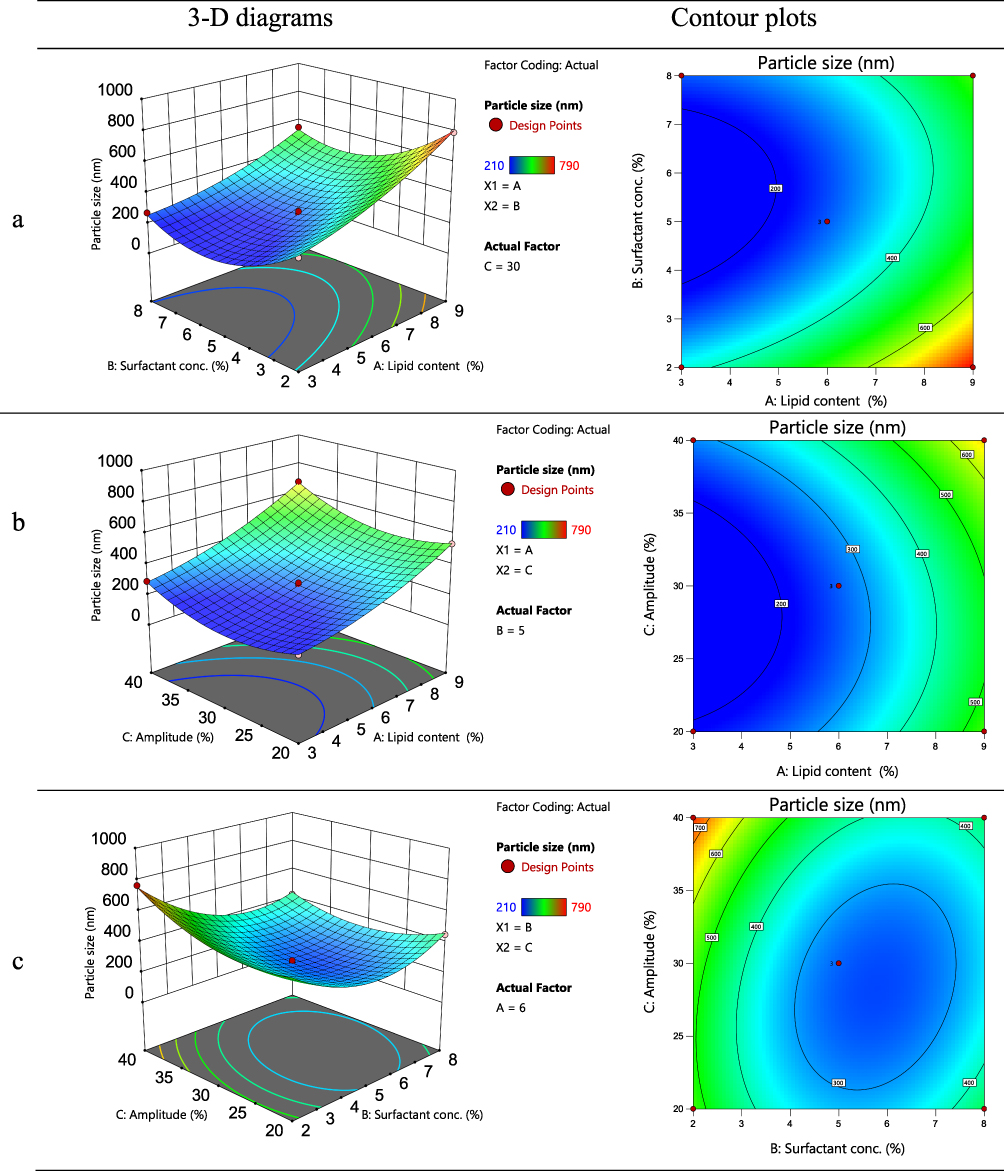 |
Figure 2 Three-dimensional surface diagrams and contour plots showing the impact of lipid content–surfactant concentration (a), lipid content–amplitude, (b) and surfactant concentration–amplitude, (c) on size of solid-lipid nanoparticles. |
Impacts on EE
EE refers to drug amounts that are trapped either within the solid-lipid matrix or adsorbed on the surface of SLNs.65 According to the results, EE of the prepared batches varied between 51%±1.7% and 83%±3.5% (Table 2). The full quadratic polynomial equation for the measured response (EE, Y2) is given in Equation 5:
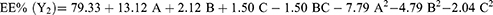
Equation 5 demonstrated a good fit to the response (Y2, EE) as R2 (0.997) and harmoniousness with the adjusted R2 (0.992), with a difference of ≤0.2 (Table 5). The model’s F=330.82 illustrates its relevance, and the lack of fit was not significant (p=2.25). A Precision ratio of 51.915 signifies an adequate signal. Model terms with p<0.05 are significant. Accordingly, A, B, C, BC, A2, B2, and C2 are significant with this assumption. Consistently with the regression equation, it was clear that EE was synergistically affected by the lipid content (A) and amplitude level (C) and inversely related to the surfactant concentration (B). The variable A (the lipid content) similarly exhibited the greatest influence with the highest EE of the generated SLNs. The 3D response surface plots (Figure 2a and b) showed a nonlinear enhancement in EE with increasing lipid content, followed by a slight decline then further increase. The increased availability of the melted CMP, with sufficient solubilization ability toward REP, can provide additional spaces to accommodate additional drug molecules. Moreover, the increased lipid content induces quick solidification of the nanoparticles and retards drug diffusion to the external phase of the medium.66 This impact, however, is suppressed at high lipid concentrations, where a marginal decrease in EE was observed. A plausible explanation is interference of the increased viscosity with the REP solubility in the lipid.6 Similarly, but to a lesser extent, an increase in surfactant concentration resulted in better drug dispersion and a growing EE curve. In addition, with increasing surfactant concentrations, portions of the REP will be incorporated in the surfactant layer at the surface of the SLNs, resulting in a high EE.67 Nonetheless, higher levels of surfactant can limit EE by promoting drug partitioning and facilitating its solubilization in both the lipid and aqueous phases.68 Alternatively, the use of high-energy amplitude during ultrasonication supported transfer of drug molecules from the aqueous phase to the lipid matrix and increased EE, though an excessive rise in sonication amplitude can expel the loosely associated drug molecules to the lipid matrix and lower the EE.69 Generally, contour plots of Figure 3 revealed that the highest degrees of EE were attained in the ranges of 6.5%–8.5% for the lipid content, 3.5%–7.5% for the surfactant concentration, and 25%–40% for the amplitude level.
 |
Figure 3 Three-dimensional surface diagrams and contour plots showing the impact of lipid content–surfactant concentration (a), lipid content–amplitude, (b) and surfactant concentration–amplitude, (c) on entrapment efficiency of solid-lipid nanoparticles. |
Optimization and Point Prediction
To achieve the optimum values of processing variables, the desirability function was explored using Design-Expert software (Figure 4). The lowest PS and maximum EE were the criteria to select the best levels of the variables. Consequently, a fresh batch of SLNs was prepared with the generated variable values to validate the optimization procedures. The optimized variables were 6.44%, 5.80%, and 29.35% for lipid content, surfactant concentration, and amplitude level, respectively. The freshly prepared SLNs showed average response values of 249±9.8 nm for PS and 77.8±2.3 for EE (Table 6). The low values of bias (<15%) signified the adequacy of the generated models’ equations.
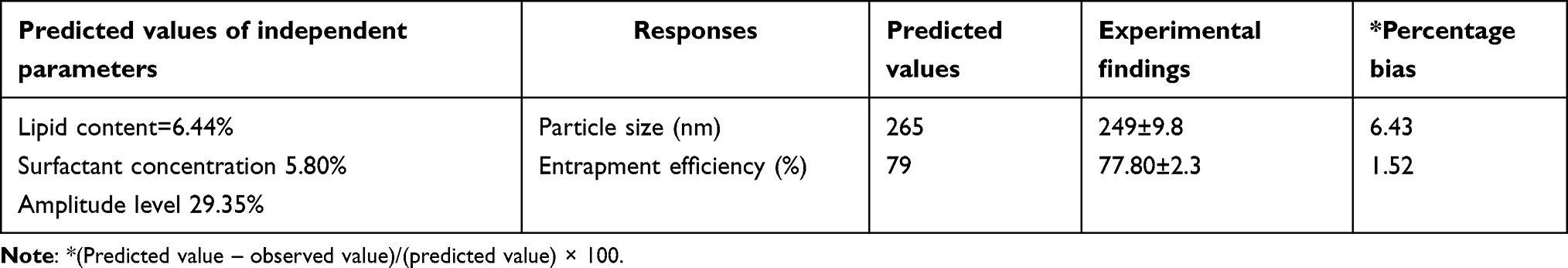 |
Table 6 Results of repaglinide solid-lipid nanoparticle optimization |
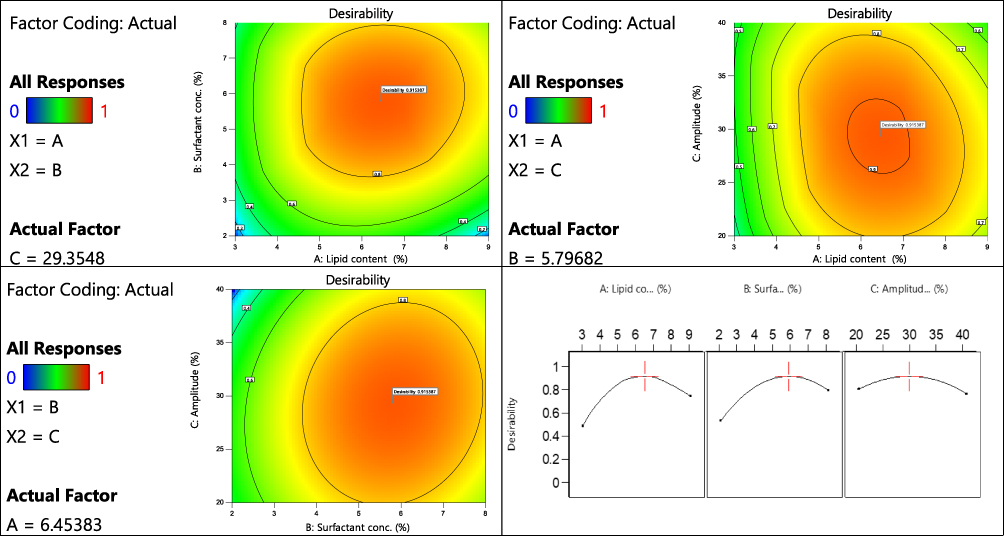 |
Figure 4 Contour plots of desirability for the optimized REP-SLNs. |
Thermal Analysis
Figure 5 displays an overview of the DSC analysis of REP-SLNs along with their raw powder ingredients. The raw REP demonstrated a melting endotherm with an onset at 130°C, a peak at 134°C, and an end at 139°C (Figure 5A). This endothermic peak agreed with the reported melting point of REP at 132.64°C.13 PLX, the surfactant, also demonstrated an obvious melting endotherm at 55°C.70 The DSC thermogram of CMP showed a sharp peak at its melting point (74°C), confirming the crystalline nature of this lipid.71 In addition, mannitol (the cryoprotectant) displayed melting at 169°C.72 The prominent peaks of PLX, CMP, and mannitol were also identified in the thermogram of REP-SLNs (Figure 5E). However, the thermogram of REP–SLNs showed a complete disappearance of the characteristic REP endothermic peak. Drug entrapment within the matrix of SLNs converts it to a molecular dispersion or an amorphous form, with a consequent absence of the melting endotherm feature of crystalline substance.6,72
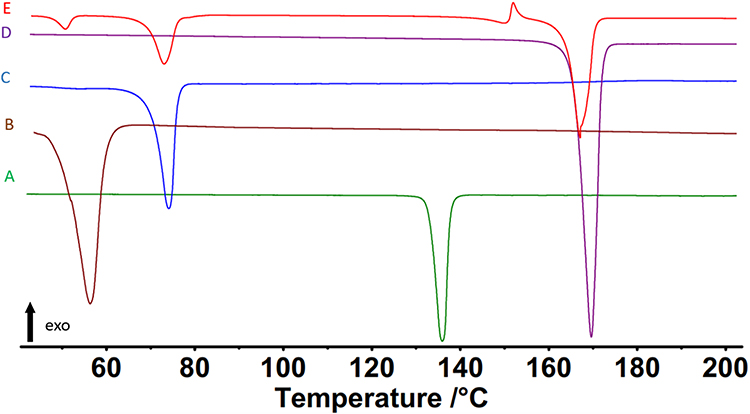 |
Figure 5 DSC thermograms of repaglinide (A), poloxamer 188 (B), Compritol 888 (C), mannitol (D), and repaglinide-loaded solid-lipid nanoparticles (E). |
TDDS Formulation and Characterization
In the current study, chitosan was selected to fabricate the TDDS because of its biocompatibility, biodegradability, good film-forming ability, and bioadhesion- and absorption-enhancing characteristics.73,74 Results of characterization of the REP-SLN-TDDS are shown in Table 7. In general, the manufactured the REP-SLN-TDDS was thin, aesthetically acceptable, and less transparent compared to the plain chitosan TDDS (Figure 6). Thickness of the generated patches was 0.12±0.04, with homogeneity confirmed by the low values of standard deviation. The REP contents estimated at different parts of the patch were 95±0.37. Uniformity of the REP load implies appropriateness of chitosan rheological and mechanical characteristics to permit homogeneous distribution of the drug throughout processing steps.48 The folding endurance is used to evaluate the mechanical strength and flexibility of the TDDS that is needed for handling. A high value of folding endurance (>310) guarantees adequate strength, elasticity, and integrity with general skin folding following administration.75 The moisture content and moisture uptake in the patches were 2.4%±0.63% and 2.9%±0.33%, respectively. Low moisture content makes patches stable, flexible, and free of brittleness. Likewise, low moisture-absorption values are advantageous for inhibiting microbiological development and reducing formulation bulkiness (Mutalik and Udupa 2004).
 |
Table 7 Physical characteristics of repaglinide solid-nanoparticle transdermal system (REP-SLN-TDDS) |
 |
Figure 6 A chitosan transdermal system loaded with REP-SLNs (A) vs a plain system (B). |
Dispersibility
Drug–nanoparticle recovery from dried formulations is a key challenge, whether in vitro or after in vivo delivery (Bhakay et al, 2013). Figure 7 depicts the PS distribution following redispersing the TDDS gently in deionized water containing 1.5%, w/v glacial acetic acid. The d50 after redispersion (258 nm) was similar to the corresponding value of the initially prepared SLNs (249 nm). This implies that there was no significant aggregation of SLNs throughout the drying procedures.
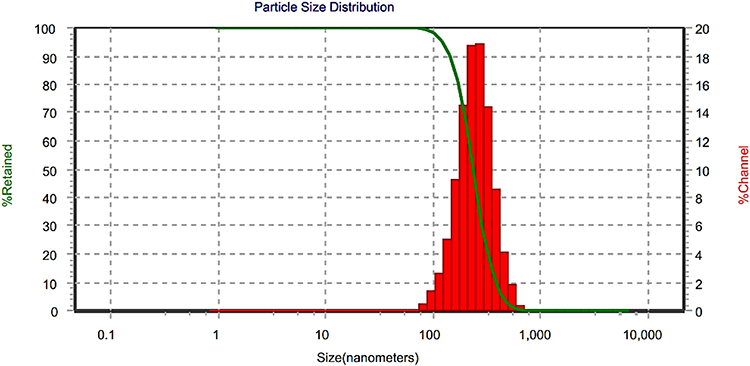 |
Figure 7 Size distribution of redispersed repaglinide solid-lipid nanoparticles from chitosan patches. |
Drug Release
Release profiles of unprocessed REP, commercial tablets, and the REP-SLN-TDDS in PBS pH 7.4 are graphically represented in Figure 8. Unprocessed REP exhibited a slow-release profile, with about 66%±5% released at 24 h. The commercial tablets showed a rapid-release pattern, with almost full release after 1 h. However, REP-SLNs demonstrated a biphasic-release pattern with an initial burst release of 45.6±3.4% of REP detected in the first 2 h followed by a sustained drug release of about (~88%±3.1%) at the 24 h time point. Desorption from the outer surface of SLNs is mainly the cause of the initial burst phase of drug release.76 The burst effect was further augmented by the large effective surface area of the nanoparticles. Subsequently, the embedded REP within the solid-lipid crystals provided a slow and prolonged release pattern. However, after incorporation within the chitosan matrix, the the REP-SLN-TDDS formulation showed a slower release pattern than REP-SLNs with a significant reduction in burst release to a value of 36.2±3.4 (p<0.05) caused by the surrounding chitosan polymer matrix.
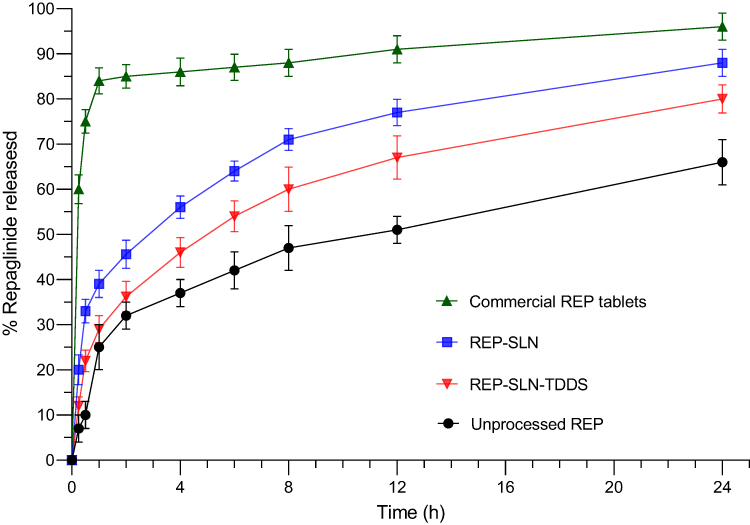 |
Figure 8 Release of repaglinide from tested formulations. Abbreviations: REP, repaglinide; SLN, solid-lipid nanoparticle; TDDS, transdermal delivery system. |
The release profiles of REP from SLN-based preparations were modeled by the equations of the zero-order, first-order, Higuchi kinetics and Korsmeyer–Peppas models (Table 8). Both REP-SLNs and the REP-SLN-TDDS were best fitted using the Korsmeyer–Peppas model, as reflected by the greater values of the regression coefficient compared to other models (r2=0.9836 and 0.9829, respectively). The release exponent (n) value was utilized to characterize the various release processes. The n values were <0.45 for both of REP-SLNs and the REP-SLN-TDDS, implying a Fickian diffusion-controlled drug-release pattern.77
 |
Table 8 Modeling of repaglinide-release kinetics from preparations of solid-lipid nanoparticles (REP-SLNs) and transdermal system (REP-SLN-TDDS) |
Ex Vivo Permeation Study
Skin-permeation patterns and the permeability metrics of the REP-SLN-TDDS and unprocessed REP suspension are shown in Figure 9 and Table 9. In this test, REP solubility in PBS pH 7.4 was augmented with methanol 20% (v/v) to ensure sink conditions.25 As seen from Figure 9, the aqueous REP suspension barely permeated the skin within 24 h of the experiment. However, the REP-SLN-TDDS showed a profile of significant permeation enhancement (p<0.01). After 24 h, the cumulative REP permeated from the REP-SLN-TDDS was found to be 233.17±8.80 μg/cm2 with a flux value (Jss) and permeability coefficient (Kp) equal to 2.481±0.22 μg/cm2/h and 1.240±0.112×10−3 cm.h−1, correspondingly. The enhancement ratio (Er) was 3.56 in favor of the REP-SLN-TDDS. The improved permeation features of REP-SLNs are mainly attributed to their ability to interaction with skin layers and modified barrier characteristics. SLNs are capable of creating a homogeneous and uniform layer on the stratum corneum, increasing residence duration and improving skin penetration.78 Being a cationic ligand, chitosan is able to interact with the negatively charged lipid and protein domains of the skin stratum.79 Such interactions could induce skin lipid disorders and modify secondary structures and loosen epithelial cell tight junctions, resulting in bigger pores and enhanced permeability via the intercellular and/or transcellular pathway.80
 |
Table 9 Permeability metrics of unprocessed repaglinide (REP) and transdermal system (REP-SLN-TDDS) |
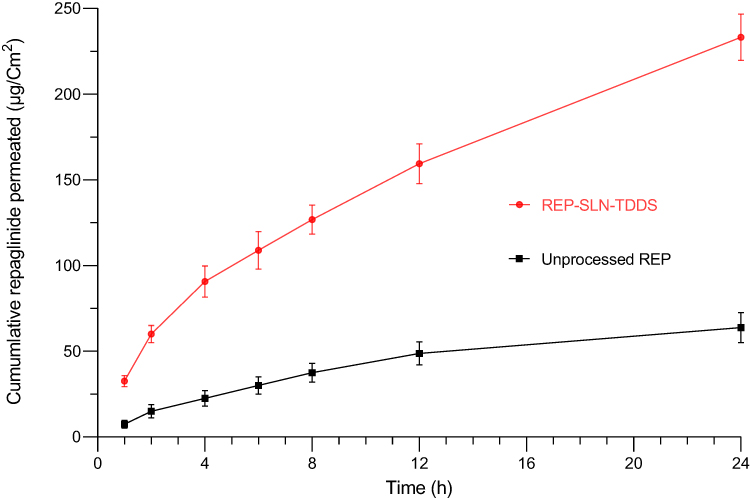 |
Figure 9 Ex vivo permeation of repaglinide from solid-lipid nanoparticle transdermal system (REP-SLN-TDDS) and unprocessed repaglinide (REP). |
In Vivo Assessment
Pharmacokinetic Profiling
Figure 10 portrays the different plasma concentration–time profiles of REP generated after oral delivery of unprocessed REP powder and commercial tablets of REP in comparison with REP-TDDS to diabetic rats. In addition, the key pharmacokinetic parameters following oral and transdermal administration of REP formulations are displayed in Table 10. As seen in Figure 10, following oral treatment with the commercial tablets, the systemic concentration of REP rapidly increased to a significantly higher sharp peak (Cmax=188.6±6.5 ng.mL−1) after 2 h, followed by rapid elimination (p<0.05). Similarly, but to much less an extent, the plasma levels of unprocessed REP powder reached a Cmax of 48.2±9.3 ng.mL−1 after 2 h. However, the transdermal flux of the REP-SLN-TDDS increased slowly and steadily to a Cmax of 153.1±3.9 ng.ml−1 at tmax of 3 h and sustained to 24 h. During the application time period, Cmax was substantially lower than the oral tablets (p<0.05) and closer to a concentration plateau, where rate of drug absorption equals the drug-elimination rate. The curves also revealed a lack of large fluctuations in plasma levels with the REP-SLN-TDDS, highlighting the possibility to reduce the drug side effects. the REP-SLN-TDDS revealed a significantly longer t½ (p<0.01) than the orally administered formulations (5.38±0.24 h vs 3.68±0.17 h for the commercial tablet). This indicates that the TDDS delivered REP for a longer periodand outperformed tablet formulations in terms of drug sustainment. the REP-SLN-TDDS also showed a significantly longer mean residence time than other tested formulations (p<0.01). The longer t½ of REP following transdermal administration is mainly attributed to extended absorption and continual replenishment of the drug from the TDDS. The value of AUC is usually taken as an indicator of drug bioavailability. The significantly greater AUC value following application of the REP-SLN-TDDS (AUC0–24·h=2141.6±96.4 ng.h.mL−1, p<0.01) confirmed the capacity to transfer REP across the skin with higher bioavailability. Moreover, the appearance of quantifiable concentrations of REP in the samples early withdrawn signified the absence of lag time of the transdermal system beside keeping drug concentrations in the pharmacologically efficacious range for longer times. The overall pharmacokinetic parameters demonstrate the effectiveness of the REP-SLN-TDDS to overcome the skin barrier and deliver REP rapidly.
 |
Table 10 Pharmacokinetic parameters of repaglinide in rat plasma for the transdermal (REP-SLN-TDDS), commercial tablet, and unprocessed formulations |
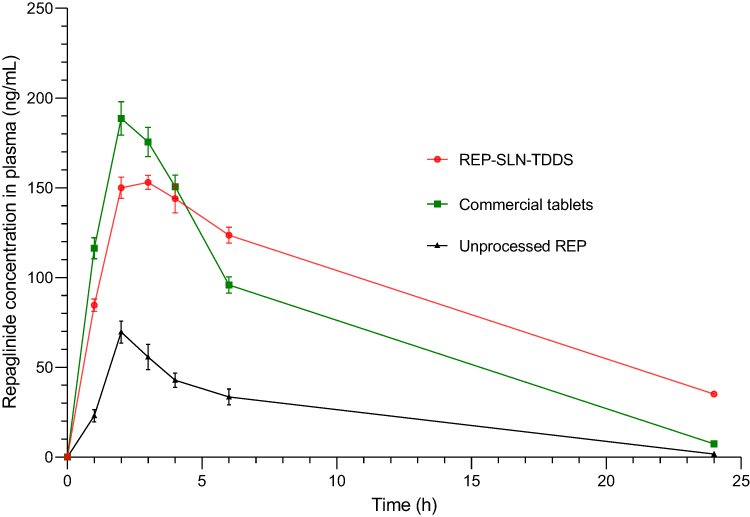 |
Figure 10 Plasma concentration–time profiles of repaglinide after oral and transdermal administration in rats. Abbreviations: REP, repaglinide; SLN, solid-lipid nanoparticle; TDDS, transdermal delivery system. |
Hypoglycemic Activity
The percentage reductions in blood glucose levels following administration of the REP-SLN-TDDS in comparison with the oral commercial tablets in diabetic rats are displayed in Figure 11. In general, the the REP-SLN-TDDS showed a better control of hyperglycemia in the treated groups. The maximum recorded blood glucose–reduction levels after 3 h were significantly lower (p<0.05) for the TDDS treatment than oral tablets (34.05%±2.098% vs 39.15±2.19%), implying a lower incidence of hypoglycemia. Of note, the reduction of blood glucose levels was maintained significantly higher at 24 h (p<0.05) for REP-TDDS, with a value of 14.32%±3.11% compared to 6.85%±2.50% for the oral commercial tablets. In light of the previous findings, the REP-SLN-TDDS demonstrated an excellent capacity for effective antidiabetic therapy. The additional advantages of lipid-based nanosized particles for transdermal application are plausibly associated to characteristics like enhanced skin hydration, adhesiveness, occlusion, lubrication, and skin emollience.9 SLNs are made up of lipids that are similar to those found in sebum and the skin itself. This could enhance the breakdown of SLNs in the sebum, followed by drug release for a topical or dermal effect. Alternatively, SLNs could permeate the skin by various routes and appendages until reaching the bloodstream. The adhesion of lipid-based nanoparticles to the skin helps keep the skin hydrated and lengthens the period of contact of the drug with the skin, permitting greater cutaneous absorption. As the particles become smaller, the more surface area is accessible for skin adherence.81 Moreover, the occlusion generated by SLN-based TDDS is also primarily owing to their nanometric size.82 With improved occlusion, SLNs diminish trans-epidermal water loss through evaporation and promote medication penetration via the skin.83 The overall combined impacts of the SLN-TDDS diminished the epidermis barrier and enabled better transdermal transfer of REP.
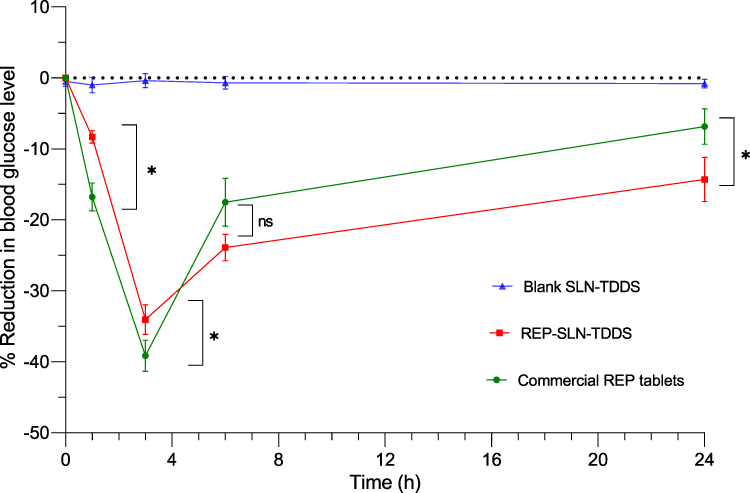 |
Figure 11 Reduction in blood glucose levels following administration of the REP-SLN-TDDS in comparison with commercial REP tablets in diabetic rats. Abbreviations: REP, repaglinide; SLN, solid-lipid nanoparticle; TDDS, transdermal delivery system; ns, not significant. Note: *p˂0.05. |
Conclusion
The BBD was effectively utilized to develop and optimize REP-SLNs employing ultrasonic hot-melt emulsification. According to the generated models, lipid content had a prominent influence on PS and EE, while surfactant concentration showed a significant negative influence on both responses. The optimized REP-SLNs were used to develop chitosan-based patches for transdermal delivery. According to the findings, the constructed transdermal system provided regulated release, improved penetration, and more efficient blood glucose management than the standard oral drug tablets. As a consequence, REP-SLNs can be considered a viable alternative for peroral REP in terms of controlled delivery and enhanced patient compliance.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through project 445-9-614.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Holm R, Kuentz M, Ilie-Spiridon A-R, Griffin BT. Lipid based formulations as supersaturating oral delivery systems: from current to future industrial applications. Eur J Pharm Sci. 2023;189:106556. doi:10.1016/j.ejps.2023.106556
2. Vasam M, Maddiboyina B, Talluri C, Alagarsamy S, Gugulothu B, Roy H. Formulation, characterization, and Taguchi design study of eplerenone lipid-based solid dispersions integrated with gelucire. BioNanoScience. 2023;13(2):576–587. doi:10.1007/s12668-023-01102-4
3. Maddiboyina B, Nakkala RK, Roy H, Roy H. Perspectives on cutting-edge nanoparticulate drug delivery technologies based on lipids and their applications. Chem Biol Drug Des. 2023;102(2):377–394. doi:10.1111/cbdd.14230
4. Patravale VB, Mirani AG. Preparation and characterization of solid lipid nanoparticles-based gel for topical delivery. In: Weissig V, Elbayoumi T, editors. Pharmaceutical Nanotechnology: Basic Protocols. New York: Springer New York; 2019:293–302.
5. Uner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int J Nanomed. 2007;2(3):289–300.
6. Khames A, Khaleel MA, El-Badawy MF, El-Nezhawy AOH. Natamycin solid lipid nanoparticles - sustained ocular delivery system of higher corneal penetration against deep fungal keratitis: preparation and optimization. Int J Nanomed. 2019;14:2515–2531. doi:10.2147/IJN.S190502
7. Alkilani AZ, McCrudden MT, Donnelly RF. Transdermal drug delivery: innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics. 2015;7(4):438–470. doi:10.3390/pharmaceutics7040438
8. Wong WF, Ang KP, Sethi G, Looi CY. Recent advancement of medical patch for transdermal drug delivery. Medicina. 2023;59(4):778. doi:10.3390/medicina59040778
9. Souto EB, Fangueiro JF, Fernandes AR, et al.. Physicochemical and biopharmaceutical aspects influencing skin permeation and role of SLN and NLC for skin drug delivery. Heliyon. 2022;8:e08938.
10. Chen MC, Ling MH, Lai KY, Pramudityo E. Chitosan microneedle patches for sustained transdermal delivery of macromolecules. Biomacromolecules. 2012;13(12):4022–4031. doi:10.1021/bm301293d
11. Ali HSM, Hanafy AF. Glibenclamide nanocrystals in a biodegradable chitosan patch for transdermal delivery: engineering, formulation, and evaluation. J Pharm Sci. 2017;106(1):402–410. doi:10.1016/j.xphs.2016.10.010
12. Karami Z, Saghatchi Zanjani MR, Nasihatsheno N, Hamidi M. Improved oral bioavailability of repaglinide, a typical BCS Class II drug, with a chitosan-coated nanoemulsion. J Biomed Mater Res B. 2020;108(3):717–728. doi:10.1002/jbm.b.34426
13. Gadadare R, Mandpe L, Pokharkar V. Ultra rapidly dissolving repaglinide nanosized crystals prepared via bottom-up and top-down approach: influence of food on pharmacokinetics behavior. AAPS Pharm Sci Tech. 2015;16(4):787–799. doi:10.1208/s12249-014-0267-8
14. Zhu Z, Yang T, Zhao Y, Gao N, Leng D, Ding P. A simple method to improve the dissolution of repaglinide and exploration of its mechanism. Asian J. Pharm. Sci. 2014;9(4):218–225. doi:10.1016/j.ajps.2014.06.004
15. Fouad SA, Teaima MH, Gebril MI, Abd Allah FI, El-Nabarawi MA, Elhabal SF. Formulation of novel niosomal repaglinide chewable tablets using coprocessed excipients: in vitro characterization, optimization and enhanced hypoglycemic activity in rats. Drug Delivery. 2023;30(1):2181747. doi:10.1080/10717544.2023.2181747
16. Liu M, Cao W, Sun Y, He Z. Preparation, characterization and in vivo evaluation of formulation of repaglinide with hydroxypropyl-β-cyclodextrin. Int J Pharm. 2014;477(1–2):159–166. doi:10.1016/j.ijpharm.2014.10.038
17. Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos. 2006;34(12):1976–1984. doi:10.1124/dmd.106.012351
18. Rawat MK, Jain A, Mishra A, Muthu MS, Singh S. Development of repaglinide loaded solid lipid nanocarrier: selection of fabrication method. Curr Drug Deliv. 2010;7(1):44–50. doi:10.2174/156720110790396472
19. Lokhande AB, Mishra S, Kulkarni RD, Naik JB. Preparation and characterization of repaglinide loaded ethylcellulose nanoparticles by solvent diffusion technique using high pressure homogenizer. J Pharm Res. 2013;7(5):421–426. doi:10.1016/j.jopr.2013.04.049
20. Navamanisubramanian R, Nerella R, Duraipandian C, Seetharaman S. Quality by design approach for optimization of repaglinide buccal tablets using box-behnken design. Future J Pharm Sci. 2018;4(2):265–272. doi:10.1016/j.fjps.2018.10.002
21. He W, Wu M, Huang S, Yin L. Matrix tablets for sustained release of repaglinide: preparation, pharmacokinetics and hypoglycemic activity in beagle dogs. Int J Pharm. 2015;478(1):297–307. doi:10.1016/j.ijpharm.2014.11.059
22. Tavakoli N, Minaiyan M, Tabbakhian M, Pendar Y. Preparation and evaluation of a controlled drug release of repaglinide through matrix pellets: in vitro and in vivo studies. J Microencapsul. 2014;31(6):529–534. doi:10.3109/02652048.2014.885604
23. Qin C, He W, Zhu C, et al.. Controlled release of metformin hydrochloride and repaglinide from sandwiched osmotic pump tablet. Int J Pharm. 2014;466(1–2):276–285. doi:10.1016/j.ijpharm.2014.03.002
24. Engla G, Soni LK, Dixit VK. Sustained release delivery of repaglinide by biodegradable microspheres. J Drug Delivery Ther. 2017;7:77–80.
25. Shinde UA, Modani SH, Singh KH. Design and development of repaglinide microemulsion gel for transdermal delivery. AAPS Pharm Sci Tech. 2018;19(1):315–325. doi:10.1208/s12249-017-0811-4
26. Beg S, Akhter S. Box–behnken designs and their applications in pharmaceutical product development. In: Beg S, editor. Design of Experiments for Pharmaceutical Product Development: Volume I : Basics and Fundamental Principles. Singapore: Springer Singapore; 2021:77–85.
27. Marasini N, Yan YD, Poudel BK, Choi HG, Yong CS, Kim JO. Development and optimization of self‐nanoemulsifying drug delivery system with enhanced bioavailability by box–behnken design and desirability function. J Pharmaceut Sci. 2012;101(12):4584–4596. doi:10.1002/jps.23333
28. Usta DY, Timur B, Teksin ZS. Formulation development, optimization by Box-Behnken design, characterization, in vitro, ex-vivo, and in vivo evaluation of bosentan-loaded self-nanoemulsifying drug delivery system: a novel alternative dosage form for pulmonary arterial hypertension treatment. Eur J Pharm Sci. 2022;174:106159. doi:10.1016/j.ejps.2022.106159
29. Parmar K, Patel J, Sheth N. Self nano-emulsifying drug delivery system for Embelin: design, characterization and in-vitro studies. Asian J Pharm Sci. 2015;10(5):396–404. doi:10.1016/j.ajps.2015.04.006
30. Abla KK, Mneimneh AT, Allam AN, Mehanna MM. Application of Box-Behnken design in the preparation, optimization, and in-vivo pharmacokinetic evaluation of oral tadalafil-loaded niosomal film. Pharmaceutics. 2023;15(1):173. doi:10.3390/pharmaceutics15010173
31. Kumar S, Narayan R, Ahammed V, Nayak Y, Naha A, Nayak UY. Development of ritonavir solid lipid nanoparticles by Box Behnken design for intestinal lymphatic targeting. J Drug Delivery Sci Technol. 2018;44:181–189. doi:10.1016/j.jddst.2017.12.014
32. Nazem Z, Firoozian F, Khodabandelou S, Mohammadi M, Mahboobian MM. Systematic optimization of solid lipid nanoparticles of silybin for improved oral drug delivery by Box-Behnken design: in vitro and in vivo evaluations. J Pharm Innovation. 2022;1:2
33. El-Say KM, Hosny KM, Ahmad A. Optimization of carvedilol solid lipid nanoparticles: an approach to control the release and enhance the oral bioavailability on rabbits. PLoS One. 2018;13(8):e0203405. doi:10.1371/journal.pone.0203405
34. Hou S, Hindle M, Byron PR. A stability-indicating HPLC assay method for budesonide. J Pharm Biomed Analysis. 2001;24(3):371–380. doi:10.1016/S0731-7085(00)00424-6
35. Das S, Ng WK, Kanaujia P, Kim S, Tan RBH. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surf b. 2011;88(1):483–489. doi:10.1016/j.colsurfb.2011.07.036
36. Nair AB, Shah J, Al-Dhubiab BE, et al.. Clarithromycin solid lipid nanoparticles for topical ocular therapy: optimization, evaluation and in vivo studies. Pharmaceutics. 2021;13:523. doi:10.3390/pharmaceutics13040523
37. Ghadiri M, Fatemi S, Vatanara A, et al.. Loading hydrophilic drug in solid lipid media as nanoparticles: statistical modeling of entrapment efficiency and particle size. Int J Pharm. 2012;424(1–2):128–137. doi:10.1016/j.ijpharm.2011.12.037
38. Yadav P, Rastogi V, Verma A. Application of Box–Behnken design and desirability function in the development and optimization of self-nanoemulsifying drug delivery system for enhanced dissolution of ezetimibe. Future J Pharm Sci. 2020;6(1):7. doi:10.1186/s43094-020-00023-3
39. Howard MD, Lu X, Jay M, Dziubla TD. Optimization of the lyophilization process for long-term stability of solid-lipid nanoparticles. Drug Dev Ind Pharm. 2012;38(10):1270–1279. doi:10.3109/03639045.2011.645835
40. Ullah W, Nawaz A, Akhlaq M, et al.. Transdermal delivery of gatifloxacin carboxymethyl cellulose-based patches: preparation and characterization. J Drug Delivery Sci Technol. 2021;66:102783. doi:10.1016/j.jddst.2021.102783
41. Kumar R, Sinha VR, Dahiya L, Sarwal A. Transdermal delivery of duloxetine-sulfobutylether-β-cyclodextrin complex for effective management of depression. Int J Pharm. 2021;594:120129. doi:10.1016/j.ijpharm.2020.120129
42. Malaiya MK, Jain A, Pooja H, Jain A, Jain D. Controlled delivery of rivastigmine using transdermal patch for effective management of Alzheimer’s disease. J Drug Delivery Sci Technol. 2018;45:408–414. doi:10.1016/j.jddst.2018.03.030
43. Mutalik S, Udupa N. Glibenclamide transdermal patches: physicochemical, pharmacodynamic, and pharmacokinetic evaluations. J Pharmaceut Sci. 2004;93(6):1577–1594. doi:10.1002/jps.20058
44. Rehman M, Ihsan A, Madni A, et al.. Solid lipid nanoparticles for thermoresponsive targeting: evidence from spectrophotometry, electrochemical, and cytotoxicity studies. Int J Nanomed. 2017;12:8325–8336. doi:10.2147/IJN.S147506
45. Zhao K, Singh J. In vitro percutaneous absorption enhancement of propranolol hydrochloride through porcine epidermis by terpenes/ethanol. J Control Release. 1999;62(3):359–366. doi:10.1016/S0168-3659(99)00171-6
46. Thakur G, Singh A, Singh I. Formulation and evaluation of transdermal composite films of chitosan-montmorillonite for the delivery of curcumin. Int J Pharm Investig. 2016;6(1):23–31. doi:10.4103/2230-973X.176468
47. Pervaiz F, Saba A, Yasin H, et al.. Fabrication of solid lipid nanoparticles-based patches of paroxetine and their ex-vivo permeation behaviour. Artif Cells Nanomed Biotechnol. 2023;51(1):108–119. doi:10.1080/21691401.2023.2179631
48. Can A, Erdal M, Güngör S, Özsoy Y. Optimization and Characterization of Chitosan Films for Transdermal Delivery of Ondansetron. Molecules. 2013;18(5):5455. doi:10.3390/molecules18055455
49. Kim JD, Kang SM, Seo BI, Choi HY, Choi HS, Ku SK. Anti-diabetic activity of SMK001, a poly herbal formula in streptozotocin induced diabetic rats: therapeutic study. Biol Pharm Bull. 2006;29(3):477–482. doi:10.1248/bpb.29.477
50. Fayyad MK, Ghanem EH. Liquid chromatography tandem mass spectrometry method for determination of anti-diabetic drug repaglinide in human plasma. Am J Anal Chem. 2014;05(04):12. doi:10.4236/ajac.2014.54035
51. Duong VA, Nguyen TTL, Maeng HJ. Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method. Molecules. 2020;25(20):4781. doi:10.3390/molecules25204781
52. Patere SN, Desai NS, Jain AS, et al.. Compritol®888 ATO a lipid excipient for sustained release of highly water soluble active: formulation, scale-up and IVIVC study. Curr Drug Deliv. 2013;10(5):548–556. doi:10.2174/1567201811310050006
53. Shah M, Chuttani K, Mishra AK, Pathak K. Oral solid compritol 888 ATO nanosuspension of simvastatin: optimization and biodistribution studies. Drug Dev. Ind. Pharm. 2011;37(5):526–537. doi:10.3109/03639045.2010.527983
54. Renukuntla J, Peterson-Sockwell S, Clark BA, et al.. Design and preclinical evaluation of nicotine–stearic acid conjugate-loaded solid lipid nanoparticles for transdermal delivery: a technical note. Pharmaceutics. 2023;15(4):1043. doi:10.3390/pharmaceutics15041043
55. Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. doi:10.1016/S0939-6411(00)00087-4
56. Azhar Shekoufeh Bahari L, Hamishehkar H. The impact of variables on particle size of solid lipid nanoparticles and nanostructured lipid carriers; a comparative literature review. Adv Pharm Bull. 2016;6(2):143–151. doi:10.15171/apb.2016.021
57. Seo JW, Kim KJ, Kim SH, Hwang KM, Seok SH, Park ES. Effect of process parameters on formation and aggregation of nanoparticles prepared with a shirasu porous glass membrane. Chem Pharm Bull. 2015;63(10):792–798. doi:10.1248/cpb.c15-00297
58. Bolla PK, Kalhapure RS, Rodriguez VA, Ramos DV, Dahl A, Renukuntla J. Preparation of solid lipid nanoparticles of furosemide-silver complex and evaluation of antibacterial activity. J Drug Delivery Sci Technol. 2019;49:6–13. doi:10.1016/j.jddst.2018.10.035
59. Emami J, Mohiti H, Hamishehkar H, Varshosaz J. Formulation and optimization of solid lipid nanoparticle formulation for pulmonary delivery of budesonide using Taguchi and Box-Behnken design. Res Pharm Sci. 2015;10:17–33.
60. Danaei M, Dehghankhold M, Ataei S, et al.. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics. 2018;4:10.
61. Thakkar HP, Desai JL, Parmar MP. Application of Box-Behnken design for optimization of formulation parameters for nanostructured lipid carriers of candesartan cilexetil. Asian J Pharm. 2014;3:8.
62. Sinsuebpol C, Changsan N. Effects of ultrasonic operating parameters and emulsifier system on sacha inchi oil nanoemulsion characteristics. J Oleo Sci. 2020;69(5):437–448. doi:10.5650/jos.ess19193
63. Cheng Q, Debnath S, Gregan E, Byrne HJ. Ultrasound-assisted SWNTs dispersion: effects of sonication parameters and solvent properties. J Phys Chem C. 2010;114(19):8821–8827. doi:10.1021/jp101431h
64. Leong TS, Wooster TJ, Kentish SE, Ashokkumar M. Minimising oil droplet size using ultrasonic emulsification. Ultrason Sonochem. 2009;16(6):721–727. doi:10.1016/j.ultsonch.2009.02.008
65. Dhiman N, Awasthi R, Sharma B, Kharkwal H, Kulkarni GT. Lipid nanoparticles as carriers for bioactive delivery. Front Chem. 2021;9:580118. doi:10.3389/fchem.2021.580118
66. Yang -Y-Y, Chung T-S, Bai X-L, Chan W-K. Effect of preparation conditions on morphology and release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion method. Chem Eng Sci. 2000;55(12):2223–2236. doi:10.1016/S0009-2509(99)00503-5
67. Hao J, Fang X, Zhou Y, et al.. Development and optimization of solid lipid nanoparticle formulation for ophthalmic delivery of chloramphenicol using a Box-Behnken design. Int J Nanomed. 2011;6:683–692. doi:10.2147/IJN.S17386
68. Qadir A, Aqil M, Ali A, et al.. Nanostructured lipidic carriers for dual drug delivery in the management of psoriasis: systematic optimization, dermatokinetic and preclinical evaluation. J Drug Delivery Sci Technol. 2020;57:101775. doi:10.1016/j.jddst.2020.101775
69. Agrawal M, Saraf S, Pradhan M, Patel RJ, Singhvi G, Alexander A. Design and optimization of curcumin loaded nano lipid carrier system using Box-Behnken design. Biomed Pharmacother. 2021;141:111919. doi:10.1016/j.biopha.2021.111919
70. Li Y, Zhao X, Zu Y, Zhang Y. Preparation and characterization of paclitaxel nanosuspension using novel emulsification method by combining high speed homogenizer and high pressure homogenization. Int J Pharm. 2015;490(1–2):324–333. doi:10.1016/j.ijpharm.2015.05.070
71. Kumar M, Sharma G, Singla D, et al.. Enhanced oral absorption of all-trans retinoic acid upon encapsulation in solid lipid nanoparticles. Pharm Nanotechnol. 2020;8(6):495–510. doi:10.2174/2211738508999201027220825
72. Avasatthi V, Pawar H, Dora CP, Bansod P, Gill MS, Suresh S. A novel nanogel formulation of methotrexate for topical treatment of psoriasis: optimization, in vitro and in vivo evaluation. Pharm Dev Technol. 2016;21(5):554–562. doi:10.3109/10837450.2015.1026605
73. Ammar H, Salama H, El-Nahhas S, Elmotasem H. Design and evaluation of chitosan films for transdermal delivery of glimepiride. Current Drug Deliv. 2008;5(4):290–298. doi:10.2174/156720108785915005
74. Roy H, Nayak BS, Nandi S. Chitosan anchored nanoparticles in current drug development utilizing computer-aided pharmacokinetic modeling: case studies for target specific cancer treatment and future prospective. Curr Pharm Des. 2020;26(15):1666–1675. doi:10.2174/1381612826666200203121241
75. Tejada G, Barrera MG, Piccirilli GN, et al.. Development and evaluation of buccal films based on chitosan for the potential treatment of oral candidiasis. AAPS Pharm Sci Tech. 2017;18(4):936–946. doi:10.1208/s12249-017-0720-6
76. Rao H, Ahmad S, Madni A, et al.. Compritol-based alprazolam solid lipid nanoparticles for sustained release of alprazolam: preparation by hot melt encapsulation. Molecules. 2022;28(1):27. doi:10.3390/molecules28010027
77. Danyuo Y, Ani CJ, Salifu AA, et al.. Anomalous release kinetics of prodigiosin from poly-n-isopropyl-acrylamid based hydrogels for the treatment of triple negative breast cancer. Sci Rep. 2019;9(1):3862. doi:10.1038/s41598-019-39578-4
78. Arora R, Katiyar SS, Kushwah V, Jain S. Solid lipid nanoparticles and nanostructured lipid carrier-based nanotherapeutics in treatment of psoriasis: a comparative study. Expert Opin Drug Deliv. 2017;14(2):165–177. doi:10.1080/17425247.2017.1264386
79. Choi WI, Lee JH, Kim J-Y, Kim J-C, Kim YH, Tae G. Tae G: efficient skin permeation of soluble proteins via flexible and functional nano-carrier. J Control Release. 2012;157(2):272–278. doi:10.1016/j.jconrel.2011.08.013
80. Elkomy MH, Ali AA, Eid HM. Chitosan on the surface of nanoparticles for enhanced drug delivery: a comprehensive review. J Control Release. 2022;351:923–940. doi:10.1016/j.jconrel.2022.10.005
81. Wissing SA. Cosmetic applications for solid lipid nanoparticles (SLN). Int J Pharm. 2003;254(1):65–68. doi:10.1016/S0378-5173(02)00684-1
82. Müller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Delivery Rev. 2002;54:S131–S155. doi:10.1016/S0169-409X(02)00118-7
83. Jenning V, Gysler A, Schäfer-Korting M, Gohla SH. Vitamin A loaded solid lipid nanoparticles for topical use: occlusive properties and drug targeting to the upper skin. Eur J Pharm Biopharm. 2000;49(3):211–218. doi:10.1016/S0939-6411(99)00075-2
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.