Back to Journals » International Journal of General Medicine » Volume 15
Relationships Among Gut Microbiota, Ischemic Stroke and Its Risk Factors: Based on Research Evidence
Authors Huang Q, Cai G, Liu T, Liu Z ![]()
Received 9 December 2021
Accepted for publication 8 February 2022
Published 23 February 2022 Volume 2022:15 Pages 2003—2023
DOI https://doi.org/10.2147/IJGM.S353276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Qinhong Huang,1 Guannan Cai,1 Ting Liu,2 Zhihua Liu3
1First Clinical School, Guangzhou Medical University, Guangzhou, 511436, People’s Republic of China; 2Guangzhou Key Laboratory of Enhanced Recovery after Abdominal Surgery, Innovation Center for Advanced Interdisciplinary Medicine, the Fifth Affiliated Hospital of Guangzhou Medical University, Guangzhou, 510700, People’s Republic of China; 3Department of Anorectal Surgery, the Fifth Affiliated Hospital of Guangzhou Medical University, Guangzhou, 510700, People’s Republic of China
Correspondence: Ting Liu; Zhihua Liu, Email [email protected]; [email protected]
Abstract: Stroke is a highly lethal disease and disabling illness while ischemic stroke accounts for the majority of stroke. It has been found that inflammation plays a key role in the initiation and progression of stroke, and atherosclerotic plaque rupture is considered to be the leading cause of ischemic stroke. Furthermore, chronic inflammatory diseases, such as obesity, type 2 diabetes mellitus (T2DM) and hypertension, are also considered as the high-risk factors for stroke. Recently, the topic on how gut microbiota affects human health has aroused great concern. The initiation and progression of ischemic stroke has been found to have close relation with gut microbiota dysbiosis. Hence, this manuscript briefly summarizes the roles of gut microbiota in ischemic stroke and its related risk factors, and the practicability of preventing and alleviating ischemic stroke by reconstructing gut microbiota.
Keywords: ischemic stroke, gut microbiota, inflammation, obesity, T2DM, hypertension, type 2 diabetes mellitus
Introduction
Stroke is the most common cause of severe disability and also the second leading cause of death worldwide after coronary heart disease.1,2 Stroke is generally divided into two types, namely ischemic and hemorrhagic stroke, and ischemic stroke accounts for 85%.3 During the initiation and progression of ischemic stroke, inflammation is an important pathological process. Preclinical stroke study has shown that inhibiting inflammatory response can significantly reduce the injuries of brain and improve neurological prognosis.4 Furthermore, atherosclerosis, obesity, diabetes, hypertension and peripheral infection are complications that most stroke patients suffer from,5 and all of these diseases are risk factors for stroke, while inflammation as a pathogenic factor plays a significant role in the initiation and progression of these diseases.6,7
Gut microbiota, which has been identified by Metagenomic analysis, mainly consists of six families, namely Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Clostridium and Verrucomicrobia, most of which are anaerobic bacteria.8 Among them, Firmicutes and Bacteroidetes are the predominant bacteria in healthy individuals, accounting for more than 90% of the total population.9 Because the gut microbiota has close relation to inflammation and many chronic diseases, it has drawn a lot of attention these years. It is found that there exist intercommunication pathways connecting the brain and the gut in the human body, known as the gut–brain axis, including sympathetic and parasympathetic activation, hypothalamus–pituitary–adrenal axis and central immune system.10,11 Changes in gut microbiota are proved to affect the physiology, behavior and cognitive function of the brain through the gut–brain axis.12–16
Gut microbiota dysbiosis can be found in gastrointestinal diseases, which are been reported to have close relation with cardiovascular and cerebrovascular diseases. Researchers conducted a retrospective study by using the Cox proportional hazard model to assess the correlation between different gastrointestinal diseases and ischemic stroke with 1,725,246 beneficiaries in average in each analysis, and concluded that several gastrointestinal diseases are associated with increased risk of ischemic stroke after adjusting the known risk factors for stroke. The most significant positive correlations included gastric diseases (risk ratio: 1.17 [95% confidence interval (CI): 1.15–1.19]), functional gastrointestinal diseases (1.16 [95% CI: 1.15–1.17]), inflammatory gastrointestinal diseases (1.13 [95% CI: 1.12–1.15]) and infectious gastrointestinal diseases (1.13 [95% CI: 1.12–1.15]).17 Further, a study shows that alteration of gut microbiota by using antibiotics, ischemic stroke is significantly alleviated through the gut–brain immune axis mechanism.18 Besides, it is found that ischemic stroke in turn leads to gut microbiota dysbiosis. Researchers have found that cerebral ischemia rapidly induces intestinal ischemia and produces excessive nitrates through free radical reactions, resulting in gut microbiota dysbiosis accompanied with Enterobacteriaceae proliferation. The proliferation of Enterobacteriaceae aggravates cerebral infarction by exacerbating systemic inflammation, which is an independent risk factor for the adverse prognosis of stroke patients.19
Obviously, the dysbiosis of gut microbiota and the initiation and progression of ischemic stroke has a mutually reinforcing effect. In this review, we summarize the relationships among gut microbiota dysbiosis and ischemic stroke and its relative risk factors, and discuss ways to alleviate ischemic stroke and improve its prognosis by reconstructing gut microbiota.
Gut Microbiota and Inflammation in Ischemic Stroke
Inflammation Induces Ischemic Stroke
It has been found that the elevated levels of inflammatory factors, such as interleukin-6, C-reactive protein and lipoprotein-associated phospholipase A2, are associated with the increased risk of stroke.20 Also, soluble lectin-like oxidized low density lipoprotein receptor-1, a kind of inflammation-induced lipid receptor, attributing to atherosclerosis, has been proved be associated with high risk of stroke.21,22 Atherosclerosis, as a kind of vascular inflammatory disease, is a high-risk factor for ischemic stroke, because thromboembolism caused by atherosclerotic plaque rupture is the most common cause of ischemic stroke.23 The cause of thromboembolic events is due to the activation of molecules and inflammatory mediators in atherosclerotic plaques, which promote the progression of plaques from relatively benign to highly unstable stages, leading to vulnerable plaque rupture. In the pathogenesis, endothelial dysfunction and activation are key events, leading to the expression of adhesion molecules, which promote the recruitment of immune cells, especially monocyte-derived macrophages and T lymphocytes. Activated macrophages and T cells secrete proteolytic enzymes, such as matrix metalloproteinases (MMPs), which destroy the stability of plaque and eventually lead to plaque rupture.7 Besides atherosclerosis, studies have shown that various inflammatory diseases are also associated with increased risk of ischemic stroke.24 All in all, these above suggest that inflammation may be a major cause of ischemic stroke.
Inflammation Affects the Progress and Prognosis of Ischemic Stroke
In many animal models and patients with stroke, rapid activation of resident cells is the feature of the cerebral inflammatory response after ischemia, primarily microglia, followed by the infiltration of circulating inflammatory cells, including granulocytes (mainly neutrophils), T cells, monocytes/macrophages, in ischemic brain regions and other regions.25–30 In the mouse model of transient middle cerebral artery occlusion (MCAO), the result of the flow cytometry showed the significantly increased activated CD11b+ microglia/macrophages and Ly6G+ neutrophils in the early stage after ischemia, while CD3+ T cells increased relatively late.31
In the acute phase of ischemic stroke (a few minutes to several hours), reactive oxygen species (ROS) and pro-inflammatory cytokines and chemokines are rapidly released from the injured tissue, leading to the brain endothelial cells activation and the up-regulation of adhesion molecules on the brain endothelial cells, which promotes the adhesion and transendothelial migration of circulating leukocytes through the blood–brain barrier (BBB) into the brain parenchyma.4,32–34 In the subacute phase (several hours to several days), the infiltrated leucocytes release cytokines and chemokines, especially the excessive production of ROS and the induction/activation of MMP (mainly MMP-9), further amplify the inflammatory response in the brain. The extensive activation of resident cells in the brain and the infiltration of leucocytes in the blood will eventually aggravate the interruption of BBB, brain edema, neuronal death and hemorrhagic transformation of ischemic stroke.33,34
Although inflammation plays a defensive role in some extent, it is generally believed that inflammatory infiltration is more harmful than beneficial in the progression of ischemic stroke. The activation of microglial cells in the brain is within a few minutes after ischemia, which exacerbates ischemic tissue damage by releasing pro-inflammatory mediators, mainly including IL-1β and TNF-α.35–37 Transcription factor NF-κB, which plays a key role in cell survival and inflammatory response by regulating various genes, is activated in neurons, astrocytes, microglia and invasive inflammatory cells after cerebral ischemia.38 Moreover, there are experimental evidences showing that inhibiting the inflammatory response after ischemic stroke effectively reduces the damage of ischemic stroke to the brain and nerves function. By the inhibition of cerebral ischemia-induced time-dependent inflammatory cell recruitment and activation, including neutrophils, T cells and monocytes/macrophages, the size of cerebral infarction and alleviates neurological impairment in experimental stroke reduces.4,39 Inhibiting the activation of NF-κB with S-nitrosoglutathione in the rat model of experimental stroke alleviates the damage of inflammation and reduce the area of large cerebral infarction.40 Further, inhibiting the activity of inflammatory mediator Mac-1 by using its antibody alleviates cells damage after transient MCAO in rats.41 Besides, anti-inflammation in animal models targeting on other inflammatory mediators also shows positive results.42–44
In addition to focal brain infiltration, inflammation outside the brain, especially systemic inflammation caused by intestinal lipopolysaccharide (LPS) leakage, has adverse effects on the progression and prognosis of ischemic stroke. Some clinical studies reported that stroke patients with more severe neurological deficits have precursor infection.45,46 Animal systemic inflammatory model also provides consistent result. Using bacterial endotoxin, mainly LPS, to systematically attack and imitate Gram-negative bacterial infection, significantly increases the severity of ischemic brain injury and neurological deficits in mice after focal cerebral ischemia.47 LPS in the circulation can pass through the impaired BBB, activating microglia in brain, and the microglia activated by LPS impairs neuronal activity by changing neuronal computing power and promotes asynchrony.48 Moreover, LPS-activated microglia promote the activation of astrocytes through the release of adenosine triphosphate (ATP) and the subsequent increase of glutamatergic synaptic transmission, which causes excitotoxicity and chemokine (mainly CCL2) secretion from damaged neurons.49 CCL2 triggers chemotaxis and activation of microglia migration by stimulating CXCR3 receptors,50 therefore, repeated LPS-induced microglia activation leads to synaptic stripping of inhibitory terminals in the cortex, which aggravates nerve damage.51
So far, we can see that inflammation increases the risk of ischemic stroke, even directly causes ischemic stroke by inducing atherosclerotic plaque rupture, causing vascular embolism/occlusion. In addition, local infiltration of inflammatory brain has adverse effects on the progression and prognosis of ischemic stroke. In view of a large number of articles have directly discussed the relationship between inflammation and ischemic stroke in details, here we only introduce briefly. Next, we will focus on how gut microbiota affects ischemic stroke through inflammatory pathways.
Gut Microbiota Dysbiosis and Ischemic Stroke Promote Each Other Through Inflammatory Pathways
Gut Microbiota Dysbiosis Affects the Occurrence and Progression of Ischemic Stroke
The digestive tract is considered to be the main organ of immune response, rich in immune cells, responsible for more than 70% of the entire immune system activities.52 Many evidences show that intestinal inflammation and immune response play an important role in the pathophysiology of stroke, which can become an important therapeutic target for the treatment of stroke.53 Studies have found that gut microbiota dysbiosis is a risk factor for stroke and affects the prognosis after stroke. Gut microbiota dysbiosis has been observed in several acute ischemic stroke (AIS) animal models, indicating that gut microbiota regulates neuroinflammation and affects brain recovery.18 It has been proved that in the first few hours/days after AIS, peripheral adaptive immune cells are activated and recruited into the brain,54 and these cells can regulate gut microbiota and be regulated by gut microbiota.55
Gut microbiota dysbiosis induces ischemic stroke, mainly because the imbalance of gut microbiota increases the abundance of opportunistic pathogens in the gut. It has been found that the opportunistic pathogens enriched in patients with stroke and transient ischemic attack, including Enterobacter, Odoribacter, Megasphaera, Oscillibacter, Akkermansia and Desulfovibrio.52,56,57 The increased levels of opportunistic pathogens in the gut are associated with increased risk of developing AIS,58 because more endotoxin is produced and released by the increased Gram-negative bacteria, which directly impairs the intestinal barrier.59 With the dysfunction/damage of intestinal barrier, some metabolites related to microorganisms enter the circulation from intestinal tract. As a response, human body produces a lot of cytokines and other media, which effectively trigger inflammatory reactions.60 Intestinal LSP leakage and the increased intestinal permeability induce macrophages to infiltrate the region, produce and activate inflammatory cytokines, leading to local inflammation.61 Moreover, the inflammation caused by intestinal permeability damage is not just limited to the intestine, because pro-inflammatory cytokines can finally reach the brain through the blood.62 Meanwhile, LPS, as an important component of Gram-negative bacteria, is a ligand of toll-like receptor 4 (TLR-4) that can form a complex with CD14 and bind to TLR-4 located on the surface of innate immune cells, activating local and distant inflammatory cascades.59,63,64 The persistence of inflammatory responses caused by microbiota dysbiosis leads to subacute or chronic inflammation, which as a result, leads to the development of relative diseases, such as inflammatory bowel disease, diabetes and cardiovascular disease (CVD).65 The inflammatory bowel disease and Crohn’s disease are proved to be associated with changes in gut microbiota and increased production of intestinal IL-17,66,67 and also have been identified as risk factors for stroke.68,69 Moreover, studies show that gut microbiota dysbiosis leads to progression of atherosclerosis.70,71 Compared with the conventionally raised controls, absence of microbiota could lead to the formation of atherosclerotic lesions, suggesting that gut microbiota dysbiosis, especially the reduction or deletion of certain categories, directly induces atherosclerosis.70 Under this condition, inflammation can induce the rupture of atherosclerotic plaque, causing vascular embolism and ischemic stroke. In addition to induce ischemic stroke, gut microbiota contributes to the progression of ischemic stroke by regulating γδ T lymphocytes,53 which are a major lymphocyte group with innate immune function, located on the epithelial surface including the intestine,72 and these lymphocytes excrete IL-17 and produce chemotactic signals for peripheral myeloid cells (such as neutrophils and monocytes), which exacerbate ischemic brain damage.73,74
TMAO and Ischemic Stroke (Figure 1)
Trimethylamine-n-oxide (TMAO) is a by-product of gut microbiota closely related to stroke.75 TMAO is formed by the trimethylamine (TMA) that enters the liver through portal circulation, oxidized by flavin monooxygenase.76–78 Microbial community plays a key role in the formation of TMA, because antibacterial research shows that TMAO will not be formed without microbial community.79 TMAO is a high risk factor and prognostic marker for stroke and CVD, because the increased generation of TMAO promotes atherosclerosis, platelet activation, and inflammation.80,81 Many studies have emphasized that TMAO participates in the development of CVD in various patient cohorts,82–84 and the association between TMAO levels and adverse clinical outcomes also has been found in many independent cohorts.77,85,86 Microbial transplantation study has shown that TMAO is a risk factor for atherosclerosis and thrombosis.87 When TMAO exists at a high serum level, it is related to harmful effects such as endothelial dysfunction, which promotes vascular inflammation, atherosclerosis and other risk factors for CVD,88–90 and one of the main reasons is that TMAO promotes atherosclerosis by increasing the expression of scavenger receptors in macrophages and the formation of foam cells in the arterial wall.77 Subsequent experiment shows that the formation of macrophage foam cells and atherosclerosis are suppressed, when the formation of TMA and TMAO by microorganisms are suppressed by small molecule inhibitor of microbial choline TMA lyase.91
 |
Figure 1 A summary on how TMAO contributes to occurrence of ischemic stroke. |
TMAO is involved in inflammatory response and thromboembolism. Researchers found the positive correlation among TMAO level, TNF-α in human92 and the number of CD14++/CD16+ monocytes, a kind of pro-inflammatory intermediate.87 Moreover, a study using cultured human aortic endothelial cells and vascular smooth muscle cells reported that TMAO induces inflammation by up-regulating the expression of various inflammatory cytokines and adhesion molecules through NF-κB signaling pathway.93,94 In this case, atherosclerotic plaque rupture resulting in vascular embolism caused by inflammation can induce ischemic stroke. In addition, it is observed that regardless of the traditional risk factors, TMAO is directly related to the adverse treatment results of patients with ischemic brain injury.75,95 The association between TMAO and poor prognosis in patients with ischemic brain injury is mainly because of the formation of thrombosis. TMAO is reported to increase the risk of thrombosis, and high-level TMAO promotes endothelial dysfunction, induces platelet activation and increases platelet reactivity.96 The activation and aggregation of platelet and the subsequent formation of occlusive intra-arterial thrombosis are critical processes in atherosclerotic thrombosis and the increase of platelet reactivity is related to the degree of end-organ injury and poor prognosis.97,98
The Reduction of SCFAs Caused by Gut Microbiota Dysbiosis Affects Ischemic Stroke
Short-chain fatty acids (SCFAs), mainly include acetate, propionate and butyrate, which are produced by gut microbiota fermenting and degrading the insoluble carbohydrates in cecum and colon.99,100 Ischemic stroke will lead to microbiota dysbiosis and intestinal barrier damage,101 resulting LPS leakage and systemic inflammatory response, which are important pathological processes of ischemic stroke. It is found that AIS induces the long-term increase of LPS and proinflammatory cytokines in circulation of cynomolgus monkeys, which is related to the increased relative abundance of Bacteroidetes, meanwhile, the gut microbiota dysbiosis and mucosal injury resulting from AIS persist for almost 12 months.102
As beneficial substances, SCFAs can arrest the progression of ischemic stroke (Figure 2). First, SCFAs provide colon an acidic environment so that the growth of pathogenic bacteria is inhibited and the balance of water and electrolyte is maintained, preventing gut dysfunction.103–106 Second, SCFAs, as the main kind of energy source of colonic epithelium,99 playing an important role in cell growth and differentiation,107,108 are important substances for maintaining intestinal epithelial barrier function.109 In colon-derived SCFAs, butyrate is the most important regulator of tight junction protein (TJP), and has been proved to enhance intestinal barrier function by increasing the expression of claudin-1 and Zonula Occludens-1 (ZO-1) and the redistribution of occludin.110 Meanwhile, butyrate can reverse the abnormal expression of ZO-1 so that repairs the dysfunctional intestinal barrier and reduces LPS translocation, thereby inhibiting macrophage activation, proinflammatory cytokine production and neutrophil infiltration.111 Last but not least, SCFAs can directly regulate inflammation. Studies have shown that SCFAs suppress the production of pro-inflammatory factors, facilitate the repair of mucosal inflammation, and inhibit the inflammatory response in the colon.112,113 The anti-inflammatory effect of SCFAs is partly due to the inhibition of NF-κB activation by binding to G protein-coupled receptors 41 and 43 (GPR41 and GPR43) of host immune cells.114–117 Between them, GPR43 plays a more important role in mediating anti-inflammatory stimulation induced by acetic acid.118 In addition, studies have shown that the activation of Th17 and Treg cells is regulated by SCFAs. SCFAs reduce the secretion of pro-inflammatory factor IL-17 by inducing the decrease of Th17, while they increase the level of Treg cells in intestinal tract.119,120 Treg cells are key brain protection regulators in acute cerebral ischemia, secreting anti-inflammatory factor IL-10 and inhibiting the neurotoxicity of pro-inflammatory factors TNF-α and IFN-γ.121 By these ways, SCFAs inhibit the progress of ischemic stroke and alleviate inflammatory injury.
 |
Figure 2 A summary on how SCFAs arrest the inflammation to alleviate ischemic stroke. |
Since SCFAs have many benefits for maintaining normal intestinal function, the decrease of SCFAs caused by gut microbiota dysbiosis significantly affects the development of ischemic stroke. A fecal microbiota transplantation experiment showed that increasing SCFAs in elderly stroke mice improves the prognosis. In the experiment, the fecal transplantation tube feeding (FTG) method from young mice was used to reconstruct the microbial community in elderly mice after stroke. Younger fecal grafts contain higher levels of SCFAs and associated bacterial strains, including Bifidobacterium longum, Clostridium symbioticum, Clostridium pratense and Lactobacillus fermentum. The result showed that SCFAs producers increased SCFAs concentrations in intestine, brain and plasma of aged stroke mice, alleviating neurological deficits and inflammation after stroke and improving functional and behavioral outcomes. Basing on this result, the researchers suggested that the decrease of SCFAs from bacteria in the elderly microbial group seemed to be partly responsible for the enhancement of immune status and poor prognosis after stroke at least.122 Another prospective study on the spectrum of gut microbiota and feces SCFAs in Chinese AIS patients reported that gut microbiota in AIS patients (especially those with increased stroke severity) was significant imbalance, accompanied by the lack of SCFAs. In AIS patients with higher severity, the abundance of SCFAs-producing bacteria was lower, while the abundance of Lactobacillus, Akmania, Enterobacteriaceae and Porphyromonas was higher. Moreover, the decreased content of SCFAs, particularly acetate, was also significantly correlated with the increase in the risk of 90-day adverse functional results.123
In summary, we know that microbiota dysbiosis can lead to the proliferation of pathogenic bacteria, SCFAs decrease and TMAO increase, resulting in atherosclerosis, systemic inflammation, and platelet hyperresponsiveness, which directly contributes to the occurrence and development of ischemic stroke (Figure 3). In turn, ischemic stroke induces gut microbiota dysbiosis and impairs intestinal barrier function, leading to systemic inflammation, which aggravates the pathological process and symptoms of ischemic stroke and adversely affects its prognosis.
 |
Figure 3 Pathogenesis of ischemic stroke caused by gut microbiota dysbiosis. |
Gut Microbiota Dysbiosis and the Risk Factors for Ischemic Stroke
Gut Microbiota Dysbiosis Induces Obesity
Obesity is characterized by excessive adipose tissue, which occurs when there is an imbalance between energy intake and consumption.124 Evidences show that gut microbiota help to collect energy and increase host fat storage.125,126 The total fat of sterile mice is shown to be 40% less than that of normal mice, although their calorie intake was 29% more than that of normal-feed littermates.126 It is generally believed that changes in gut microbiota is one of the main causes of obesity.127–129 When compared with lean subjects, obese subjects show significantly lower diversity of gut microbiota, lower proportion of Bacteroidetes, and higher abundance of Firmicutes. Moreover, after dietary treatment, the relative abundance of Bacteroides increased, while that of Firmicutes decreased.130 Animal model also support this view. The number of Bacteroides in abnormal leptin gene homozygous mice (ob/ob) decreased by 50% compared with that in wild-type (+/+) or hybrid (ob/+) mice, and the number of Firmicutes increased proportionally (p < 0.05).130 Using bird-gun metagenomic sequencing technology to detect the cecum microbial DNA of mice with different degrees of obesity (ob/ob, ob/+, +/+), the result shows that compared with the lean mice, the ratio of Firmicutes to Bacteroidetes in obese mice increased.126 However, there are different results about the changes of Bacteroides and Firmicutes. A study reported that when compared with lean people, the proportion of Bacteroides in obese people is lower, the proportion of actinomycetes is higher, but there is no difference in the proportion of Firmicutes.131 Evaluating the gene expression profiles of gut microbiota in different subjects by using real-time PCR found the remarkably lower concentrations of Bacteroides in lean (p < 0.01) or anorexia (p < 0.05) subjects than those in obese subjects, while the concentrations of Firmicutes in three groups were quite similar.132 Other studies even found that the proportion of Bacteroides in overweight and obese subjects increased significantly.133,134 Besides Bacteroides and Firmicutes, other strains may also be involved in the occurrence of obesity, but opinions are still inconsistent. A prospective study found infants between three weeks and one year old with low levels of Staphylococcus and high levels of Bacteroides fragilis were correlated with high risk of developing obesity in future life.135 Moreover, a self-controlled study show that the level of Bifidobacterium of children, who become obese at the age of 10, is much lower they were in 3 months old (p = 0.087).136 Also, there is analysis showing that the composition of bacteria cultured in the obese group was remarkably lower than the normal weight group, mainly caused by Clostridium perfringens (p = 0.001) and Bacteroides (p = 0.012).137 But interestingly, it has been reported that the levels of Clostridium and Bifidobacterium in obese adolescents decreased significantly after receiving obesity treatment plans.138 The study of Methanoberebacter smithii (M. smithii) also yielded two opposite results. Studies found that compared with lean subjects, the proportion of M. smithii in obese subjects was low and the number was significantly decrease.134,139 However, Zhang et al140 found that M. smithii was more abundant in obese individuals than in lean individuals, which is consistent with Armougom’s finding.132
LPS leakage mediated inflammation caused by microbiota dysbiosis involves in the occurrence of obesity. It was found that there was a large amount of LPS in the blood circulation of mice fed with high-fat diet, causing endotoxemia, but when using antibiotics, no increase in LPS was found.141 On the one hand, LPS through combining with CD14, activate the TLR inflammation pathway.64 Infusion of LPS in mice fed with normal diet causes metabolic disorder that are similar to high-fat diet feeding mice, while the metabolic disorder caused by high-fat diet was alleviated in CD14 deficient mice. Further, it is observed that macrophage infiltration in adipose tissue increases in both wild-type mice fed with high-fat diet and mice injected with LPS, but no such phenomenon was observed in CD14 deficient mice.142 On the other hand, LPS triggers the expression of prostaglandin-endoperoxidase 2/ cyclooxygenase 2 (Ptgs2/COX2) mRNA, which leads to the increase of COX2 level and activity.143 COX2 is a pro-inflammatory enzyme that catalyzes the conversion of arachidonic acid to prostaglandin, while prostaglandin dilated vessels leading to the increase of lymphocyte migration and TLR4-dependent inflammation heavier, which is related to leptin resistance.144,145 These supported that microbiota dysbiosis through LPS mediated inflammation pathway affects obesity. In addition to LPS, the SCFAs level of obese individuals and lean individuals are also different. A study found that there exists a 20% higher average levels of total SCFAs in obese individuals than those of lean individuals (p = 0.024) by fecal samples detection.134 A molecular analysis experiment for Indian individuals also shown that SCFAs are significantly overproduced in obese subjects through microbial glycolysis.146 As a result, excessive SCFAs provide the host with additional energy substrate, and then contribute to the formation of lipid or glucose, leading to obesity.147 More directly, changes in gut microbiota composition in obesity reduce fasting-induced adipokines, a kind of circulation lipoprotein lipase inhibitor, which increase white fat storage.148
Obesity Increases the Risk of Developing Ischemic Stroke
Obesity, a complex disease concerned with genetic and environmental factors, is often correlated with the development of several chronic complications, such as hyperglycemia, hypertriglyceridemia, dyslipidemia and hypertension,149 and these obesity complications are also risk factors for ischemic stroke. In terms of obesity itself, studies have shown that central obesity increases the risk of ischemic stroke and other cardiovascular phenotypes.150,151 Compared with people of normal weight, obese people have about twice the risk of stroke.152 At present, the definition of obesity is mainly based on waist–hip ratio (WHR) and BMI, both show a strong correlation with the risk of ischemic stroke. Obesity (BMI > 30 kg/m2) was correlated with increased stroke risk (odds ratio, 1.57,95% CI = 1.28–1.94).153 WHR showed a stronger correlation than BMI.154 For every 10% increase in WHR, the risk of ischemic stroke in large arteries increases by 75% (95% CI = 44% − 113%), and the risk of ischemic stroke in small vessels increases by 57% (CI = 29% − 91%).155 In addition, obesity increases the risk of ischemic stroke by inducing complications such as type 2 diabetes mellitus (T2DM) and hypertension. About a tenth of the observed WHR effect was SBP-mediated ischemic stroke (12%; CI = 4% – 20%).155 Case-control study also reported that obesity is a risk factor for ischemic stroke in young adults, and that this correlation may be partially mediated by hypertension, diabetes and/or other variables correlated with these diseases.153
From these above, we can know that gut microbiota disturbance is involved in the occurrence of obesity through a variety of ways, increasing the risk of ischemic stroke (Figure 4). However, there is still controversy about the content change of specific strains in obesity. Even so, when compared with normal individuals, the content of these bacteria in obese patients is not normal and the common features, such as inflammation and SCFAs increase, are found in obese patients, which suggesting that gut microbiota dysbiosis do affect the occurrence of obesity, but the causes of different results need further study.
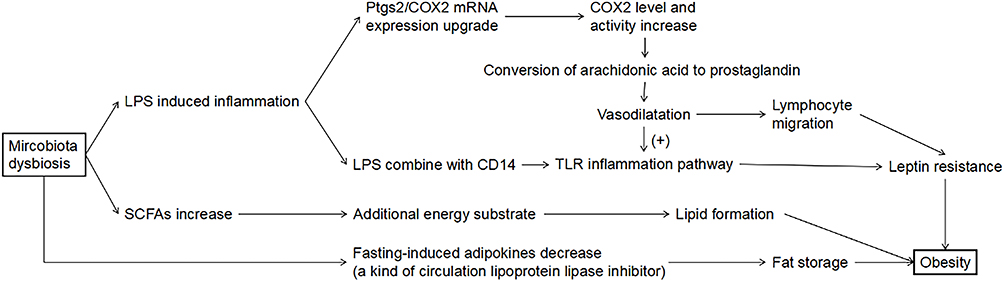 |
Figure 4 Pathogenesis of obesity caused by gut microbiota dysbiosis. |
Microbiota Dysbiosis Leads to T2DM
T2DM is distinguished by insulin resistance (IR) and hyperglycemia. It has been proved that microbiota dysbiosis are involved in the initiation and progression of T2DM.156 In T2DM rat models, researchers found that the abundance of Bacteroides decreased, while the abundance of Firmicutes increased, which means the ratio of Firmicutes/Bacteroides increased.157,158 Also, it has proved that the enhanced relative abundance of Firmicutes and Actinobacteria was associated with fasting blood glucose, while the decreased relative abundance of Bacteroides and Proteus was associated with fasting blood glucose.159 In the feces of patients with diabetes, lower Bacillus content and higher Enterococcus content were also found when compared with those of normal people.160,161 Further, it is reported that T2DM is related to the decreased abundance of butyrate-producing bacteria and the increased abundance of Lactobacillus.156,162,163 Consistently, the decreased percentage of butyrate-producing bacteria in T2DM patients, including Roseburia and Faecalibacterium prausnitzii, has been found when compared with healthy subjects.164,165 Decreasing the level of butyrate-producing bacteria in patients with metabolic syndrome by using vancomycin results in the reduced of insulin sensitivity,166 which has a negative effect on T2DM.
As an inflammation-related disease, diabetes is affected by intestinal disorder-induced inflammation. Intestinal disorders cause increased abundance of opportunistic pathogens, including Gram-negative bacteria, resulting in increased release of LPS, which follows that LPS directly impairs the intestinal barrier and triggers the release of pro-inflammatory cytokines, inducing inflammatory response through TLR signaling pathway.59 Inflammation destroys the structure and function of endothelial cells, leading to the imbalance of insulin transport, thereby inducing IR.167,168 In addition, the activation of mitogen-activated protein kinase (MAPK) signaling pathway inhibits the activation of insulin receptor substrate, inducing the death of β cell and IR.169 Besides LPS leakage, diabetic patients showed low SCFAs content and the proportion of SCFAs-producing bacteria,170 while the decrease of SCFAs attributes to the reduce amount of islet cells and the decreased sensitivity of insulin, which leads to IR.171 It has been mentioned above that SCFAs maintain the normal function of intestinal barrier and regulate Th17 and Treg cells.119,120 The decrease of SCFAs makes the intestinal barrier lack of protection, and reduces the intestinal anti-inflammatory ability, which promote the occurrence and development of IR. Experiments have shown that increasing Treg cells reduces the infiltration of macrophages in adipose tissue, which is important for alleviating IR.172 In addition, the damage to the diversity and amount of SCFAs-producing bacteria in the gut increases the glycated hemoglobin (HbA1c) level in T2DM patients,173 a kind of protein that represents the average plasma glucose levels over the past 2 to 3 months,174 suggesting that the poor glycemic control.
It is worth noting that T2DM, as a complication of obesity, is also affected by obesity to some extent.175,176 Obesity individuals are usually characterized by IR, which is related to low-grade subclinical inflammation leading to hyperglycemia and is conducive to the occurrence of T2DM.177 Studies have shown that when compared with healthy people, obese people with IR are distinguished by changed composition of gut microbiota, especially the increased ratio of Firmicutes/Bacteroidetes.178,179 Moreover, it is reported that the percentage of Akkermansia muciniphila decreases in obesity, and reconstruct this kind of bacterial improves insulin function and glucose tolerance.180 Animal study also has some findings. Sterile mice transplanted with gut microbiota of obese mice (ob/ob) increased obesity related to IR.126 Transplanting fecal microbiota from lean individuals to IR patients with metabolic syndrome showed that insulin sensitivity of IR patients was improved, related to the increase number of butyrate-producing bacteria.181 IR in obesity induced by high-fat diet is also largely affected by gut microbiota. Obesity caused by high-fat diets increases the proportion of LPS-containing microbial populations and plasma LPS levels, leading to endotoxemia-induced IR.142,182 An experiment proved that the loss of Toll-4 receptors prevents IR induced by high-fat diet,183 showing that inflammation, obesity and IR exist certain relation. Another study found that when treating diet-induced obese mice with norfloxacin and ampicillin, blood glucose was controlled accompanied by the decreased level of TNF-α in jejunum,184 indicating that the regulation of antibiotics on gut microbiota reduces inflammation and increase glucose tolerance in obese mice. In addition, the stimulation of extracellular signal-regulated kinases 1 and 2, IκB kinase complex and c-Jun N-terminal kinases reduces the tyrosine phosphorylation of insulin receptor substrate protein in inflammatory regions of obese individuals, resulting in weakening insulin signaling.185 These examples show that there is a causal relationship between microbial groups, obesity and IR.
Diabetes is a Major Risk Factor for Ischemic Stroke
Animal and clinical studies have shown that diabetes is an important independent risk factor for stroke.186–189 Compared with non-diabetic patients, diabetic patients are more prone to stroke. Studies estimate that about 20–33% of hospitalized patients with acute stroke have diabetes.190–192 Compared with hemorrhagic stroke, ischemic stroke is more associated with diabetes.193,194 Moreover, the size of cerebral infarction is larger, and mortality and disability rate are higher in patients with hyperglycemia or diabetes after stroke.195,196 In addition, study finds that the level of HbA1c can predict the risk of stroke,197 and there is a correlation between elevated HbA1c and poor clinical outcomes after stroke.198
Diabetes is distinguished by IR and hyperglycemia, both of which leads to increased expression and secretion of plasminogen activator inhibitor-1 (PAI-1), while PAI-1 inhibits fibrinolysis by inhibiting plasminogen activator, causing coagulation-fibrinolysis imbalance.199 Besides, through IR and hyperglycemia, diabetes also leads to vascular endothelial dysfunction and platelet hyperresponsiveness, thereby, diabetic patients are usually in prethrombotic state,200 which increases the risk of ischemic stroke caused by thromboembolism. In addition to prethrombotic state, hyperglycemia leads to revascularization damage, reperfusion reduce, reperfusion injury increase and direct tissue damage.201 Animal studies reported that hyperglycemia expands the volume of cerebral infarction and attributes to hemorrhagic transformation of infarction region, resulting in worse prognosis.202,203 The negative prognosis is largely due to the adverse effects of hyperglycemia on multiple components involved in the development of stroke. First, hyperglycemia inhibits immune response and tissue repair during reperfusion by inhibiting microglia activation and proliferation whose activation has been shown to regulate the proliferation of brain endothelial cells.204,205 Second, hyperglycemia prolonged the duration of astrocytes activation.205 The activated astrocytes promote neuronal survival and plasticity following ischemia by releasing nutrients and extracellular matrix molecules.206–208 Also, the persistent activation of astrocytes form glial scar tissue around the infarct cerebral tissue, which provides a physical barrier for axon growth, thereby inhibiting the recovery of neurological function after stroke.209,210 Third, hyperglycemia induces an abnormal increase in the number of neutrophils in the cerebral membrane after ischemia.205 Neutrophils lead to increased production of free radicals, increased neuroinflammation, destruction of BBB and hemorrhagic transformation after stroke.211 Most importantly, hyperglycemia directly disrupting the BBB component, including inhibition of TJP and damage to basement membrane, explains why the transition from ischemic stroke to hemorrhagic stroke increases in animal models of stroke and clinical stroke patients.212,213
Thus, we can see that intestinal disorders induces hyperglycemia (Figure 5), thereby increasing the risk of ischemic stroke and aggravating ischemic stroke damage. Moreover, diabetes as a complication of obesity, obesity indirectly affects the occurrence and development of ischemic stroke by affecting the progress of diabetes.
 |
Figure 5 Pathogenesis of T2DM directly caused by gut microbiota dysbiosis. |
Microbiota Dysbiosis Affects Hypertension
When compared the gut microbiota composition of Wistar-Kyoto rats (WKY) with spontaneously hypertensive rats (SHR) and chronic angiotensin II perfusion hypertensive rats (Ang II), the abundance, diversity and uniformity of gut microbiota in SHR and Ang II rats were lower, with the significant increase of Firmicutes to Bacteroidetes ratio and lactobacillus but the significant reduce of acetate-producing and butyrate-producing bacteria, and the decreased abundance and diversity of gut microbiota were also found in hypertensive patients. Besides, this experiment also found that the use of broad-spectrum antibiotic minocycline lowers the blood pressure of Ang II rats and reverses the gut microbiota dysbiosis mentioned above.214 Further, rats experiment showed that the blood pressure decreased in SHR when the gut microbiota in normal blood pressure rats was transplanted to them by oral gavage. In turn, the blood pressure of normal blood pressure rats increased when the gut microbiota in stroke prone SHR was transplanted to them by oral gavage accompanied by a significant increased abundance of Firmicutes and a significant decreased abundance of Bacteroidetes.215 Other animal studies showed that the gut microbiota in the hypertensive animal model group was significantly different from that in the control group, and these differences include low abundance of SCFAs-producing bacteria, high abundance of lactic acid-producing bacteria, low abundance of Bacteroides, and high abundance of Proteobacteria and Cyanobacteria.214,216
As a disease related to chronic inflammation,217 hypertension can be affected by inflammation mediated by intestinal LSP leakage. An experiment showed that LPS application to rats increases heart rate and norepinephrine level, reduces pressure reflex sensitivity, and increases neuroinflammation, such as TLR and TNF-α expression in PVN, a region playing a key role in blood pressure regulation.218 In addition to the effect of LPS, SCFAs can combine with orphan G protein-coupled receptors 41 (Gpr41) and olfactory receptor 78 (Olfr78) to control the rise and fall of blood pressure. Between them, Olfr78 is affected by propionic acid and acetate, inducing the release of renin and increasing blood pressure, while Gpr41 is affected by propionate, reducing blood pressure.219,220 Generally, in healthy gut, the effect of SCFAs is more likely to lower blood pressure.221–223 Besides, as one of SCFAs, butyrate inhibits histone deacetylase (HDAC) to exert anti-inflammatory effect, which is also effective in lowering blood pressure.224,225 Studies have shown that the activation of HDAC is associated with hypertension in SHR, and using butyrate reduces blood pressure and renal inflammation by inhibiting HDAC in mice.226,227 Also, in the intervention tests of patients with metabolic syndrome, the effect of butyrate on lowering blood pressure was observed.228,229 Therefore, the decrease of SCFAs-producing bacteria could be one of the reasons of developing hypertension.214
In addition, gut microbiota dysbiosis attributes to high-salt diet induced hypertension. The positive correlation between high-salt diet and hypertension has been supported by many epidemiological studies around the world.230 Among which, a study showed that high-salt diet affected the gut microbiota of mice, significantly reducing the abundance of Lactobacillus and their tryptophan metabolite indole-3-lactic acid, and increasing the number of Th17 cells in the lamina propria of the small intestine, spleen and spinal cord, which promote the inflammation. In turn, supplementary Lactobacillus in mice fed with high-salt diet increases the content of indole-3-lactic acid, reducing the blood pressure and the number of Th17 in small intestine colon and spleen. Similar phenomenon is also observed in human.231 Another study showed that high-salt diet breaks the immune balance of the gut, which was manifested as increasing Th17 cells and their reactivity in the intestinal lamina propria of mice, reducing the IL-10 secretion level of Treg cells and their inhibitory effect on T cell proliferation, worsening the inflammation.232
Hypertension is a High-Risk Factor for Ischemic Stroke
Stroke is a disease affected by multiple factors, and hypertension is the primary risk factor for stroke.233 The incidence of stroke was proportional to systolic blood pressure and diastolic blood pressure, and the relative risk of stroke increased 3.1 times in males and 2.9 times in females.234,235 In women with preeclampsia (a common pregnancy-induced hypertension), it is found to be 4–5 times higher the long-term risk for stroke.236–238 Further, reducing the blood pressure of hypertensive patients is found to be a very effective in preventing ischemic stroke, because for every 10 mmHg decrease in systolic blood pressure and 5 mmHg decrease in diastolic blood pressure, the stroke risk can be reduced by 41% (95% CI: 33%–48%).239 Hypertension also worsens stroke results. Compared with patients with normal blood pressure, patients with hypertension in the past had a smaller amount of recoverable tissue and larger infarction.240–243 There are researchers who have reported that hypertension affects the intestinal barrier and increase intestinal permeability,244 which contribute to the systemic inflammatory response mediated by LSP leakage. Other researchers have found that high-salt diet induces the multiplication of Th17 lymphocytes in the small intestine of mice, and IL-17 in the plasma is increased. IL-17 in the circulatory system reduces NO by inhibiting the phosphorylation of NO synthase in endothelial cells through Rho kinase pathway, promoting neurovascular injury and cognitive dysfunction.245
Therefore, we can know that gut microbiota dysbiosis, especially the increase in the ratio of Firmicutes to Bacteroides and the decrease of Lactobacillus, are major features of hypertension. Hypertension induced by gut microbiota dysbiosis through inflammation, immune and anabolism, directly increases the risk of ischemic stroke and deteriorates the outcome of ischemic stroke (Figure 6).
 |
Figure 6 Pathogenesis of hypertension caused by gut microbiota dysbiosis. |
Discussion
From the above expound, we can know that gut microbiota dysbiosis directly leads to ischemic stroke by inducing inflammation, atherosclerosis and thromboembolism, through increasing TMAO, reducing SCFAs and LPS leakage, and has a negative impact on the outcome of ischemic stroke. Moreover, gut microbiota dysbiosis is also involved in the occurrence and development of high-risk factors for ischemic stroke such as obesity, T2DM and hypertension, which increases the risk of developing ischemic stroke. Also, it is shown that the ratio of Firmicutes to Bacteroides increase and the LPS-induced inflammation are the common feature of obesity, T2DM and hypertension caused by gut microbiota dysbiosis.
In view of the above facts, therapeutic intervention measures for ischemic stroke target on regulating gut microbiota are feasible.246 By regulating gut microbiota, alleviates inflammation, atherosclerosis and thrombosis, to prevent the occurrence of ischemic stroke. Also, by intervening and regulating gut microbiota, alleviates obesity, diabetes and hypertension to reduce the risk of developing ischemic stroke. Even after ischemic stroke, gut microbiota can be adjusted to promote recovery and improve prognosis. A mice experiment published in Nature Medicine found that the volume of cerebral infarction in the antibiotic treatment group was 60% less than that in the control group after experimental ischemic stroke. The use of antibiotic reduces diversity of gut microbiota, mainly including the retraction of Clostridiaceae (a kind of Firmicutes) and S24-7 spp (a kind of Bacteroidetes), and the increase of Proteobacteria. Moreover, transplanting the gut microbiota of the antibiotic treatment group into the control group significantly reduces the volume of cerebral infarction after ischemic stroke.18 Also, an experiment reported that improving intestinal barrier function is helpful to the recovery of BBB function after ischemic stroke.101
In addition, the intestinal regulatory therapy for high-risk factors for ischemic stroke has shown a significant effect. Supplementing probiotics as a convenient method has shown high efficacy in alleviating obesity, hyperglycemia and hypertension. In adults, probiotic supplement including strains of Lactobacillus and Bifidobacterium, alone or in combination, as well as Pediococcus pentosaceus resulted in significant reductions in body weight, waist circumference, BMI and fat mass.247–252 Probiotic therapy of Lactobacillus paracasei reduced and reversed LPS-related pathological effects in rodents, especially alleviated T2DM-related β-cell dysfunction.253 Intervention with Lactobacillus casei in diabetic mice for 15 days found a sharp decline in blood glucose levels.8 Supplementing Lactobacillus rhamnosus GG for 2 months to streptozotocin-induced diabetic rats improves the glucose tolerance and decreases the level of HbA1c significantly.254 Also, adding Lactobacillus to high-salt diet mice increases the content of indole-3-lactic acid, lowers the blood pressure and reduces the amount Th17 in intestine, colon and spleen.231 As a more thorough method, fecal microbial transplantation also shows positive effect. Transplantation of fecal microbiota from obese, steatosis or T2DM patients partially replicate the donors’ metabolic phenotype in mice recipients.126,179,255,256 Also, the transplantation of fecal microbiota from lean individuals to IR patients with metabolic syndrome improved insulin sensitivity.181 Moreover, diabetic patients receiving fecal microbial transplantation showed stable blood glucose.257 All these above supports that preventing and treating ischemic stroke by regulating gut microbiota through supplementing probiotics or FMT could be a practicable way.
Conclusion
In this review, we have discussed the relationships among gut microbiota, ischemic stroke and the high-risk factors for ischemic stroke, pointing that regulating gut microbiota could be a feasible way to prevent and treat ischemic stroke. By reconstructing gut microbiota, we can alleviate the high-risk factors for ischemic stroke, such as obesity, diabetes and hypertension, and inhibit the inflammation and vascular embolization, lowering the risk of developing ischemic stroke to a great extent. Moreover, reconstructing gut microbiota after ischemic stroke can also arrest the progression of the disease and improve the prognosis. However, the consist of gut microbiota is so complex that we cannot totally figure out what exactly to do to prevent the gut microbiota dysbiosis and reform the gut microbiota thoroughly, and these aspects need further exploring. For now, supplementing probiotics or FMT can be a good way for some extent.
Consent for Publication
All authors have read and approved the manuscript.
Acknowledgments
This work was financially supported by Guangzhou Science and Technology Plan Joint Foundation of City and University (202102010085).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work is supported by grants 20212137 from Traditional Chinese Medicine Bureau of Guangdong Province, grants 20211A010077 from the Guangzhou Municipal Health Commission.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cassidy JM, Cramer SC. Spontaneous and therapeutic-induced mechanisms of functional recovery after stroke. Transl Stroke Res. 2017;8(1):33–46. doi:10.1007/s12975-016-0467-5
2. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi:10.1161/CIR.0b013e31823ac046
3. Rammal SA, Almekhlafi MA. Diabetes mellitus and stroke in the Arab countries. J Taibah Univ Med Sci. 2016;11:295–300. doi:10.1016/j.jtumed.2016.05.001
4. Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30(8):783–793. doi:10.1179/174313208X341085
5. McColl BW, Allan SM, Rothwell NJ. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. 2009;158(3):1049–1061. doi:10.1016/j.neuroscience.2008.08.019
6. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25(1):4–7. doi:10.1016/j.it.2003.10.013
7. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. doi:10.1038/nri1882
8. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi:10.1126/science.1110591
9. Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. doi:10.1126/science.1124234
10. Houlden A, Goldrick M, Brough D, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016;57:10–20. doi:10.1016/j.bbi.2016.04.003
11. Barugh AJ, Gray P, Shenkin SD, MacLullich AM, Mead GE. Cortisol levels and the severity and outcomes of acute stroke: a systematic review. J Neurol. 2014;261(3):533–545. doi:10.1007/s00415-013-7231-5
12. Diaz Heijtz R, Wang S, Anuar F, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A. 2011;108(7):3047–3052. doi:10.1073/pnas.1010529108
13. Jenkins TA, Nguyen JC, Polglaze KE, Bertrand PP. Influence of tryptophan and serotonin on mood and cognition with a possible role of the gut-brain axis. Nutrients. 2016;8(1):56. doi:10.3390/nu8010056
14. Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34(46):15490–15496. doi:10.1523/JNEUROSCI.3299-14.2014
15. Schmidt C. Mental health: thinking from the gut. Nature. 2015;518(7540):S12–15. doi:10.1038/518S13a
16. Smith PA. The tantalizing links between gut microbes and the brain. Nature. 2015;526(7573):312–314. doi:10.1038/526312a
17. Roth WH, Cai A, Zhang C, Chen ML, Merkler AE, Kamel H. Gastrointestinal Disorders and Risk of First-Ever Ischemic Stroke. Stroke. 2020;51(12):3577–3583. doi:10.1161/STROKEAHA.120.030643
18. Benakis C, Brea D, Caballero S, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22(5):516–523. doi:10.1038/nm.4068
19. Xu K, Gao X, Xia G, et al. Rapid gut dysbiosis induced by stroke exacerbates brain infarction in turn. Gut. 2021;70(8):1486–1494. doi:10.1136/gutjnl-2020-323263
20. Esenwa CC, Elkind MS. Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol. 2016;12(10):594–604. doi:10.1038/nrneurol.2016.125
21. Markstad H, Edsfeldt A, Yao Mattison I, et al. High levels of soluble lectinlike oxidized low-density lipoprotein receptor-1 are associated with carotid plaque inflammation and increased risk of ischemic stroke. J Am Heart Assoc. 2019;8(4):e009874. doi:10.1161/JAHA.118.009874
22. Skarpengland T, Skjelland M, Kong XY, et al. Increased Levels of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 in Ischemic Stroke and Transient Ischemic Attack. J Am Heart Assoc. 2018;7(2). doi:10.1161/JAHA.117.006479.
23. Warlow C, Sudlow C, Dennis M, Wardlaw J, Sandercock P. Stroke. Lancet. 2003;362(9391):1211–1224. doi:10.1016/S0140-6736(03)14544-8
24. Parikh NS, Merkler AE, Iadecola C. Inflammation, Autoimmunity, Infection, and Stroke: epidemiology and Lessons From Therapeutic Intervention. Stroke. 2020;51(3):711–718. doi:10.1161/STROKEAHA.119.024157
25. Buck BH, Liebeskind DS, Saver JL, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39(2):355–360. doi:10.1161/STROKEAHA.107.490128
26. Gerhard A, Neumaier B, Elitok E, et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11(13):2957–2960. doi:10.1097/00001756-200009110-00025
27. Lindsberg PJ, Carpén O, Paetau A, Karjalainen-Lindsberg ML, Kaste M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation. 1996;94(5):939–945. doi:10.1161/01.CIR.94.5.939
28. Price CJ, Menon DK, Peters AM, et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke. 2004;35(7):1659–1664. doi:10.1161/01.STR.0000130592.71028.92
29. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183(1):25–33. doi:10.1016/S0014-4886(03)00082-7
30. Tanaka R, Komine-Kobayashi M, Mochizuki H, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117(3):531–539. doi:10.1016/S0306-4522(02)00954-5
31. Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932(1–2):110–119. doi:10.1016/S0006-8993(02)02292-8
32. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. doi:10.1189/jlb.1109766
33. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. Febs j. 2009;276(1):13–26. doi:10.1111/j.1742-4658.2008.06766.x
34. Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. 2006;18(1–2):145–157. doi:10.1615/CritRevNeurobiol.v18.i1-2.150
35. Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7(1):111–118. doi:10.1002/glia.440070117
36. Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28(6):1233–1244. doi:10.1161/01.STR.28.6.1233
37. Rothwell N, Allan S, Toulmond S. The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100(11):2648–2652. doi:10.1172/JCI119808
38. Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995–1006. doi:10.1016/j.neuroscience.2008.07.007
39. Wang X. Investigational anti-inflammatory agents for the treatment of ischaemic brain injury. Expert Opin Investig Drugs. 2005;14(4):393–409. doi:10.1517/13543784.14.4.393
40. Khan M, Sekhon B, Giri S, et al. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25(2):177–192. doi:10.1038/sj.jcbfm.9600012
41. Chopp M, Zhang RL, Chen H, Li Y, Jiang N, Rusche JR. Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke. 1994;25(4):
42. Prestigiacomo CJ, Kim SC, Connolly ES, Liao H, Yan SF, Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke. 1999;30(5):1110–1117. doi:10.1161/01.STR.30.5.1110
43. Zhang L, Zhang ZG, Zhang RL, Lu M, Krams M, Chopp M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke. 2003;34(7):1790–1795. doi:10.1161/01.STR.0000077016.55891.2E
44. Zhang RL, Chopp M, Jiang N, et al. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26(8):
45. Palasik W, Fiszer U, Lechowicz W, Czartoryska B, Krzesiewicz M, Lugowska A. Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur Neurol. 2005;53(4):188–193. doi:10.1159/000086355
46. Smith CJ, Emsley HC, Vail A, et al. Variability of the systemic acute phase response after ischemic stroke. J Neurol Sci. 2006;251(1–2):77–81. doi:10.1016/j.jns.2006.09.011
47. McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28(38):9451–9462. doi:10.1523/JNEUROSCI.2674-08.2008
48. Akiyoshi R, Wake H, Kato D, et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro. 2018;5(5):
49. Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109(4):E197–205. doi:10.1073/pnas.1111098109
50. Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW. Ischemia-induced neuronal expression of the microglia attracting chemokine Secondary Lymphoid-tissue Chemokine (SLC). Glia. 2001;34(2):121–133. doi:10.1002/glia.1047
51. Chen Z, Jalabi W, Hu W, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun. 2014;5:4486. doi:10.1038/ncomms5486
52. Li N, Wang X, Sun C, et al. Change of intestinal microbiota in cerebral ischemic stroke patients. BMC Microbiol. 2019;19(1):191. doi:10.1186/s12866-019-1552-1
53. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi:10.1038/nm.2399
54. Santos Samary C, Pelosi P, Leme Silva P, Rieken Macedo Rocco P. Immunomodulation after ischemic stroke: potential mechanisms and implications for therapy. Crit Care. 2016;20(1):391. doi:10.1186/s13054-016-1573-1
55. Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–455. doi:10.1038/nature12726
56. Xia GH, You C, Gao XX, et al. Stroke Dysbiosis Index (SDI) in Gut Microbiome Are Associated With Brain Injury and Prognosis of Stroke. Front Neurol. 2019;10:397. doi:10.3389/fneur.2019.00397
57. Yin J, Liao SX, He Y, et al. Dysbiosis of Gut Microbiota With Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. J Am Heart Assoc. 2015;4(11). doi:10.1161/JAHA.115.002699.
58. Zeng X, Gao X, Peng Y, et al. Higher Risk of Stroke Is Correlated With Increased Opportunistic Pathogen Load and Reduced Levels of Butyrate-Producing Bacteria in the Gut. Front Cell Infect Microbiol. 2019;9:4. doi:10.3389/fcimb.2019.00004
59. Gupta S, Maratha A, Siednienko J, et al. Analysis of inflammatory cytokine and TLR expression levels in Type 2 Diabetes with complications. Sci Rep. 2017;7(1):7633. doi:10.1038/s41598-017-07230-8
60. Brandsma E, Kloosterhuis NJ, Koster M, et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ Res. 2019;124(1):94–100. doi:10.1161/CIRCRESAHA.118.313234
61. Ghosh SS, Wang J, Yannie PJ, Ghosh S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J Endocr Soc. 2020;4(2):bvz039. doi:10.1210/jendso/bvz039
62. Kelly JR, Kennedy PJ, Cryan JF, Dinan TG, Clarke G, Hyland NP. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci. 2015;9:392. doi:10.3389/fncel.2015.00392
63. Cani PD, Neyrinck AM, Fava F, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50(11):2374–2383. doi:10.1007/s00125-007-0791-0
64. Janssen AW, Kersten S. Potential mediators linking gut bacteria to metabolic health: a critical view. J Physiol. 2017;595(2):477–487. doi:10.1113/JP272476
65. Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3(6):637–682. doi:10.3390/nu3060637
66. Abraham C, Cho J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm Bowel Dis. 2009;15(7):1090–1100. doi:10.1002/ibd.20894
67. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–323. doi:10.1038/nri2515
68. Keller JJ, Wang J, Hwang YL, et al. Increased risk of stroke among patients with Crohn’s disease: a population-based matched cohort study. Int J Colorectal Dis. 2015;30(5):645–653. doi:10.1007/s00384-015-2132-y
69. Singh S, Kullo IJ, Pardi DS, Loftus EV. Epidemiology, risk factors and management of cardiovascular diseases in IBD. Nat Rev Gastroenterol Hepatol. 2015;12(1):26–35. doi:10.1038/nrgastro.2014.202
70. Kasahara K, Tanoue T, Yamashita T, et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res. 2017;58(3):519–528. doi:10.1194/jlr.M072165
71. Stepankova R, Tonar Z, Bartova J, et al. Absence of microbiota (germ-free conditions) accelerates the atherosclerosis in ApoE-deficient mice fed standard low cholesterol diet. J Atheroscler Thromb. 2010;17(8):796–804. doi:10.5551/jat.3285
72. Prinz I, Silva-Santos B, Pennington DJ. Functional development of γδ T cells. Eur J Immunol. 2013;43(8):1988–1994. doi:10.1002/eji.201343759
73. Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793–3802. doi:10.1182/blood-2012-02-412726
74. Shichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15(8):946–950. doi:10.1038/nm.1999
75. Aron-Wisnewsky J, Clement K. Dietary choline derived TMAO: new role in thrombosis. Ame Medical Journal. 2017;2:112. doi:10.21037/amj.2017.07.10
76. Bennett BJ, de Aguiar Vallim TQ, Wang Z, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49–60. doi:10.1016/j.cmet.2012.12.011
77. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi:10.1038/nature09922
78. Warrier M, Shih DM, Burrows AC, et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015;10(3):326–338. doi:10.1016/j.celrep.2014.12.036
79. Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–1584. doi:10.1056/NEJMoa1109400
80. Nam HS. Gut Microbiota and Ischemic Stroke: the Role of Trimethylamine N-Oxide. J Stroke. 2019;21(2):151–159. doi:10.5853/jos.2019.00472
81. Sharma V, Sharma V, Shahjouei S, et al. At the Intersection of Gut Microbiome and Stroke: a Systematic Review of the Literature. Front Neurol. 2021;12:729399. doi:10.3389/fneur.2021.729399
82. Haghikia A, Li XS, Liman TG, et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients With Stroke and Is Related to Proinflammatory Monocytes. Arterioscler Thromb Vasc Biol. 2018;38(9):2225–2235. doi:10.1161/ATVBAHA.118.311023
83. Schiattarella GG, Sannino A, Toscano E, et al. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J. 2017;38(39):2948–2956. doi:10.1093/eurheartj/ehx342
84. Senthong V, Wang Z, Li XS, et al. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: the Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J Am Heart Assoc. 2016;5(6). doi:10.1161/JAHA.115.002816.
85. Tang WH, Wang Z, Fan Y, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64(18):1908–1914. doi:10.1016/j.jacc.2014.02.617
86. Tang WH, Wang Z, Shrestha K, et al. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21(2):91–96. doi:10.1016/j.cardfail.2014.11.006
87. Komaroff AL. The Microbiome and Risk for Atherosclerosis. JAMA. 2018;319(23):2381–2382. doi:10.1001/jama.2018.5240
88. Al-Obaide MAI, Singh R, Datta P, et al. Gut Microbiota-Dependent Trimethylamine-N-oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J Clin Med. 2017;6(9):86. doi:10.3390/jcm6090086
89. Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi:10.1038/nm.3145
90. Ma G, Pan B, Chen Y, et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci Rep. 2017;37(2). doi:10.1042/BSR20160244.
91. Wang Z, Roberts AB, Buffa JA, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163(7):1585–1595. doi:10.1016/j.cell.2015.11.055
92. Rohrmann S, Linseisen J, Allenspach M, von Eckardstein A, Müller D. Plasma Concentrations of Trimethylamine-N-oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J Nutr. 2016;146(2):283–289. doi:10.3945/jn.115.220103
93. Seldin MM, Meng Y, Qi H, et al. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J Am Heart Assoc. 2016;5(2). doi:10.1161/JAHA.115.002767.
94. Sun X, Jiao X, Ma Y, et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res Commun. 2016;481(1–2):63–70. doi:10.1016/j.bbrc.2016.11.017
95. Wu C, Li C, Zhao W, et al. Elevated trimethylamine N-oxide related to ischemic brain lesions after carotid artery stenting. Neurology. 2018;90(15):e1283–e1290. doi:10.1212/WNL.0000000000005298
96. Zhu W, Gregory JC, Org E, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165(1):111–124. doi:10.1016/j.cell.2016.02.011
97. Frossard M, Fuchs I, Leitner JM, et al. Platelet function predicts myocardial damage in patients with acute myocardial infarction. Circulation. 2004;110(11):1392–1397. doi:10.1161/01.CIR.0000141575.92958.9C
98. Tantry US, Bonello L, Aradi D, et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62(24):2261–2273. doi:10.1016/j.jacc.2013.07.101
99. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28(10):1221–1227. doi:10.1136/gut.28.10.1221
100. Macfarlane GT, Macfarlane S. Fermentation in the human large intestine: its physiologic consequences and the potential contribution of prebiotics. J Clin Gastroenterol. 2011;45(Suppl):S120–127. doi:10.1097/MCG.0b013e31822fecfe
101. Chen R, Wu P, Cai Z, et al. Puerariae Lobatae Radix with chuanxiong Rhizoma for treatment of cerebral ischemic stroke by remodeling gut microbiota to regulate the brain-gut barriers. J Nutr Biochem. 2019;65:101–114. doi:10.1016/j.jnutbio.2018.12.004
102. Chen Y, Liang J, Ouyang F, et al. Persistence of Gut Microbiota Dysbiosis and Chronic Systemic Inflammation After Cerebral Infarction in Cynomolgus Monkeys. Front Neurol. 2019;10:661. doi:10.3389/fneur.2019.00661
103. Chambers ES, Viardot A, Psichas A, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. 2015;64(11):1744–1754. doi:10.1136/gutjnl-2014-307913
104. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to Host Physiology: short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165(6):1332–1345. doi:10.1016/j.cell.2016.05.041
105. Sawicki CM, Livingston KA, Obin M, Roberts SB, Chung M, McKeown NM. Dietary Fiber and the Human Gut Microbiota: application of Evidence Mapping Methodology. Nutrients. 2017;9(2):125. doi:10.3390/nu9020125
106. Tolhurst G, Heffron H, Lam YS, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61(2):364–371. doi:10.2337/db11-1019
107. Ardawi MS, Newsholme EA. Fuel utilization in colonocytes of the rat. Biochem J. 1985;231(3):713–719. doi:10.1042/bj2310713
108. Roediger WE. Short chain fatty acids as metabolic regulators of ion absorption in the colon. Acta Vet Scand Suppl. 1989;86:116–125.
109. Barcelo A, Claustre J, Moro F, Chayvialle JA, Cuber JC, Plaisancié P. Mucin secretion is modulated by luminal factors in the isolated vascularly perfused rat colon. Gut. 2000;46(2):218–224. doi:10.1136/gut.46.2.218
110. Wang HB, Wang PY, Wang X, Wan YL, Liu YC. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig Dis Sci. 2012;57(12):3126–3135. doi:10.1007/s10620-012-2259-4
111. Liu B, Qian J, Wang Q, Wang F, Ma Z, Qiao Y. Butyrate protects rat liver against total hepatic ischemia reperfusion injury with bowel congestion. PLoS One. 2014;9(8):e106184. doi:10.1371/journal.pone.0106184
112. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–119. doi:10.1111/j.1365-2036.2007.03562.x
113. Sivaprakasam S, Prasad PD, Singh N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol Ther. 2016;164:144–151. doi:10.1016/j.pharmthera.2016.04.007
114. Al-Lahham SH, Roelofsen H, Priebe M, et al. Regulation of adipokine production in human adipose tissue by propionic acid. Eur J Clin Invest. 2010;40(5):401–407. doi:10.1111/j.1365-2362.2010.02278.x
115. Fukae J, Amasaki Y, Yamashita Y, et al. Butyrate suppresses tumor necrosis factor alpha production by regulating specific messenger RNA degradation mediated through a cis-acting AU-rich element. Arthritis Rheum. 2005;52(9):2697–2707. doi:10.1002/art.21258
116. Säemann MD, Böhmig GA, Osterreicher CH, et al. Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB j. 2000;14(15):2380–2382. doi:10.1096/fj.00-0359fje
117. Tedelind S, Westberg F, Kjerrulf M, Vidal A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J Gastroenterol. 2007;13(20):2826–2832. doi:10.3748/wjg.v13.i20.2826
118. Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–1286. doi:10.1038/nature08530
119. Haase S, Haghikia A, Wilck N, Müller DN, Linker RA. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology. 2018;154(2):230–238. doi:10.1111/imm.12933
120. Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol. 2017;52(1):1–8. doi:10.1007/s00535-016-1242-9
121. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117(4):433–442. doi:10.1111/j.1365-2567.2006.02321.x
122. Lee J, d’Aigle J, Atadja L, et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ Res. 2020;127(4):453–465. doi:10.1161/CIRCRESAHA.119.316448
123. Tan C, Wu Q, Wang H, et al. Dysbiosis of Gut Microbiota and Short-Chain Fatty Acids in Acute Ischemic Stroke and the Subsequent Risk for Poor Functional Outcomes. JPEN J Parenter Enteral Nutr. 2021;45(3):518–529. doi:10.1002/jpen.1861
124. WHO. Obesity and Overweight. WHO; 2015.
125. Samuel BS, Shaito A, Motoike T, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105(43):16767–16772. doi:10.1073/pnas.0808567105
126. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi:10.1038/nature05414
127. Castaner O, Goday A, Park YM, et al. The Gut Microbiome Profile in Obesity: a Systematic Review. Int J Endocrinol. 2018;2018:4095789. doi:10.1155/2018/4095789
128. Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol. 2014;20(43):16079–16094. doi:10.3748/wjg.v20.i43.16079
129. Kobyliak N, Virchenko O, Falalyeyeva T. Pathophysiological role of host microbiota in the development of obesity. Nutr J. 2016;15:43. doi:10.1186/s12937-016-0166-9
130. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. doi:10.1073/pnas.0504978102
131. Turnbaugh PJ, Quince C, Faith JJ, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci U S A. 2010;107(16):7503–7508. doi:10.1073/pnas.1002355107
132. Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One. 2009;4(9):e7125. doi:10.1371/journal.pone.0007125
133. Collado MC, Isolauri E, Laitinen K, Salminen S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr. 2008;88(4):894–899. doi:10.1093/ajcn/88.4.894
134. Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–195. doi:10.1038/oby.2009.167
135. Vael C, Verhulst SL, Nelen V, Goossens H, Desager KN. Intestinal microflora and body mass index during the first three years of life: an observational study. Gut Pathog. 2011;3(1):8. doi:10.1186/1757-4749-3-8
136. Luoto R, Kalliomäki M, Laitinen K, et al. Initial dietary and microbiological environments deviate in normal-weight compared to overweight children at 10 years of age. J Pediatr Gastroenterol Nutr. 2011;52(1):90–95. doi:10.1097/MPG.0b013e3181f3457f
137. Zuo HJ, Xie ZM, Zhang WW, et al. Gut bacteria alteration in obese people and its relationship with gene polymorphism. World J Gastroenterol. 2011;17(8):1076–1081. doi:10.3748/wjg.v17.i8.1076
138. Santacruz A, Marcos A, Wärnberg J, et al. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity. 2009;17(10):1906–1915. doi:10.1038/oby.2009.112
139. Million M, Maraninchi M, Henry M, et al. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int J Obes. 2012;36(6):817–825. doi:10.1038/ijo.2011.153
140. Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106(7):2365–2370. doi:10.1073/pnas.0812600106
141. Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi:10.2337/db07-1403
142. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
143. Quan N, Whiteside M, Herkenham M. Cyclooxygenase 2 mRNA expression in rat brain after peripheral injection of lipopolysaccharide. Brain Res. 1998;802(1–2):189–197. doi:10.1016/S0006-8993(98)00402-8
144. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi:10.1126/science.282.5396.2085
145. Wauman J, Zabeau L, Tavernier J. The Leptin Receptor Complex: heavier Than Expected? Front Endocrinol (Lausanne). 2017;8:30. doi:10.3389/fendo.2017.00030
146. Patil DP, Dhotre DP, Chavan SG, et al. Molecular analysis of gut microbiota in obesity among Indian individuals. J Biosci. 2012;37(4):647–657. doi:10.1007/s12038-012-9244-0
147. Muscogiuri G, Cantone E, Cassarano S, et al. Gut microbiota: a new path to treat obesity. Int J Obes Suppl. 2019;9(1):10–19. doi:10.1038/s41367-019-0011-7
148. Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi:10.1073/pnas.0407076101
149. Alberti KG, Zimmet P, Shaw J. The metabolic syndrome–a new worldwide definition. Lancet. 2005;366(9491):1059–1062. doi:10.1016/S0140-6736(05)67402-8
150. Dale CE, Fatemifar G, Palmer TM, et al. Causal Associations of Adiposity and Body Fat Distribution With Coronary Heart Disease, Stroke Subtypes, and Type 2 Diabetes Mellitus: a Mendelian Randomization Analysis. Circulation. 2017;135(24):2373–2388. doi:10.1161/CIRCULATIONAHA.116.026560
151. Larsson SC, Bäck M, Rees JMB, Mason AM, Burgess S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J. 2020;41(2):221–226. doi:10.1093/eurheartj/ehz388
152. Guo Y, Yue XJ, Li HH, et al. Overweight and Obesity in Young Adulthood and the Risk of Stroke: a Meta-analysis. J Stroke Cerebrovasc Dis. 2016;25(12):2995–3004. doi:10.1016/j.jstrokecerebrovasdis.2016.08.018
153. Mitchell AB, Cole JW, McArdle PF, et al. Obesity increases risk of ischemic stroke in young adults. Stroke. 2015;46(6):1690–1692. doi:10.1161/STROKEAHA.115.008940
154. Walker SP, Rimm EB, Ascherio A, Kawachi I, Stampfer MJ, Willett WC. Body size and fat distribution as predictors of stroke among US men. Am J Epidemiol. 1996;144(12):1143–1150. doi:10.1093/oxfordjournals.aje.a008892
155. Marini S, Merino J, Montgomery BE, et al. Mendelian Randomization Study of Obesity and Cerebrovascular Disease. Ann Neurol. 2020;87(4):516–524. doi:10.1002/ana.25686
156. Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi:10.1038/nature11450
157. Zhang Q, Yu H, Xiao X, Hu L, Xin F, Yu X. Inulin-type fructan improves diabetic phenotype and gut microbiota profiles in rats. PeerJ. 2018;6:e4446. doi:10.7717/peerj.4446
158. Zhu L, Sha L, Li K, et al. Dietary flaxseed oil rich in omega-3 suppresses severity of type 2 diabetes mellitus via anti-inflammation and modulating gut microbiota in rats. Lipids Health Dis. 2020;19(1):20. doi:10.1186/s12944-019-1167-4
159. Ahmad A, Yang W, Chen G, et al. Analysis of gut microbiota of obese individuals with type 2 diabetes and healthy individuals. PLoS One. 2019;14(12):e0226372. doi:10.1371/journal.pone.0226372
160. Lê KA, Li Y, Xu X, et al. Alterations in fecal Lactobacillus and Bifidobacterium species in type 2 diabetic patients in Southern China population. Front Physiol. 2012;3:496. doi:10.3389/fphys.2012.00496
161. Wu X, Ma C, Han L, et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol. 2010;61(1):69–78. doi:10.1007/s00284-010-9582-9
162. Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103. doi:10.1038/nature12198
163. Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi:10.1371/journal.pone.0009085
164. Jumpertz R, Le DS, Turnbaugh PJ, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. 2011;94(1):58–65. doi:10.3945/ajcn.110.010132
165. Pauli JR, Ropelle ER, Cintra DE, et al. Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. J Physiol. 2008;586(2):659–671. doi:10.1113/jphysiol.2007.142414
166. Vrieze A, Out C, Fuentes S, et al. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol. 2014;60(4):824–831. doi:10.1016/j.jhep.2013.11.034
167. Lawan A, Min K, Zhang L, et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes. 2018;67(4):624–635. doi:10.2337/db17-0826
168. Song X, Sun X, Oh SF, et al. Microbial bile acid metabolites modulate gut RORγ(+) regulatory T cell homeostasis. Nature. 2020;577(7790):410–415. doi:10.1038/s41586-019-1865-0
169. Torkamandi S, Bastami M, Ghaedi H, Moghadam F, Mirfakhraie R, Omrani MD. MAP3K1 May be a Promising Susceptibility Gene for Type 2 Diabetes Mellitus in an Iranian Population. Int J Mol Cell Med. 2016;5(3):134–140.
170. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2014;7(1):17–44. doi:10.3390/nu7010017
171. Sun Y, Huang Y, Ye F, et al. Effects of probiotics on glycemic control and intestinal dominant flora in patients with type 2 diabetes mellitus: a protocol for systematic review and meta-analysis. Medicine. 2020;99(46):e23039. doi:10.1097/MD.0000000000023039
172. Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. doi:10.1038/nm.2002
173. Zhao L, Zhang F, Ding X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359(6380):1151–1156. doi:10.1126/science.aao5774
174. Lin H, Yi J. Current Status of HbA1c Biosensors. Sensors. 2017;17(8). doi:10.3390/s17081798
175. Nguyen NT, Magno CP, Lane KT, Hinojosa MW, Lane JS. Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: findings from the National Health and Nutrition Examination Survey, 1999 to 2004. J Am Coll Surg. 2008;207(6):928–934. doi:10.1016/j.jamcollsurg.2008.08.022
176. Zhang L, Zhang WH, Zhang L, Wang PY. Prevalence of overweight/obesity and its associations with hypertension, diabetes, dyslipidemia, and metabolic syndrome: a survey in the suburban area of Beijing, 2007. Obes Facts. 2011;4(4):284–289. doi:10.1159/000331014
177. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582(1):97–105. doi:10.1016/j.febslet.2007.11.057
178. Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121(6):2126–2132. doi:10.1172/JCI58109
179. Tremaroli V, Karlsson F, Werling M, et al. Roux-en-Y Gastric Bypass and Vertical Banded Gastroplasty Induce Long-Term Changes on the Human Gut Microbiome Contributing to Fat Mass Regulation. Cell Metab. 2015;22(2):228–238. doi:10.1016/j.cmet.2015.07.009
180. Everard A, Belzer C, Geurts L, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110(22):9066–9071. doi:10.1073/pnas.1219451110
181. Sharma M, Thandassery RB, Bhargava N. Double pylorus: an optical illusion or reality? Gastroenterology. 2012;143(2):e7–8. doi:10.1053/j.gastro.2012.01.053
182. Jialal I, Rajamani U. Endotoxemia of metabolic syndrome: a pivotal mediator of meta-inflammation. Metab Syndr Relat Disord. 2014;12(9):454–456. doi:10.1089/met.2014.1504
183. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi:10.1172/JCI28898
184. Rankin LC, Artis D. Beyond Host Defense: emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell. 2018;173(3):554–567. doi:10.1016/j.cell.2018.03.013
185. Tanti JF, Ceppo F, Jager J, Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol (Lausanne). 2012;3:181. doi:10.3389/fendo.2012.00181
186. Ankolekar S, Rewell S, Howells DW, Bath PM. The influence of stroke risk factors and comorbidities on assessment of stroke therapies in humans and animals. Int J Stroke. 2012;7(5):386–397. doi:10.1111/j.1747-4949.2012.00802.x
187. Barrett-Connor E, Khaw KT. Diabetes mellitus: an independent risk factor for stroke? Am J Epidemiol. 1988;128(1):116–123. doi:10.1093/oxfordjournals.aje.a114934
188. Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11(3):261–271. doi:10.1016/S1474-4422(12)70005-4
189. van Sloten TT, Sedaghat S, Carnethon MR, Launer LJ, Stehouwer CDA. Cerebral microvascular complications of type 2 diabetes: stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. 2020;8(4):325–336. doi:10.1016/S2213-8587(19)30405-X
190. Zahra F, Kidwai SS, Siddiqi SA, Khan RM. Frequency of newly diagnosed diabetes mellitus in acute ischaemic stroke patients. J Coll Physicians Surg Pak. 2012;22(4):226–229.
191. Gray CS, Scott JF, French JM, Alberti KG, O’Connell JE. Prevalence and prediction of unrecognised diabetes mellitus and impaired glucose tolerance following acute stroke. Age Ageing. 2004;33(1):71–77. doi:10.1093/ageing/afh026
192. O’Donnell MJ, Chin SL, Rangarajan S, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. 2016;388(10046):761–775. doi:10.1016/S0140-6736(16)30506-2
193. Lau LH, Lew J, Borschmann K, Thijs V, Ekinci EI. Prevalence of diabetes and its effects on stroke outcomes: a meta-analysis and literature review. J Diabetes Investig. 2019;10(3):780–792. doi:10.1111/jdi.12932
194. Tsai CF, Anderson N, Thomas B, Sudlow CL. Comparing Risk Factor Profiles between Intracerebral Hemorrhage and Ischemic Stroke in Chinese and White Populations: systematic Review and Meta-Analysis. PLoS One. 2016;11(3):e0151743. doi:10.1371/journal.pone.0151743
195. Ergul A, Kelly-Cobbs A, Abdalla M, Fagan SC. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets. 2012;12(2):148–158. doi:10.2174/187153012800493477
196. Reeves MJ, Vaidya RS, Fonarow GC, et al. Quality of care and outcomes in patients with diabetes hospitalized with ischemic stroke: findings from Get With the Guidelines-Stroke. Stroke. 2010;41(5):e409–417. doi:10.1161/STROKEAHA.109.572693
197. Selvin E, Coresh J, Shahar E, Zhang L, Steffes M, Sharrett AR. Glycaemia (haemoglobin A1c) and incident ischaemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol. 2005;4(12):821–826. doi:10.1016/S1474-4422(05)70227-1
198. Lattanzi S, Bartolini M, Provinciali L, Silvestrini M. Glycosylated Hemoglobin and Functional Outcome after Acute Ischemic Stroke. J Stroke Cerebrovasc Dis. 2016;25(7):1786–1791. doi:10.1016/j.jstrokecerebrovasdis.2016.03.018
199. Grant PJ. Diabetes mellitus as a prothrombotic condition. J Intern Med. 2007;262(2):157–172. doi:10.1111/j.1365-2796.2007.01824.x
200. Vazzana N, Ranalli P, Cuccurullo C, Davì G. Diabetes mellitus and thrombosis. Thromb Res. 2012;129(3):371–377. doi:10.1016/j.thromres.2011.11.052
201. Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat Rev Neurol. 2010;6(3):145–155. doi:10.1038/nrneurol.2009.231
202. Hu Q, Manaenko A, Bian H, et al. Hyperbaric Oxygen Reduces Infarction Volume and Hemorrhagic Transformation Through ATP/NAD(+)/Sirt1 Pathway in Hyperglycemic Middle Cerebral Artery Occlusion Rats. Stroke. 2017;48(6):1655–1664. doi:10.1161/STROKEAHA.116.015753
203. Li W, Qu Z, Prakash R, et al. Comparative analysis of the neurovascular injury and functional outcomes in experimental stroke models in diabetic Goto-Kakizaki rats. Brain Res. 2013;1541:106–114. doi:10.1016/j.brainres.2013.10.021
204. Bannister JV, Bellavite P, Davoli A, Thornalley PJ, Rossi F. The generation of hydroxyl radicals following superoxide production by neutrophil NADPH oxidase. FEBS Lett. 1982;150(2):300–302.
205. Guo Y, Dong L, Gong A, et al. Damage to the blood‑brain barrier and activation of neuroinflammation by focal cerebral ischemia under hyperglycemic condition. Int J Mol Med. 2021;48(1). doi:10.3892/ijmm.2021.4975.
206. Overman JJ, Clarkson AN, Wanner IB, et al. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109(33):E2230–2239. doi:10.1073/pnas.1204386109
207. Liauw J, Hoang S, Choi M, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28(10):1722–1732. doi:10.1038/jcbfm.2008.65
208. Anderson MA, Burda JE, Ren Y, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532(7598):195–200. doi:10.1038/nature17623
209. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5(5):347–360. doi:10.1038/nrn1387
210. Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209(2):294–301. doi:10.1016/j.expneurol.2007.05.014
211. Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888–901. doi:10.1038/jcbfm.2015.45
212. Kazmierski R, Michalak S, Wencel-Warot A, Nowinski WL. Serum tight-junction proteins predict hemorrhagic transformation in ischemic stroke patients. Neurology. 2012;79(16):1677–1685. doi:10.1212/WNL.0b013e31826e9a83
213. Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. 2012;79(13 Suppl 1):S52–57. doi:10.1212/WNL.0b013e3182697e70
214. Yang T, Santisteban MM, Rodriguez V, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65(6):1331–1340. doi:10.1161/HYPERTENSIONAHA.115.05315
215. Adnan S, Nelson JW, Ajami NJ, et al. Alterations in the gut microbiota can elicit hypertension in rats. Physiol Genomics. 2017;49(2):96–104. doi:10.1152/physiolgenomics.00081.2016
216. Marques FZ, Nelson E, Chu PY, et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135(10):964–977. doi:10.1161/CIRCULATIONAHA.116.024545
217. Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46(5):1118–1122. doi:10.1161/01.HYP.0000185463.27209.b0
218. Masson GS, Nair AR, Dange RB, Silva-Soares PP, Michelini LC, Francis J. Toll-like receptor 4 promotes autonomic dysfunction, inflammation and microglia activation in the hypothalamic paraventricular nucleus: role of endoplasmic reticulum stress. PLoS One. 2015;10(3):e0122850. doi:10.1371/journal.pone.0122850
219. Goffredo M, Mass K, Parks EJ, et al. Role of Gut Microbiota and Short Chain Fatty Acids in Modulating Energy Harvest and Fat Partitioning in Youth. J Clin Endocrinol Metab. 2016;101(11):4367–4376. doi:10.1210/jc.2016-1797
220. Pluznick JL, Protzko RJ, Gevorgyan H, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110(11):4410–4415. doi:10.1073/pnas.1215927110
221. Huart J, Leenders J, Taminiau B, et al. Gut Microbiota and Fecal Levels of Short-Chain Fatty Acids Differ Upon 24-Hour Blood Pressure Levels in Men. Hypertension. 2019;74(4):1005–1013. doi:10.1161/HYPERTENSIONAHA.118.12588
222. Sun S, Lulla A, Sioda M, et al. Gut Microbiota Composition and Blood Pressure. Hypertension. 2019;73(5):998–1006. doi:10.1161/HYPERTENSIONAHA.118.12109
223. Yan Q, Gu Y, Li X, et al. Alterations of the Gut Microbiome in Hypertension. Front Cell Infect Microbiol. 2017;7:381. doi:10.3389/fcimb.2017.00381
224. Aguilar EC, Leonel AJ, Teixeira LG, et al. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metab Cardiovasc Dis. 2014;24(6):606–613. doi:10.1016/j.numecd.2014.01.002
225. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. doi:10.1073/pnas.1322269111
226. Cardinale JP, Sriramula S, Pariaut R, et al. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension. 2010;56(3):437–444. doi:10.1161/HYPERTENSIONAHA.110.154567
227. Kumar P, Gogulamudi VR, Periasamy R, Raghavaraju G, Subramanian U, Pandey KN. Inhibition of HDAC enhances STAT acetylation, blocks NF-κB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am J Physiol Renal Physiol. 2017;313(3):F781–f795. doi:10.1152/ajprenal.00166.2017
228. Bouter K, Bakker GJ, Levin E, et al. Differential metabolic effects of oral butyrate treatment in lean versus metabolic syndrome subjects. Clin Transl Gastroenterol. 2018;9(5):155. doi:10.1038/s41424-018-0025-4
229. Roshanravan N, Mahdavi R, Alizadeh E, et al. Effect of Butyrate and Inulin Supplementation on Glycemic Status, Lipid Profile and Glucagon-Like Peptide 1 Level in Patients with Type 2 Diabetes: a Randomized Double-Blind, Placebo-Controlled Trial. Horm Metab Res. 2017;49(11):886–891. doi:10.1055/s-0043-119089
230. Graudal NA, Hubeck-Graudal T, Jurgens G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev. 2017;4(4):Cd004022. doi:10.1002/14651858.CD004022.pub4
231. Wilck N, Matus MG, Kearney SM, et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature. 2017;551(7682):585–589. doi:10.1038/nature24628
232. Wei Y, Lu C, Chen J, et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget. 2017;8(1):70–82. doi:10.18632/oncotarget.13783
233. Kim AS, Johnston SC. Temporal and geographic trends in the global stroke epidemic. Stroke. 2013;44(6 Suppl 1):S123–125. doi:10.1161/STROKEAHA.111.000067
234. Kannel WB, Wolf PA, McGee DL, Dawber TR, McNamara P, Castelli WP. Systolic blood pressure, arterial rigidity, and risk of stroke. The Framingham study. JAMA. 1981;245(12):1225–1229. doi:10.1001/jama.1981.03310370017013
235. Kannel WB, Wolf PA, Verter J, McNamara PM. Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study. 1970. JAMA. 1996;276(15):1269–1278. doi:10.1001/jama.1996.03540150071040
236. Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. 2007;335(7627):974. doi:10.1136/bmj.39335.385301.BE
237. Brown DW, Dueker N, Jamieson DJ, et al. Preeclampsia and the risk of ischemic stroke among young women: results from the Stroke Prevention in Young Women Study. Stroke. 2006;37(4):1055–1059. doi:10.1161/01.STR.0000206284.96739.ee
238. McDonald SD, Malinowski A, Zhou Q, Yusuf S, Devereaux PJ. Cardiovascular sequelae of preeclampsia/eclampsia: a systematic review and meta-analyses. Am Heart J. 2008;156(5):918–930. doi:10.1016/j.ahj.2008.06.042
239. Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. doi:10.1136/bmj.b1665
240. Ahmed N, Wahlgren N, Brainin M, et al. Relationship of blood pressure, antihypertensive therapy, and outcome in ischemic stroke treated with intravenous thrombolysis: retrospective analysis from Safe Implementation of Thrombolysis in Stroke-International Stroke Thrombolysis Register (SITS-ISTR). Stroke. 2009;40(7):2442–2449. doi:10.1161/STROKEAHA.109.548602
241. Leonardi-Bee J, Bath PM, Phillips SJ, Sandercock PA. Blood pressure and clinical outcomes in the International Stroke Trial. Stroke. 2002;33(5):1315–1320. doi:10.1161/01.STR.0000014509.11540.66
242. Lima FO, Furie KL, Silva GS, et al. The pattern of leptomeningeal collaterals on CT angiography is a strong predictor of long-term functional outcome in stroke patients with large vessel intracranial occlusion. Stroke. 2010;41(10):2316–2322. doi:10.1161/STROKEAHA.110.592303
243. Menon BK, Smith EE, Coutts SB, et al. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann Neurol. 2013;74(2):241–248. doi:10.1002/ana.23906
244. Santisteban MM, Qi Y, Zubcevic J, et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ Res. 2017;120(2):312–323. doi:10.1161/CIRCRESAHA.116.309006
245. Faraco G, Brea D, Garcia-Bonilla L, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018;21(2):240–249. doi:10.1038/s41593-017-0059-z
246. Yamashiro K, Kurita N, Urabe T, Hattori N. Role of the Gut Microbiota in Stroke Pathogenesis and Potential Therapeutic Implications. Ann Nutr Metab. 2021;77(Suppl2):36–44. doi:10.1159/000516398
247. Gomes AC, de Sousa RG, Botelho PB, Gomes TL, Prada PO, Mota JF. The additional effects of a probiotic mix on abdominal adiposity and antioxidant Status: a double-blind, randomized trial. Obesity. 2017;25(1):30–38. doi:10.1002/oby.21671
248. Higashikawa F, Noda M, Awaya T, et al. Antiobesity effect of Pediococcus pentosaceus LP28 on overweight subjects: a randomized, double-blind, placebo-controlled clinical trial. Eur J Clin Nutr. 2016;70(5):582–587. doi:10.1038/ejcn.2016.17
249. Jung S, Lee YJ, Kim M, et al. Supplementation with two probiotic strains, Lactobacillus curvatus HY7601 and Lactobacillus plantarum KY1032, reduced body adiposity and Lp-PLA2 activity in overweight subjects. J Funct Foods. 2015;19:744–752. doi:10.1016/j.jff.2015.10.006
250. Kim J, Yun JM, Kim MK, Kwon O, Cho B. Lactobacillus gasseri BNR17 Supplementation Reduces the Visceral Fat Accumulation and Waist Circumference in Obese Adults: a Randomized, Double-Blind, Placebo-Controlled Trial. J Med Food. 2018;21(5):454–461. doi:10.1089/jmf.2017.3937
251. Minami J, Iwabuchi N, Tanaka M, et al. Effects of Bifidobacterium breve B-3 on body fat reductions in pre-obese adults: a randomized, double-blind, placebo-controlled trial. Biosci Microbiota Food Health. 2018;37(3):67–75. doi:10.12938/bmfh.18-001
252. Pedret A, Valls RM, Calderón-Pérez L, et al. Effects of daily consumption of the probiotic Bifidobacterium animalis subsp. lactis CECT 8145 on anthropometric adiposity biomarkers in abdominally obese subjects: a randomized controlled trial. Int J Obes. 2019;43(9):1863–1868. doi:10.1038/s41366-018-0220-0
253. Tian P, Li B, He C, et al. Antidiabetic (type 2) effects of Lactobacillus G15 and Q14 in rats through regulation of intestinal permeability and microbiota. Food Funct. 2016;7(9):3789–3797. doi:10.1039/C6FO00831C
254. Tabuchi M, Ozaki M, Tamura A, et al. Antidiabetic effect of Lactobacillus GG in streptozotocin-induced diabetic rats. Biosci Biotechnol Biochem. 2003;67(6):1421–1424. doi:10.1271/bbb.67.1421
255. Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi:10.1126/science.1241214
256. Zhou D, Pan Q, Shen F, et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep. 2017;7(1):1529. doi:10.1038/s41598-017-01751-y
257. Cai TT, Ye XL, Yong HJ, et al. Fecal microbiota transplantation relieve painful diabetic neuropathy: a case report. Medicine. 2018;97(50):e13543. doi:10.1097/MD.0000000000013543
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.