")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Renal transplantation in patients with Alport syndrome: patient selection, outcomes, and donor evaluation
Authors Kashtan CE
Received 2 June 2018
Accepted for publication 15 August 2018
Published 16 October 2018 Volume 2018:11 Pages 267—270
DOI https://doi.org/10.2147/IJNRD.S150539
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Pravin Singhal
Clifford E Kashtan
Division of Nephrology, Department of Pediatrics, University of Minnesota Medical School, Minneapolis, MN 55454, USA
Abstract: Alport syndrome is an inherited disorder of basement membrane collagen IV that frequently results in end-stage renal disease. Patients with Alport syndrome who undergo renal transplantation have generally excellent outcomes. Posttransplant antiglomerular basement membrane nephritis is a rare complication of renal transplantation for Alport syndrome. Because Alport syndrome is a genetic disorder, potential related donors must be carefully evaluated in order to minimize harm.
Keywords: Alport syndrome, kidney transplantation, collagen IV, posttransplant anti-GBM nephritis
Introduction
Alport syndrome is an inherited cause of end-stage renal disease (ESRD) that results from mutations that affect the collagen IV a345 network of glomerular basement membranes (GBM) as well as basement membranes in the cochlea and eye.1 Although early treatment with angiotensin-converting enzyme inhibitors appears to delay the onset of ESRD, many patients require renal replacement therapy.
The risk and timing of ESRD is heavily influenced by sex and genotype, making some subsets of Alport patients more likely to require dialysis and kidney transplantation than others. Alport syndrome arises from mutations in the COL4A3, COL4A4, and COL4A5 genes. The majority of individuals with Alport syndrome (approximately 60% according to recent studies utilizing next-generation sequencing) have the X-linked form of the disease because of mutations in COL4A5.2,3 Autosomal recessive Alport syndrome (ARAS) is caused by mutations in both alleles of COL4A3 or COL4A4, and accounts for about 15% of people with the disease.2,3 Finally, about 25% of individuals with Alport syndrome have autosomal dominant disease, resulting from mutation in one allele of COL4A3 or COL4A4.2,3
Sex has a marked impact on the prognosis of X-linked Alport syndrome (XLAS). Fifty percent of untreated males with XLAS reach ESRD by age 25 and 90% reach ESRD by age 40.4 While only about 12% of females with XLAS develop ESRD before age 40, the probability of ESRD increases to about 30% by age 60 and 40% by age 80.4 The nature of the underlying mutation in COL4A5 is an important determinant of the rate of progression to ESRD in XLAS males. Truncating mutations (deletions, frameshift, and nonsense mutations) are associated with an earlier onset of ESRD in comparison with splice-site and missense mutations.4,5
Patients with ARAS typically reach ESRD by age 40, with some exceptions, regardless of sex.6 Autosomal dominant Alport syndrome (ADAS) tends to advance less aggressively than XLAS or ARAS. While an early study of men with ADAS reported a median renal survival of 50 years, more recent studies have reported a median renal survival of 70–80 years in ADAS patients.7–9
In this review, I examine the factors that influence ESRD risk and timing in Alport syndrome. In addition, I describe transplant outcomes in Alport patients and the rare, but important, phenomenon of posttransplant anti-GBM nephritis. Finally, I discuss the selection of living related kidney donors in Alport families.
Patient and graft survival after transplantation
Studies carried out over the past several decades have consistently demonstrated that patient and graft survival rates in Alport patients match or exceed patient and graft survival in patients with other causes of ESRD. A 1995 analysis of data collected by the North American Pediatric Renal Transplant Cooperative Study showed a 2-year graft survival rate of 95% in Alport patients who received a kidney from a living donor and 70% in cadaver kidney recipients, compared with 89% and 74%, respectively, in patients with structural urinary tract disorders.10 A more recent study of data from The European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) Registry showed that male Alport patients had superior patient and graft survival rates compared with controls matched for age, year of transplantation, and kidney donor source.11 These and other single-center studies demonstrate that the outcome of kidney transplantation for Alport syndrome is generally quite favorable.
Although preemptive kidney transplantation is the preferred mode of therapy for Alport patients approaching ESRD, it is worth noting that Alport patients who require chronic dialysis exhibit relatively good outcomes. Analysis of ERA-EDTA data showed that the survival of male Alport patients on dialysis was superior to matched contols, for both hemodialysis and peritoneal dialysis.11
Posttransplant anti-GBM nephritis
Posttransplant anti-GBM nephritis is a rare but potentially devastating complication of kidney transplantation in patients with Alport syndrome. Affected patients have been almost exclusively males with the X-linked form of Alport syndrome, although rare cases in females with ARAS have been reported.12 The incidence of posttransplant anti-GBM nephritis in males with XLAS has been estimated to be about 2%–3% and does not appear to have changed significantly since the phenomenon was first described.12,13
In males with XLAS who develop posttransplant anti-GBM nephritis, the primary target of anti-GBM antibodies is the collagen IV α5 chain.14,15 These patients are thought to be immunologically naïve for the collagen IV α5 chain, prompting an immunologic response following transplantation of a normal kidney. Some of these patients, as well as patients with ARAS who experience posttransplant anti-GBM nephritis, express antibodies against the collagen IV α3 chain, the target epitope associated with idiopathic anti-GBM nephritis and Goodpasture syndrome.14,16,17
Those patients who develop posttransplant anti-GBM nephritis are typically male, progress to ESRD before age 40, and exhibit sensorineural deafness.12,18 Patients exhibiting this phenotype frequently have a truncating mutation in the COL4A5 gene, such as a deletion or nonsense mutation.4 However, neither the Alport phenotype nor the COL4A5 genotype is particularly helpful in predicting who will develop posttransplant anti-GBM nephritis. Antignac et al19 showed that males with truncating COL4A5 mutations could be transplanted without the development of posttransplant anti-GBM nephritis; this observation was recently confirmed by Gillion et al.13 The risk of developing posttransplant anti-GBM nephritis appears to be very low for patients with missense mutations in COL4A5 and in patients with ADAS.
Most cases of posttransplant anti-GBM nephritis are diagnosed within the first 12 months after transplant; however, intervals of several years between transplant and diagnosis have been reported.12 Clinical features range from isolated serum creatinine elevation to florid rapidly progressive glomerulonephritis. Biopsies of affected kidneys typically reveal a crescentic glomerulonephritis with intense linear GBM fixation of IgG and C3 by direct immunofluorescence. Some Alport patients who undergo transplant biopsy show a linear deposition of IgG along the GBM without other histological features of anti-GBM nephritis;20 the reported patients maintained good allograft function without specific intervention.20 Since the onset of posttransplant anti-GBM nephritis is typically within the first year after transplant, monitoring for circulating anti-GBM antibodies by monthly ELISA for the first 12 months following transplantation, in addition to routine surveillance for allograft dysfunction, would be a reasonable approach in male patients with truncating mutations in COL4A5 and in patients with ARAS. However, it has not been demonstrated that such monitoring results in an earlier diagnosis of posttransplant anti-GBM nephritis or in improved outcomes.
Transplant nephrologists should have a low threshold for allograft biopsy in males with XLAS who exhibit allograft dysfunction. On the basis of reported cases, about 75% of allografts exhibiting posttransplant anti-GBM nephritis fail irreversibly, usually within a few weeks to months. Plasmapheresis and cyclophosphamide administration are reasonable interventions but have often failed to salvage graft function. Most patients with posttransplant anti-GBM nephritis in a first allograft have had recurrence in subsequent transplants, despite prolonged intervals between transplants and the disappearance of detectable circulating anti-GBM antibodies prior to retransplantation.
Donor selection
As with other familial renal disorders, the genetics of Alport syndrome should be considered during the living donor evaluation process. Depending upon the gene involved, family members with heterozygous mutations in collagen IV genes have differing risks for chronic kidney disease (CKD) and ESRD.
Women with XLAS (COL4A5 heterozygotes)
As noted previously, there is a significant risk of ESRD in women with XLAS increasing from about 12% at age 40 to about 30% by age 60.18 Consequently, women with XLAS should be donors of last resort, to be considered only when no donor can be found among the unaffected male and female members of the family and the prospective donor is highly motivated. Women with overt proteinuria and those who display a hearing deficit should be rejected as donors, since both are risk factors for progression to ESRD.18,21 I have recommended elsewhere that only women with XLAS who are over 45 years of age should be considered as potential donors, since the prevalence of proteinuria increases with age, eventually developing in 75% of one large cohort of women with XLAS.18,22 A history of gross hematuria in childhood may be an additional risk factor for progression in heterozygous women with XLAS.21 There is a limited amount of outcome data for women with XLAS who serve as kidney donors. A report describing five such women found that most exhibited declining glomerular filtration rates, proteinuria, and hypertension in the years following nephrectomy.23
Heterozygotes for COL4A3 and COL4A4 mutations
The phenotypes associated with heterozygous mutations in COL4A3 and COL4A4 are variable, ranging from asymptomatic individuals to isolated hematuria to CKD and ESRD.1 Evaluation of these individuals as potential kidney donors should be based on genotype–phenotype correlations in the family and/or mutation databases, as depicted in Table 1. When the phenotype associated with a particular mutation is unclear, the default mode should be exclusion of the heterozygous individuals as kidney donors.
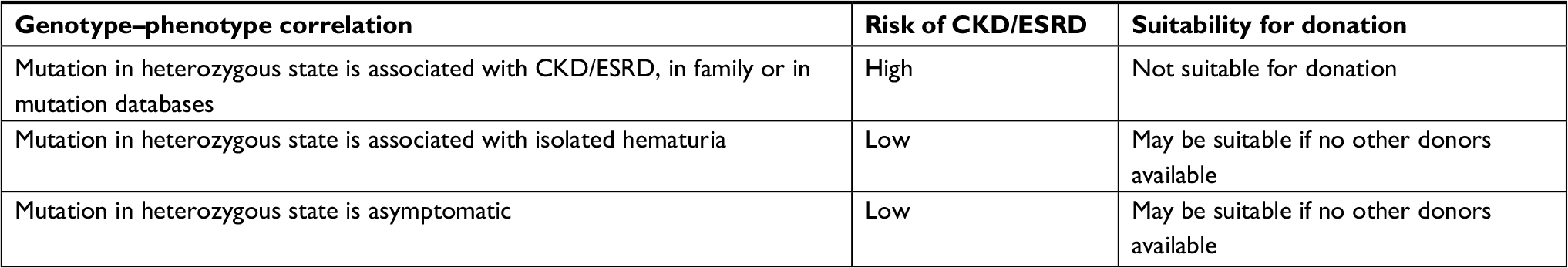 | Table 1 Donor suitability of individuals who are heterozygous for a mutation in COL4A3 or COL4A4 based on the phenotype of the mutation in the heterozygous state Abbreviations: CKD, chronic kidney disease; ESRD, end-stage renal disease. |
Conclusion
Patients with Alport syndrome are excellent candidates for kidney transplantation, with patient and graft survival rates that are equal to or better than those for patients with other causes of ESRD. Preemptive kidney transplantation is the treatment of choice for ESRD resulting from Alport syndrome.
Selection of living related donors for patients with Alport syndrome requires careful consideration of the risk for CKD/ESRD on the basis of the genotype. Although molecular genetic analysis is not always possible, mutation identification is very helpful in making informed decisions about living kidney donation.
Should those rare patients who have lost a kidney because of posttransplant anti-GBM nephritis be eligible for retransplantation? In my view, these patients should be eligible, although the pros and cons of a retransplant should be assessed on an individual-by-individual basis.
Acknowledgment
Dr Kashtan is the executive director of the Alport Syndrome Treatments and Outcomes Registry (alportregistry.org, NCT00481130), which receives financial support from the Alport Syndrome Foundation (alportsyndrome.org) and the Schuman and Pedersen families. Dr Kashtan is a site investigator for the CARDINAL trial (NCT03019185) sponsored by Reata Pharmaceuticals, Inc. and the HERA trial (NCT02855268) sponsored by Regulus Therapeutics, Inc. Dr Kashtan has also recently received research support from the Novartis Institute for Biomedical Research (NCT03074357).
Disclosure
The author reports no conflicts of interest in this work.
References
Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018;93(5):1045–1051. | ||
Fallerini C, Dosa L, Tita R, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet. 2014;86(3):252–257. | ||
Morinière V, Dahan K, Hilbert P, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. 2014;25(12):2740–2751. | ||
Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649–657. | ||
Bekheirnia MR, Reed B, Gregory MC, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010;21(5):876–883. | ||
Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol. 2013;24(12):1945–1954. | ||
Pochet JM, Bobrie G, Landais P, Goldfarb B, Grünfeld JP. Renal prognosis in Alport’s and related syndromes: influence of the mode of inheritance. Nephrol Dial Transplant. 1989;4(12):1016–1021. | ||
Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant. 2013;28(12):2946–2960. | ||
Kamiyoshi N, Nozu K, Fu XJ, et al. Genetic, Clinical, and Pathologic Backgrounds of Patients with Autosomal Dominant Alport Syndrome. Clin J Am Soc Nephrol. 2016;11(8):1441–1449. | ||
Kashtan CE, Mcenery PT, Tejani A, Stablein DM. Renal allograft survival according to primary diagnosis: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol. 1995;9(6):679–684. | ||
Temme J, Kramer A, Jager KJ, et al. Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin J Am Soc Nephrol. 2012;7(12):1969–1976. | ||
Kashtan CE. Renal transplantation in patients with Alport syndrome. Pediatr Transplant. 2006;10(6):651–657. | ||
Gillion V, Dahan K, Cosyns JP, et al. Genotype and outcome after kidney transplantation in Alport syndrome. Kidney Int Rep. 2018;3(3):652–660. | ||
Brainwood D, Kashtan C, Gubler MC, Turner AN. Targets of alloantibodies in Alport anti-glomerular basement membrane disease after renal transplantation. Kidney Int. 1998;53(3):762–766. | ||
Borza DB. Autoepitopes and alloepitopes of type IV collagen: role in the molecular pathogenesis of anti-GBM antibody glomerulonephritis. Nephron Exp Nephrol. 2007;106(2):e37–e43. | ||
Kalluri R, Torre A, Shield CF, et al. Identification of alpha3, alpha4, and alpha5 chains of type IV collagen as alloantigens for Alport posttransplant anti-glomerular basement membrane antibodies. Transplantation. 2000;69(4):679–684. | ||
Kalluri R, van den Heuvel LP, Smeets HJ, et al. A COL4A3 gene mutation and post-transplant anti-alpha 3(IV) collagen alloantibodies in Alport syndrome. Kidney Int. 1995;47(4):1199–1204. | ||
Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14(10):2603–2610. | ||
Antignac C, Knebelmann B, Drouot L, et al. Deletions in the COL4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. J Clin Invest. 1994;93(3):1195–1207. | ||
Quérin S, Noël LH, Grünfeld JP, et al. Linear glomerular IgG fixation in renal allografts: incidence and significance in Alport’s syndrome. Clin Nephrol. 1986;25(3):134–140. | ||
Grünfeld JP, Noël LH, Hafez S, Droz D. Renal prognosis in women with hereditary nephritis. Clin Nephrol. 1985;23(6):267–271. | ||
Kashtan CE. Women with Alport syndrome: risks and rewards of kidney donation. Nephrol Dial Transplant. 2009;24(5):1369–1370. | ||
Gross O, Weber M, Fries JW, Müller GA. Living donor kidney transplantation from relatives with mild urinary abnormalities in Alport syndrome: long-term risk, benefit and outcome. Nephrol Dial Transplant. 2009;24(5):1626–1630. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.