Back to Archived Journals » Hypoxia » Volume 2
Regulation of obesity and insulin resistance by hypoxia-inducible factors
Authors Ban J, Ruthenborg R, Cho K, Kim J ![]()
Received 3 June 2014
Accepted for publication 25 July 2014
Published 13 November 2014 Volume 2014:2 Pages 171—183
DOI https://doi.org/10.2147/HP.S68771
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Jae-Jun Ban, Robin J Ruthenborg, Kevin W Cho, Jung-whan Kim
Department of Biological Sciences, The University of Texas at Dallas, Richardson, TX, USA
Abstract: In obesity, dysregulated metabolism and aberrant expansion of adipose tissue lead to the development of tissue hypoxia that plays an important role in contributing to obesity-associated metabolic disorders. Recent studies utilizing adipocyte-specific hypoxia-inducible factor-α (HIF-α) gain- or loss-of-function animal models highlight the pivotal involvement of hypoxic responses in the pathogenesis of obesity-associated inflammation and insulin resistance. HIF-1α, a master transcription factor of oxygen homeostasis, induces inflammation and insulin resistance in obesity, whereas its isoform, HIF-2α, exerts opposing functions in these obesity-associated metabolic phenotypes. In this review, recent evidence elucidating functional implications of adipocyte HIFs in obesity and, more importantly, how these regulate obesity-associated inflammation, fibrosis, and insulin resistance will be discussed. Further, we propose that modulation of HIF-1 could be a potential novel therapeutic strategy for antidiabetic treatment.
Keywords: hypoxia-inducible factor-1, inflammation, oxygen
Introduction
Hypoxia has been the focus of intensive investigation mainly due to its functional and clinical implication in numerous pathological conditions including cancer, inflammation, tissue injury, and ischemic diseases.1 It has been known for many decades that hypoxia is one of the prominent factors that influence energy balance and obesity from initial metabolic studies among high-altitude population and obstructive sleep apnea patients.2 Intriguingly, there is a significant inverse relationship between body mass index and altitude of residence, whereas obstructive sleep apnea patients display oxidative stress, systemic hypertension, and hypoxia-inducible factor (HIF) dependent inflammation. Although these seemingly contradictory effects of hypoxia highlight the important and complicated role of hypoxia on energy balance and metabolism, little is known about key molecules and signaling pathways in the relationship between hypoxia and obesity.
Hypoxia occurs when oxygen demand exceeds supply, which results in insufficient oxygen supply into tissues or cells. Recent animal studies have demonstrated that adipose tissues become hypoxic in obesity.3–5 Potential contributors to the onset of adipose tissue hypoxia in obesity include: 1) inadequate blood flow to white adipose tissues;6 2) overall increase in adipocyte size, which is larger than oxygen’s diffusion capacity of 150–200 μm;7 or 3) enhanced oxygen consumption by adipocytes or inflammatory cells infiltrated into the obese microenvironment.8–10 Yet, molecular and cellular mechanisms that initiate and maintain the hypoxic microenvironment in obese adipose tissues remain to be elucidated.
Glucose intolerance is characterized by systemic hyperglycemia due to impaired glucose absorption and utilization by cells, and is considered as a prediabetic state associated with insulin resistance.11 About 80%–90% of people diagnosed with type 2 diabetes are also diagnosed as obese.12 A number of preclinical and clinical studies have suggested that chronic inflammation may be the crucial link between obesity and insulin resistance.13–15 Virtually all obese animals and humans display low-grade chronic inflammation in their adipose tissues, which is characterized by proinflammatory macrophage infiltration and oxidative stress. Given that hypoxia is one of the prominent regulators for inflammation and reactive oxygen species production,16–19 hypoxia and cellular hypoxic responses may provide mechanistic insight into causal relationships between obesity, inflammation, and insulin resistance.20
HIFs, oxygen-sensitive transcriptional regulators, mediate cellular and microenvironmental hypoxic remodeling.21 Upon oxygen deprivation, alpha subunits of HIFs are stabilized, translocated to the nucleus, and bound to hypoxia responsive elements to induce hypoxia-regulated gene expression. Recently, we and other groups, utilizing animal model systems, reported that adipocyte-specific constitutively active HIF-1α expression aggravates diabetic phenotypes with adipose tissue inflammation and fibrosis. Furthermore, loss of HIF-1α in adipocytes significantly attenuates obesity-associated inflammation and improves insulin sensitivity.22–26 These findings argue for critical involvement of hypoxic HIF-1 signaling in metabolic disorders in obese mice. However, little is known about how the hypoxic adipose microenvironment affects the biology of adipocytes and other critical cell components in obesity such as macrophages. Moreover, recent studies by our group and others utilizing animal models with cell type-specific knocking-out or overexpression of genes in hypoxic signaling pathways demonstrated that different types of cells differentially respond to hypoxia, and further, exhibit unique hypoxic responses and phenotypes.27–35 This suggests that there may be multidirectional interaction among adipocytes and surrounding non-adipocytes including macrophages, endothelial cells, and various mesenchymal cells. Additionally, it should be noted that independently established aP2-cre mice display differential temporal/spatial Cre expression in adipocytes as well as in non-adipocytic lineage cells including myeloid and neuronal cells.36 This may be responsible for seemingly contradictory phenotypes among the various HIFs gain- and loss-of-function mouse models (Table 1).
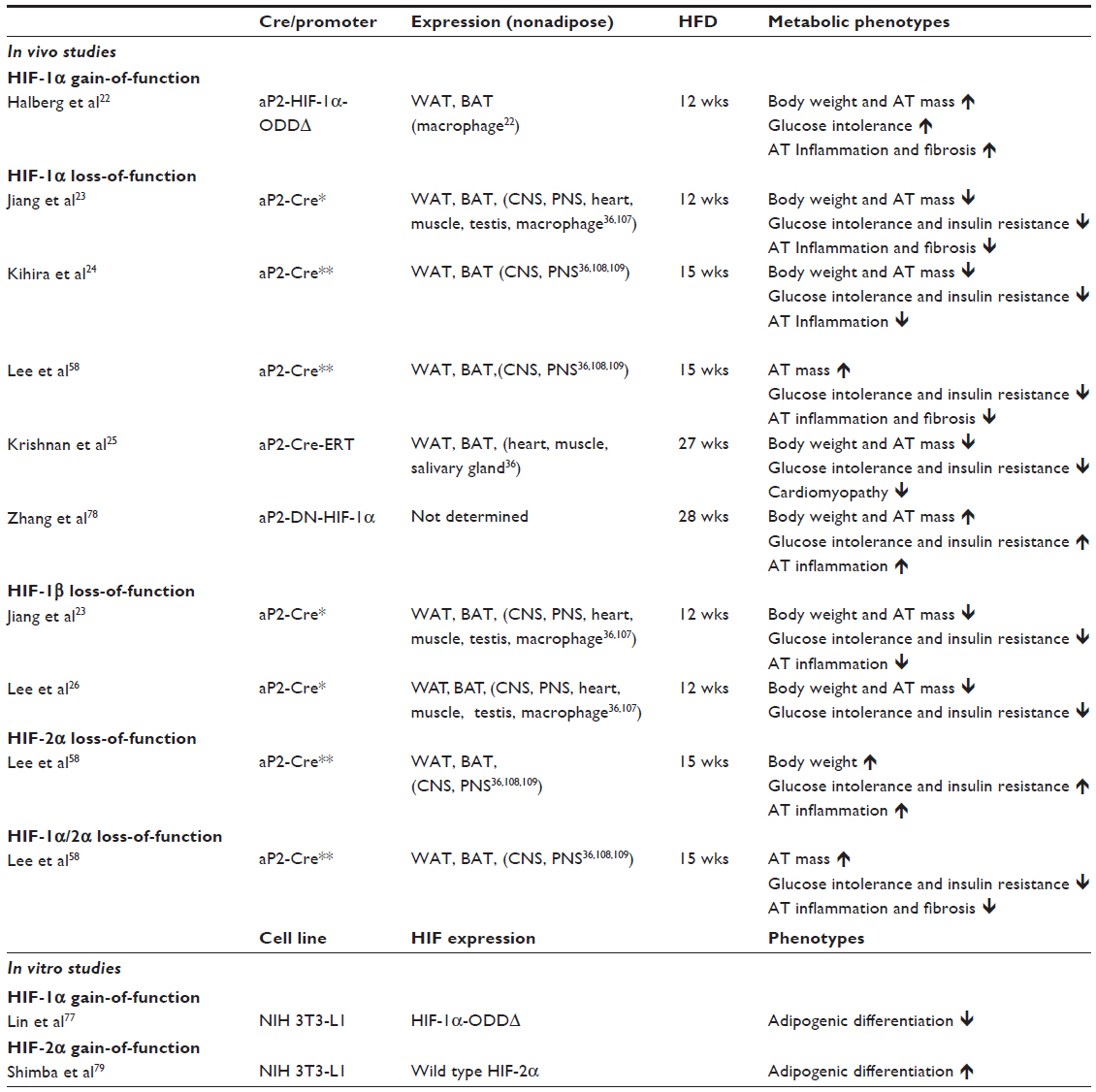 | Table 1 In vivo and in vitro models for adipocyte HIFs |
In this review, we will describe the functional effects of HIF signaling on metabolically dysfunctional adipocytes and how this modulates obesity and insulin resistance. Although non-adipose tissue HIF signaling has been implicated in obesity and other metabolic disorders, this has been elegantly reviewed elsewhere37 and will not be discussed here. We will also discuss inhibition of HIF signaling as a potential therapeutic target for obesity and metabolic diseases.
HIFs
The HIF proteins are central transcription factors of oxygen homeostasis. HIFs are a heterodimeric complex composed of an α and a β subunit. Both the subunits belong to the basic helix-loop-helix Per/Arnt/Sim (PAS) family of transcription factors and are constitutively expressed in the cell. The HIF-α subunit is oxygen-sensitive and is rapidly degraded in nonhypoxic conditions with a half-life of less than 10 minutes,38 whereas the HIF-β subunit is constitutively expressed.39 In the presence of ample oxygen, the prolyl and asparaginyl hydroxylases hydroxylate the oxygen-dependent domain (ODD) of HIF-α on specific proline and asparagine residues, respectively.40–42 Prolyl hydroxylases (PHDs) require molecular oxygen, Fe(II), ascorbate, and the Krebs cycle intermediate, 2-oxoglutarate, as substrates.40,41 Proline hydroxylation allows the von Hippel–Lindau protein, a tumor suppressor E3 ubiquitin ligase, to bind and target HIF-α for proteasome degradation via polyubiquitination, whereas asparaginyl hydroxylation inhibits transcription activities of HIFs by blocking HIF interaction with the transcriptional cofactor, p300.42–45 In response to hypoxic conditions, the lack of available molecular oxygen, an absolute requirement for HIF hydroxylation, stabilizes HIF-α increasing cellular levels of the protein and facilitating its translocation into the nucleus. In the nucleus, HIF-α heterodimerizes with HIF-β and forms complexes with other coactivators, such as p300, in order to bind to hypoxia-responsive elements of genomic DNA and to transactivate its target genes (Figure 1). In addition to oxygen availability, inducible changes in the cellular abundance of the PHDs (PHD1–3) provide an additional interface that regulates the oxygen-sensitive HIF signaling.46
 | Figure 1 Oxygen-dependent regulation of HIF-1α. |
There are three HIF-α subunits identified: HIF-1α, -2α, and -3α. HIF-1α has been most-extensively characterized. HIF-1α mediates adaptation and survival to hypoxia through activating genes involved in angiogenesis (eg, vascular endothelial growth factor [VEGF]), glucose uptake (eg, glucose transporter 1), and glycolysis (eg, lactate dehydrogenase and pyruvate dehydrogenase kinase 1).47 Although another isoform, HIF-2α, also termed endothelial PAS domain-containing protein 1, shares similar functional homology and common target genes to HIF-1α,48 recent studies have demonstrated that they have non-overlapping target genes that are uniquely involved in HIF-2α-mediated cellular processes such as erythropoietin and the stem cell factor octamer-binding transcription factor 4.49–52 Furthermore, HIF-1α and HIF-2α appear to have opposing activities in certain contexts.53 In von Hippel–Lindau tumor suppressor (VHL)-null renal cell carcinoma cells, where both HIF-1α and HIF-2α are constitutively active, HIF-2α may function as a tumor-promoting player whereas HIF-1α exhibits tumor-suppressing activities.54,55 This counteraction of the two isoforms may be mediated in part by functional crosstalk between HIF-αs and the proto-oncogene c-Myc.56 When HIF-1α is available, it attenuates c-Myc transcriptional activity by disrupting c-Myc/Max complexes. On the other hand, HIF-2α associates with Max, stabilizing the c-Myc/Max complexes and enhancing c-Myc transcriptional activity. More opposing functions of HIF-1α and HIF-2α have been shown to be critical in nitric oxide (NO) homeostasis of macrophages, keratinocytes, and endothelial cells.28,33,57 HIF-1α induces the expression of inducible nitric oxide synthase (iNOS), which increases the production of NO. HIF-2α, on the other hand, induces arginase expression, which inhibits NO production by removing L-arginine, a substrate for NO production. Consistent with these previous findings, our recent study suggested that these opposing functions of HIF-1α and HIF-2α are critically involved in the pathogenesis of adipocyte dysfunction in obesity.58 We will discuss in more detail the functional effects of HIFs on obesity and insulin resistance in this review.
Adipose tissue hypoxia in obesity
While atmosphere oxygen tension is 160 mmHg (21%), physiological oxygen levels vary significantly between the tissues from 150 mmHg (17%) in the upper respiratory tract to 1–10 mmHg in the retina.20 Although human clinical studies have reported contradictory results in the levels of adipose tissue blood flow and oxygenation in obese subjects,59–62 direct measurement of oxygen tension by oxygen-meters revealed that oxygen tension in white adipose tissue in obese mice (leptin-deficient mutant ob/ob mice) and high-fat fed mice is approximately 15 mmHg (<2%) as compared to lean mice that have oxygen tensions ranging from 45 to 55 mmHg.5 Recent studies from our group and others have further shown that in obese adipose tissues, hypoxia can be detected by the hypoxia-specific chemical probe, pimonidazole, as well as by immunohistochemical detection of HIF-1α expression (Figure 2).3,5,22 In relatively severe hypoxic conditions, pimonidazole becomes reduced and binds to sulfhydryl groups of various molecules forming pimonidazole adducts in the hypoxic cells.63 Although this method is not quantitative and it is unable to detect mildly hypoxic tissues (above 10 mmHg pO2), it has been widely used and shown to be effective in consistently detecting tissue hypoxia of cancer and ischemia.
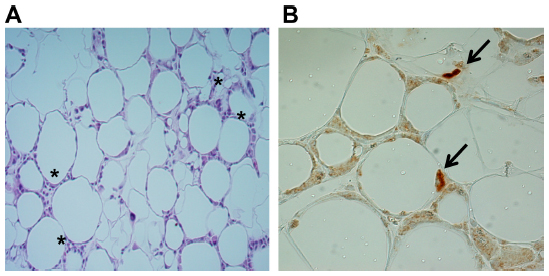 | Figure 2 HIF-1α expression in obese adipose tissues. |
Despite compelling evidence of hypoxia in obese adipose tissues, the onset mechanisms of adipose tissue hypoxia in obesity are not fully understood. Obesity is characterized by hyperplasia (cell number increase) as well as hypertrophy (cell size increase) of obese adipocytes resulting in a rapid enlargement of adipose tissue. Given that the diffusion capacity of oxygen is limited to 150–200 μm, aberrantly enlarged adipose tissue may have insufficient intra-adipose blood perfusion contributing to hypoxia in these tissues.7 One can then reason that restoring blood perfusion into adipose tissue may attenuate adipose tissue hypoxia and adipocyte dysfunction in obesity. Several groups attempted to address this by modulating vasculature and VEGF expression and activity. Rupnick et al have shown that angiogenesis inhibitor TNP-470 treatment in ob/ob mice has a profound weight-loss effect that is associated with vascular remodeling, decreased endothelial cell proliferation, and increased endothelial cell apoptosis without any apparent toxicity in 3T3-L1 preadipocytes.64 This suggests that weight loss is due to an endothelial-mediated antiangiogenic mechanism. A subsequent study by Bråkenhielm et al has demonstrated that TNP-470 prevents obesity and lowers insulin level suggesting that antiangiogenic therapy may improve insulin sensitivity.65 In contrast to these initial observations, a recent study by White et al showed that TNP-470 treatment induces glucose intolerance indicating that, despite a significant weight loss, antiangiogenic vascular suppression may result in adipocyte dysfunction and systemic diabetic phenotypes.66 More direct observations have been reported from studies utilizing adipocyte-specific gain-of-function or loss-of-function VEGF transgenic animals.6 Several studies reported that overexpression of VEGF in adipocytes significantly attenuated obesity and insulin resistance in high-fat diet-fed mice, whereas another study showed that β-actin-mediated partial suppression of systemic VEGF expression displayed resistance to high-fat diet-induced obesity.67–70 One can speculate that VEGF derived from non-adipocytes, particularly macrophages, may have differential effects on obesity and insulin resistance. Although the exact nature of discrepancy still remains to be determined, these studies highlight the complexity of adipose tissue VEGF and vasculature that appear to play a critical role in adipose blood perfusion and hypoxia, which in turn, regulate metabolic homeostasis.
In addition to oxygen supply through vasculature, tissue oxygen tension or oxygen availability is determined by oxygen consumption rate of local cellular components in various physiologic and pathologic conditions.8–10 Our recent study provided evidence for the increased oxygen consumption of adipocytes as an initial contributor to adipose tissue hypoxia in high-fat diet-induced obesity.58 Intriguingly, short-term exposure to a high-fat diet (less than 3 days) was sufficient to create adipose tissue hypoxia as determined by adipose tissue accumulation of hypoxia probe pimonidazole, HIF-1α and its target gene expression, and elevated levels of glycolysis and lactate. It should be noted that mice as early as 3 days after high-fat diet did not show any significant adipose tissue remodeling suggesting that this is independent of adipose tissue oxygen perfusion. Although mechanisms have not been fully elucidated, saturated free fatty acids increase mitochondrial oxygen consumption through the adenine nucleotide translocator (ANT)-dependent uncoupling of mitochondrial respiration.71,72 ANTs, localized in the mitochondrial inner membrane, induce proton leakage by pumping protons into the mitochondrial matrix. We showed that the oxygen consumption rate of primary adipocytes isolated from 3 days high-fat diet-fed mice was significantly elevated, even in the presence of oligomycin, indicating the uncoupling mechanism. Further, cultured 3T3-L1 adipocytes treated with saturated free fatty acids displayed increased oxygen consumption, which is inhibited by ANT inhibitor carboxyatractyloside. In high-fat diet-fed mice, both pharmacological (carboxyatractyloside treatment) and genetic (viral ANT2 short hairpin RNA knock-down) inhibitions of ANT successfully alleviated adipose tissue hypoxia without changes in systemic oxygen saturation, which are associated with improved glucose tolerance and insulin sensitivity. These observations indicate that adipose tissue oxygen consumption rate is a critical determinant for adipose tissue hypoxia and HIF-1α expression promoting obesity-associated metabolic dysfunction.
Taken together, these studies provide evidence for a model of adipose tissue hypoxia in obesity. In prolonged or chronic obesity, uncontrolled expansion of adipose tissue may cause insufficient blood perfusion and oxygen delivery leading to perhaps more persistent adipose tissue hypoxia. In the short-term or transient manner, prior to adipocyte hypertrophy and hyperplasia, elevated saturated free fatty acids increase adipocyte oxygen consumption resulting in a state of relative hypoxia via ANT-mediated uncoupled mitochondrial respiration. Although biological significance of the relationship between these two distinct mechanisms is not fully understood, HIF-mediated hypoxic signaling appears to be invariably activated in hypoxic obese adipose tissue and exerts a broad and deep influence on adipocyte biology and metabolic disorders.
Regulation of adipogenesis and adiposity by HIFs
As nutritional intake exceeds energy expenditure, the remaining energy is stored in white adipocytes leading to hypertrophy. While hypertrophy prevails and rapidly occurs in obesity, the link between hyperplasia and obesity is still controversial.73 Considering childhood obesity influences adipocyte number in adulthood, it is conceivable that there is some kind of positive relevance between excessive food intake and hyperplasia.73 One possible mechanism is the induction of adipogenesis factors by severe and chronic hypertrophy of adipocytes.74 However, the precise mechanism of how obesity affects induction of hyperplasia needs to be further studied. In the case of hyperplasia, appropriate recruitment of angiogenesis factors needs to be accompanied with fat-tissue enlargement.75 During fat-pad expansion, adipocytes communicate with endothelial cells via paracrine signaling and hypoxia seems to be a critical regulator of angiogenesis.75 Given that hypoxia is a prominent feature in obese adipose tissues, functional roles of HIFs in adipogenic activation and differentiation have been of particular interest.
HIF-1α
Early studies have demonstrated that HIFs play an important role in in vitro adipogenic differentiation of 3T3-L1 preadipocytes (Table 1). He et al reported that HIF-1α protein levels were elevated by 3T3-L1 adipocyte differentiation.76 Lin et al showed that hypoxia prevents differentiation of 3T3-L1 by sustaining the expression of pref-1, a preadipocyte-secreted inhibitor of adipocyte differentiation.77 Induction of HIF-1α appears to be a key contributor to the hypoxia-mediated maintenance of the undifferentiated state. These early in vitro studies suggest that HIFs may play an important role in adipogenic differentiation.
Adipocyte-specific ablation of HIF-1α activity in obesity animal models provides evidence that HIF-1α may promote adipogenesis in high-fat diet-mediated obesity. Adipocyte Hif-1α-null mice exhibited less body weight, reduced white adipose tissue mass, and reduced adipocyte size on high-fat diet.23–25 One study showed that adipocyte-specific overexpression of a truncated dominant-negative form of HIF-1α resulted in more severe obesity with increased white adipose tissue mass.78 However, the biological implication of truncated HIF-1α used in this study has not been characterized and obese phenotypes appear to be due to brown adipose tissue-dependent mechanisms. Studies conducted by Jiang et al and Lee et al demonstrated that adipocyte-specific deletion of HIF-1β, an obligate partner of HIF-1α, results in a lean phenotype and smaller adipocyte size.23,26 In support of these studies, transgenic overexpression of the constitutively active form of HIF-1α (ΔODD-HIF-1α) in adipocytes led to increased body weight and adiposity.22 Our recent study, however, showed that adipocyte Hif-1α-null mice exhibit normal body weight on both normal chow and high-fat diet.58 Furthermore, there was a significant increase in white adipose tissue mass and adipocyte size in adipocyte Hif-1α-null mice as compared to wild type control mice. Yet, in this study, Hif-1α-null mice exhibited a significant improvement in glucose tolerance and insulin sensitivity suggesting that adipocyte HIF-1 signaling may exert its antidiabetic effects through systemic metabolic regulation (eg, fat mobilization or inflammation) rather than regulation of adipogenic differentiation.
HIF-2α
In contrast to HIF-1α, HIF-2α overexpression in 3T3-L1 significantly enhanced the peroxisome proliferator activator γ2-mediated adipogenesis that is associated with increased glucose uptake and lipid biosynthesis, suggesting that HIF-2α promotes in vitro adipogenic differentiation.79 However, a recent study by Park et al has demonstrated that hypoxic induction of Wnt10b suppresses adipogenesis of 3T3-L1.80 HIF-2α, not HIF-1α, appears to be responsible for transactivation of the Wnt10b gene, which maintains preadipocytes in an undifferentiated state in hypoxia. We recently generated adipocyte-specific HIF-2α knockout mice and revealed that HIF-2α ablation in adipocytes aggravates high-fat diet-induced obesity and insulin resistance, suggesting that HIF-2α may play a role in suppressing adipogenic differentiation and obesity when fed a high-fat diet or it may counteract HIF-1α activities as proposed previously.58
Despite contradictory observations on functional contributions of HIFs to adiposity, it illuminates multifaceted roles of HIFs in the regulation of adipogenic differentiation in obesity. However, it remains unclear whether aP2, also termed fabp4, promoter-driven adipocyte-specific Cre mice used in the above-mentioned studies display recombination in preadipocytes.36 Thus, it will be of critical importance to characterize HIF signaling in conjunction with the adipocyte cellular lineage trace. For example, adipocyte precursor-specific Cre lines can be employed to elucidate specific roles of HIFs in the differentiation of preadipocytes into adipocytes.81
HIFs and insulin resistance
Association of obstructive sleep apnea that is characterized by repetitive cycles of intermittent hypoxia with metabolic disorders including obesity and insulin resistance has been reported in a number of clinical studies in humans, suggesting that oxygen homeostasis is one of the key contributors to glucose metabolism and insulin sensitivity.2,82 Moreover, lean mice as well as ob/ob mice exposed to intermittent hypoxia have been shown to develop systemic insulin resistance.83 Regazzetti et al have demonstrated that hypoxia is sufficient to suppress the insulin signaling pathway in human and murine adipocytes.84 Hypoxia (1% O2) or hypoxia mimetic CoCl2 treatment significantly reduced phosphorylation of insulin receptor (IR) and protein kinase B in 3T3-L1 and differentiated human adipocytes. Moreover, inhibited insulin signaling was associated with the reduction of insulin-induced glucose uptake. Overexpression of HIF-1α or HIF-2α mimicked hypoxia-induced insulin signaling inhibition whereas siRNA knockdown of HIF-1α or HIF-2α partially restored insulin signaling in hypoxia indicating that HIF signaling is an important component of adipocyte insulin signaling. Although molecular and cellular mechanisms underlying the links between adipose tissue hypoxia and insulin resistance is still poorly understood, recent studies from our group and others provide insight into how hypoxic adipose microenvironment in high-fat diet-induced obese mice affects adipocyte functions. Further, these studies provide insight on how adipocyte hypoxic HIF signaling influences systemic metabolic homeostasis.
HIF-1α
Adipocyte-specific Hif-1α deletion in high-fat diet-induced obese mice consistently exhibits improved glucose tolerance and insulin sensitivity. Jiang et al further demonstrated that adipocyte Hif-1α-null mice have enhanced systemic insulin signaling pathways such as phosphorylation of Akt in white adipose tissue, liver, and skeletal muscle.23 Consistently, adipocyte Hif-1α-null mice show increased expression of adiponectin, which is known to promote energy expenditure and insulin sensitivity, and decreased expression of SOCS3 that inhibits IR signaling by preventing autophosphorylation of IR. Another study by Krishnan et al employed aP2 Cre-ERT2 mice allowing the inducible deletion of Hif-1α in adipocytes to avoid potential Cre recombination/Hif-1α deletion during embryonic and early postnatal development.25 Upon tamoxifen-induced Hif-1α ablation, high-fat diet-induced obese mice exhibited a significant improvement in systemic metabolism including lean phenotype, increased glucose tolerance and insulin sensitivity, and attenuated obesity-associated cardiomyopathy. Indirect calorimetric analysis revealed that adipocyte loss of HIF-1α promotes systemic energy expenditure and fatty acid β-oxidation in visceral adipose tissue of high-fat diet-fed mice. The authors further showed that peroxisome proliferator-activator receptor gamma coactivator-1 alpha (PGC-1α), a central transcriptional coactivator of mitochondrial biogenesis and energy metabolism, is critically involved in Hif-1α-mediated fatty acid oxidation in adipose tissue.85 PGC-1α activity is negatively regulated by acetylation at the protein level. Intriguingly, PGC-1α was heavily acetylated in the high-fat diet-fed wild type mice whereas virtually no acetylation was detected in adipocyte Hif-1α-null mice. This suggests that loss of Hif-1α results in PGC-1α deacetylation, which in turn promotes transcriptional activity of PGC-1α on its target genes involved in mitochondrial biogenesis and energy expenditure. Accordingly, Hif-1α-null adipocytes contain more mitochondria with increased expression of numerous PGC-1α target genes. Sirtuin 2, a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase that can target PGC-1α,86 was identified to be bound and transcriptionally repressed by HIF-1α. Taken together, this study demonstrated that HIF-1α/sirtuin 2/PGC-1α axis plays a crucial role in regulating fatty acid oxidation and energy expenditure in white adipose tissue. In support of the above loss-of-function studies, Halberg et al reported that overexpression of a constitutively active form of HIF-1α (ΔODD-HIF-1α) in adipocytes exhibited increased body weight and decreased glucose tolerance, demonstrating that aberrant HIF-1 activity leads to an exacerbated diabetic phenotype.22
Our recent study further provides evidence for the functional contribution of HIF-1α to the hypoxia-associated insulin resistance. Adipocyte Hif-1α deletion improved glucose tolerance and systemic insulin sensitivity in muscle, liver, and adipose tissue on a high-fat diet.58 At the molecular level, we found that HIF-1α-induced iNOS leads to NO production in obese adipose tissue. NO has been shown to inhibit insulin signaling by S-nitrosylation-dependent inactivation of Akt.87
Adipocyte-specific HIF-1β deletion has also been analyzed to evaluate the role of the HIF signaling pathway in obesity.23,26 Adipocyte Hif-1β-null mice were lean and resistant to age- and high-fat diet-mediated insulin resistance. These results indicate that mice lacking adipocyte HIF-1β exhibit similar phenotypes to HIF-1α knockout mice. However, it should be noted that the HIF-1β subunit is an obligate partner protein for DNA binding and transcription of both HIF-1α and HIF-2α, as well as other PAS-domain containing proteins such as aryl hydrocarbon receptor.88 Indeed, some differential effects of HIF-1α and HIF-1β deletions have been indicated. For example, there was a significant difference in the temporal course of body weight gain and onset of improvement of glucose metabolism.23 Nonetheless, these studies support the critical roles for hypoxia/HIF signaling in adipocytes by regulating metabolic homeostasis.
HIF-2α
Given recently proposed antagonistic roles of HIF-1α and HIF-2α in NO homeostasis, HIF-1α promotes NO production by inducing its target iNOS, while HIF-2α suppresses NO production by inducing its target, arginase, removing NO precursor L-arginine.28,33,57 As a result, we sought to determine the effects of adipocyte Hif-2α deletion on obesity and glucose metabolism. Adipocyte Hif-2α-null mice exhibit increased body weight, reduced glucose tolerance, and insulin sensitivity on a high-fat diet that is associated with increased NO levels and arginase expression in adipose tissue.58 This suggests that HIF-2α plays a protective role against obesity-induced insulin resistance and further supports opposing roles of HIF-1α and HIF-2α in adipocyte glucose metabolism. In order to better understand the interplay of the two HIF-α isoforms in obesity, we created adipocyte-specific HIF-1α/HIF-2α double-knockout mice and, intriguingly, found that double-knockout mice appear to recapitulate virtually all phenotypes that HIF-1α knockout mice exhibit: improved glucose tolerance and insulin sensitivity, reduced NO production, and attenuated adipose tissue inflammation and fibrosis. These results suggest that HIF-1α exerts predominant roles in contributing to the development of obesity-induced insulin resistance, while HIF-2α prevents the pathological metabolic progress by suppressing HIF-1α activity. Yet, further studies will be required to better elucidate the molecular mechanisms underlying the opposing interaction between HIF-1α and HIF-2α in dysfunctional adipocytes. A possibility of HIF-2α having parallel or HIF-1α-independent protective mechanisms should not be excluded as well.
HIFs and obesity-associated inflammation
One of the hallmarks of obesity is low-grade chronic inflammation in metabolic tissues including adipose tissue.13–15 In response to imbalanced metabolic stimuli, adipocytes initiate inflammatory signaling pathways that lead to the induction of inflammatory cytokines such as tumor necrosis factor-α (TNF-α).89–91 Subsequently, various inflammatory cell populations are infiltrated into adipose tissues. In particular, proinflammatory (M1) macrophages have been shown to be a prominent immune cell population recruited into obese adipose tissues of obese humans as well as genetic or dietary-induced obese mice, which further contributes to the adipose tissue inflammatory response.15 Although metabolic imbalance has been proposed to be a major initial cause of the inflammatory response in obesity, molecular and cellular processes that regulate inflammatory signaling, cytokine expression, and inflammatory cell recruitment remain to be elucidated.
HIF-1α
Obesity-associated inflammation contributes to insulin resistance and metabolic dysfunction in obesity.13–15 Accordingly, Hif-1α knockout mice exhibiting improved glucose tolerance and insulin sensitivity invariably have less inflammation characterized by decreased macrophage infiltration and inflammatory cytokine expression as well as reduced adipose tissue fibrosis. Kihira et al showed a significant decrease in the expression of a macrophage marker, F4/80, and crown-like structures resulting from clustered macrophages surrounding dying or dead adipocytes in high-fat diet adipocyte Hif-1α knockout mice.24 Moreover, the expression of obesity-associated inflammatory cytokines, TNF-α and monocyte chemoattractant protein-1 (MCP-1), was decreased in adipose tissues of Hif-1α knockout mice. Our recent study also demonstrated a reduction of M1 macrophage infiltration in adipose tissues in the adipocyte Hif-1α knockout mice.58 Consistent with these results, macrophage markers, such as F4/80 and CD11b, and inflammatory cytokines, such as TNF-α, MCP-1 (CCL2), RANTES (CCL5), interleukin-6, macrophage inflammatory protein-1α, and CYR62, were significantly reduced in adipose tissues of Hif-1α knockout mice. Conditioned medium of Hif-1α-null adipocytes showed reduced chemotactic capacity in vitro. The diminished chemotaxis of Hif-1α-null adipocytes was confirmed in vivo by showing decreased migration of fluorescently labeled primary macrophages into the adipose tissues of Hif-1α knockout mice. Consistently, the mice overexpressing a constitutively active form of HIF-1α (ΔODD-HIF-1α) in adipocytes showed elevated local adipose tissue inflammation.22 By gene expression profiling, adipose tissue-specific HIF-1α activation led to induction of macrophage markers such as F4/80, CD68, and CSF1R, as well as monocyte chemoattractants such as CCL2, CCL7, and CCL8, along with alteration of extracellular matrix (ECM) remodeling genes. Although further follow-up studies should be warranted to delineate the exact role of HIF-1α, the above-noted studies highlight HIF-1α as an essential inducer for obesity-associated inflammation.
There is increasing evidence that adipose tissue fibrosis and ECM remodeling in obesity may be a key factor that induces adipose tissue inflammation and insulin resistance.22,92 It has been suggested that limiting expansion of adipose tissues by fibrosis may cause adipose tissue dysfunction in the obese condition.22,93 An original study by Halberg et al demonstrated that adipocyte HIF-1α activation characteristically induces adipose tissue fibrosis that is associated with a general upregulation of a number of ECM molecules and their regulators including lysyl oxidase (LOX).22 Consistently, genetic or pharmacological inhibition of HIF-1 led to less fibrotic accumulation in adipose tissues.93
HIF-2α
Considering opposing roles of HIF-1α and HIF-2α, adipocyte Hif-2α knockout mice exhibited increased adipose tissue inflammation on high-fat diet characterized by more M1 macrophage infiltration and higher expression of inflammatory genes including TNF-α, IL-1, and 5-lipoxygenase-activating protein.58 This suggests that HIF-2α suppresses obesity-associated inflammation in adipose tissues. However, it remains to be determined whether HIF-2α contributes directly to anti-inflammatory processes or indirectly through suppressing proinflammatory activities of HIF-1α.
Targeting hypoxia and HIFs for treatment of obesity and insulin resistance
For many decades, the focus on treating obesity has been nonpharmacological management such as changes in diet and degree of physical exercise. Although surgical procedures such as laparoscopic gastric bypass have been effectively used for severely obese patients for over a decade without significant adverse effects, and administration of sympathomimetic drugs has been popular, these treatments can be invasive and dangerous to the patients.94,95 Recently, there has been an effort to reduce and reverse the hyperplasia in adipose tissue by inhibiting angiogenesis based on the hypothesis that the hyperplasia of white adipose tissue is dependent on angiogenesis.96–98 Angiostatin and endostatin, the endogenous inhibitors that act via inhibition of Col1 expression and c-Jun N-terminal kinase (JNK) pathway, were shown to reduce the body weight of obese mice. TNP-470 and VEGFR2-specific inhibitors have also been shown to prevent obesity in ob/ob mice subjected to a high-fat diet.65,66 TNP-470 is an antibiotic, which has antineoplastic effect in mammalian tissues. Its mechanism has been proposed to be an inactivation of methionine aminopeptidase 2 and an induction of p53 pathway.99 Alongside the normalization of white adipose tissue, TNP-470 also alleviates insulin insensitivity. However, it has also been shown to be neurotoxic which may alter appetite in test subjects and requires further investigation. Other potential therapeutic agents, such as a specific peptide motif that binds a specific vascular marker in white adipose tissue, were shown to have a positive effect but do not induce apoptosis. Given that antiangiogenic approaches may promote hypoxia and HIF-1 activity, further investigation will be required to evaluate the therapeutic potential of antiangiogenesis.
Given profound contributions of hypoxia to the development of obesity and insulin resistance as well as a significant attenuation of obesity and/or insulin resistance phenotypes in mice lacking HIF-1α in adipose tissue, one can argue for HIF-1 inhibition as a potential therapeutic strategy for obesity and insulin resistance. Two of these therapeutic agents, PX-478 and digoxin, have been recently tested on high-fat diet-induced obesity animal model by Sun et al.93 PX-478 (S-2-amino-3-[4’-N,N,-bis(chloroethyl)amino]phenyl propionic acid N-oxide dihydrochloride) is a potent HIF-1α inhibitor that was initially identified via high-throughput screening for compounds that inhibit HIF-1α expression and tumor growth.100 Although its mechanisms of inhibiting HIF-1α protein accumulation are not fully understood, recent studies suggest that PX-478 may inhibit HIF-1α via multiple mechanisms including transcription, translation, and protein degradation.101 HIF-1α inhibition by systemic PX-478 administration normalized high-fat diet-induced body weight gain and fat mass, indicating that overall adiposity was reduced. The fasting glucose levels and insulin sensitivity were also improved and energy expenditure was increased by PX-478 administration. Aberrant plasma levels of cholesterol, triglycerides, and leptin, as well as local adipose tissue leptin expression, were normalized in PX-478-treated mice. Furthermore, PX-478 treatment led to the reduction of inflammatory cytokines and adipose tissue macrophage infiltration. This is associated with decreased expression of fibrotic genes including Col1, Col3, and lysyl oxidase as well as reduced ECM accumulation.
A cardiac glycoside, digoxin, was identified through a library screening for a potential small-molecule HIF-1 inhibitor.102 The administration of digoxin and other cardiac glycosides showed a significant anticancer effect by inhibiting HIF-1α translation. Digoxin also showed potent beneficial anti-obese and antidiabetic effects in high-fat diet mice.93 Digoxin may have molecular targets other than HIF-1 and has a relatively narrow therapeutic index. Despite this limitation, digoxin has been a commonly prescribed drug for the treatment of heart failure. If the efficacy on obesity and insulin resistance could be guaranteed, further studies for safe use of this drug should be considered.
Taken together, these studies provide a proof-of-concept for HIF-1α inhibition as a novel therapeutic strategy to modify the detrimental obesity-induced tissue inflammation and insulin resistance. Intriguingly, a recent study by Rahtu-Korpela et al has shown that prolyl 4-hydroxylase-2 (P4H-2) inhibition, which stabilizes HIFs, protected against obesity and insulin resistance, indicating that activation of HIF could be used as a therapeutic strategy for obesity and associated phenotypes.103 Although, this study utilized whole-body p4h2-deficient mice and pharmacological administration of P4H inhibitor which may exert antiobesity and antidiabetic effects through modulating nonadipose tissues such as liver and muscle and/or through HIF-independent mechanisms. This accentuates the potential adverse outcomes in targeting HIF-1α in obesity and metabolic dysfunction.
Conclusion
Despite that a number of preclinical and clinical studies have repeatedly demonstrated the causal association of adipose tissue inflammation with insulin resistance, molecular cues that link between obesity, inflammation, and insulin resistance still remain to be delineated. Recent advancements identifying hypoxia as a prominent microenvironmental component of obese adipose tissue directs us toward the missing link (Figure 3): HIF-1 signaling that leads to the development of obesity-associated inflammation and insulin resistance. A number of small-molecule compounds and antisense oligonucleotides that inhibit HIF-1 activity in novel ways are currently in development or clinical trial.104–106 Thus, it is imperative that we gain a better understanding of how HIF-1 signaling exerts adipose tissue inflammation and insulin resistance.
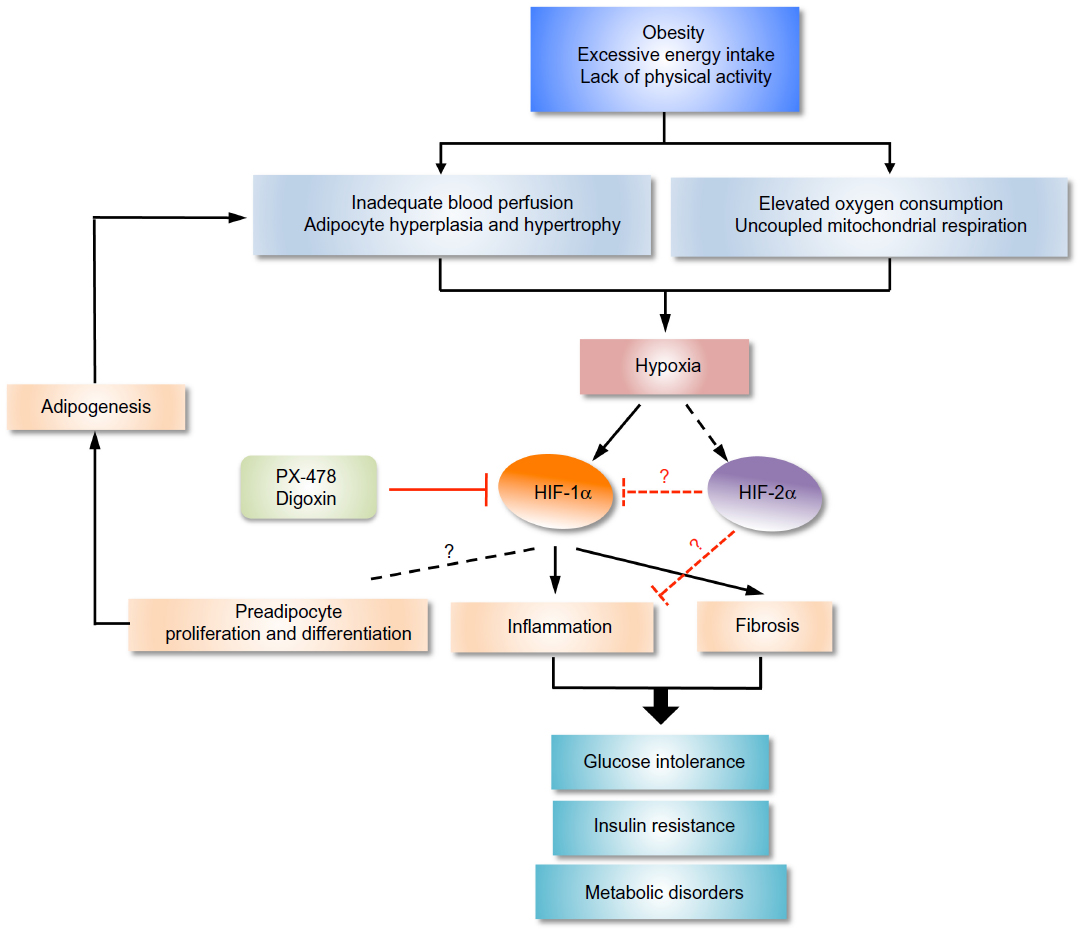 | Figure 3 Regulation of obesity and diabetes by HIFs. |
Acknowledgments
We thank the Kim Lab members for valuable discussions. This work was supported by the University of Texas at Dallas start-up grant. We sincerely apologize for inadvertent omission of any pertinent original references owing to space constraints.
Disclosure
The authors report no conflicts of interest in this work.
References
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. | |
Kayser B, Verges S. Hypoxia, energy balance and obesity: from pathophysiological mechanisms to new treatment strategies. Obes Rev. 2013;14(7):579–592. | |
Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. | |
Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond). 2008;32(3):451–463. | |
Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293(4):E1118–E1128. | |
Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18(4):478–489. | |
Jo J, Gavrilova O, Pack S, et al. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput Biol. 2009;5(3):e1000324. | |
Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106(7):2311–2317. | |
Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302(5652):1975–1978. | |
Sato Y, Endo H, Okuyama H, et al. Cellular hypoxia of pancreatic beta-cells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem. 2011;286(14):12524–12532. | |
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. | |
Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12(1):75–80. | |
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. | |
Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. | |
Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. | |
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. | |
Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9(9):609–617. | |
Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. 2013;13(4):646–653. | |
Tormos KV, Chandel NS. Inter-connection between mitochondria and HIFs. J Cell Mol Med. 2010;14(4):795–804. | |
Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. | |
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. | |
Halberg N, Khan T, Trujillo ME, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29(16):4467–4483. | |
Jiang C, Qu A, Matsubara T, et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes. 2011;60(10):2484–2495. | |
Kihira Y, Miyake M, Hirata M, et al. Deletion of hypoxia-inducible factor-1α in adipocytes enhances glucagon-like peptide-1 secretion and reduces adipose tissue inflammation. PloS One. 2014;9(4):e93856. | |
Krishnan J, Danzer C, Simka T, et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26(3):259–270. | |
Lee KY, Gesta S, Boucher J, Wang XL, Kahn CR. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 2011;14(4):491–503. | |
Boutin AT, Weidemann A, Fu ZX, et al. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell. 2008;133(2):223–234. | |
Cowburn AS, Takeda N, Boutin AT, et al. HIF isoforms in the skin differentially regulate systemic arterial pressure. Proc Natl Acad Sci U S A. 2013;110(43):17570–17575. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1 alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465–7475. | |
Kim JW, Evans C, Weidemann A, et al. Loss of fibroblast HIF-1alpha accelerates tumorigenesis. Cancer Res. 2012;72(13):3187–3195. | |
Stockmann C, Doedens A, Weidemann A, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456(7223):814–818. | |
Takeda N, O’Dea EL, Doedens A, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. | |
Tang N, Wang L, Esko J, et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6(5):485–495. | |
Weidemann A, Kerdiles YM, Knaup KX, et al. The glial cell response is an essential component of hypoxia-induced erythropoiesis in mice. J Clin Invest. 2009;119(11):3373–3383. | |
Lee KY, Russell SJ, Ussar S, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62(3):864–874. | |
Girgis CM, Cheng K, Scott CH, Gunton JE. Novel links between HIFs, type 2 diabetes, and metabolic syndrome. Trends Endocrinol Metab. 2012;23(8):372–380. | |
Berra E, Roux D, Richard DE, Pouysségur J. Hypoxia-inducible factor-1 alpha (HIF-1 alpha) escapes O(2)-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep. 2001;2(7):615–620. | |
Hoffman EC, Reyes H, Chu FF, et al. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252(5008):954–958. | |
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. | |
Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. | |
Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–427. | |
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. | |
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | |
Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15(4):621–627. | |
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11(1):72–82. | |
Covello KL, Kehler J, Yu H, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20(5):557–570. | |
Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26(9):3514–3526. | |
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23(24):9361–9374. | |
Warnecke C, Zaborowska Z, Kurreck J, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004;18(12):1462–1464. | |
Keith B, Johnson RS, Simon MC. HIF1 alpha and HIF2 alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. | |
Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25(13):5675–5686. | |
Shen C, Beroukhim R, Schumacher SE, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1(3):222–235. | |
Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14(6):435–446. | |
Branco-Price C, Zhang N, Schnelle M, et al. Endothelial cell HIF-1alpha and HIF-2alpha differentially regulate metastatic success. Cancer Cell. 2012;21(1):52–65. | |
Lee YS, Kim JW, Osborne O, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell. 2014;157(6):1339–1352. | |
Goossens GH, Bizzarri A, Venteclef N, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. 2011;124(1):67–76. | |
Hodson L, Humphreys SM, Karpe F, Frayn KN. Metabolic signatures of human adipose tissue hypoxia in obesity. Diabetes. 2013;62(5):1417–1425. | |
Kabon B, Nagele A, Reddy D, et al. Obesity decreases perioperative tissue oxygenation. Anesthesiology. 2004;100(2):274–280. | |
Pasarica M, Sereda OR, Redman LM, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 2009;58(3):718–725. | |
Ljungkvist AS, Bussink J, Rijken PF, Raleigh JA, Denekamp J, Van Der Kogel AJ. Changes in tumor hypoxia measured with a double hypoxic marker technique. Int J Radiat Oncol Biol Phys. 2000;48(5):1529–1538. | |
Rupnick MA, Panigrahy D, Zhang CY, et al. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99(16):10730–10735. | |
Bråkenhielm E, Cao R, Gao B, et al. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res. 2004;94(12):1579–1588. | |
White HM, Acton AJ, Considine RV. The angiogenic inhibitor TNP-470 decreases caloric intake and weight gain in high-fat fed mice. Obesity (Silver Spring). 2012;20(10):2003–2009. | |
Elias I, Franckhauser S, Ferré T, et al. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61(7):1801–1813. | |
Lu X, Ji Y, Zhang L, et al. Resistance to obesity by repression of VEGF gene expression through induction of brown-like adipocyte differentiation. Endocrinology. 2012;153(7):3123–3132. | |
Sun K, Wernstedt Asterholm I, Kusminski CM, et al. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109(15):5874–5879. | |
Sung HK, Doh KO, Son JE, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17(1):61–72. | |
Andreyev AYu, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP, Volkov NI. Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett. 1988;226(2):265–269. | |
Brand MD, Pakay JL, Ocloo A, et al. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392(Pt 2):353–362. | |
Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. | |
Marques BG, Hausman DB, Martin RJ. Association of fat cell size and paracrine growth factors in development of hyperplastic obesity. Am J Physiol. 1998;275(6 Pt 2):R1898–R1908. | |
Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87(3):377–389. | |
He Q, Gao Z, Yin J, Zhang J, Yun Z, Ye J. Regulation of HIF-1{alpha} activity in adipose tissue by obesity-associated factors: adipogenesis, insulin, and hypoxia. Am J Physiol Endocrinol Metab. 2011;300(5):E877–E885. | |
Lin Q, Lee YJ, Yun Z. Differentiation arrest by hypoxia. J Biol Chem. 2006;281(41):30678–30683. | |
Zhang X, Lam KS, Ye H, et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. J Biol Chem. 2010;285(43):32869–32877. | |
Shimba S, Wada T, Hara S, Tezuka M. EPAS1 promotes adipose differentiation in 3T3-L1 cells. J Biol Chem. 2004;279(39):40946–40953. | |
Park YK, Park B, Lee S, Choi K, Moon Y, Park H. Hypoxia-inducible factor-2α-dependent hypoxic induction of Wnt10b expression in adipogenic cells. J Biol Chem. 2013;288(36):26311–26322. | |
Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem. 2012;81:715–736. | |
Aurora RN, Punjabi NM. Obstructive sleep apnoea and type 2 diabetes mellitus: a bidirectional association. Lancet Respir Med. 2013;1(4):329–338. | |
Iiyori N, Alonso LC, Li J, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175(8):851–857. | |
Regazzetti C, Peraldi P, Gremeaux T, et al. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes. 2009;58(1):95–103. | |
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. | |
Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582(1):46–53. | |
Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280(9):7511–7518. | |
Labrecque MP, Prefontaine GG, Beischlag TV. The aryl hydrocarbon receptor nuclear translocator (ARNT) family of proteins: transcriptional modifiers with multi-functional protein interfaces. Curr Mol Med. 2013;13(7):1047–1065. | |
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. | |
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. | |
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–614. | |
Henegar C, Tordjman J, Achard V, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008;9(1):R14. | |
Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol. 2013;33(5):904–917. | |
Edelman S, Ng-Mak DS, Fusco M, et al. Control of type 2 diabetes after 1 year of laparoscopic adjustable gastric banding in the helping evaluate reduction in obesity (HERO) study. Diabetes Obes Metab. Epub May 13, 2014. | |
O’Brien PE, Sawyer SM, Laurie C, et al. Laparoscopic adjustable gastric banding in severely obese adolescents: a randomized trial. JAMA. 2010;303(6):519–526. | |
Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest. 2007;117(9):2362–2368. | |
Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov. 2010;9(2):107–115. | |
Cao Y. Angiogenesis as a therapeutic target for obesity and metabolic diseases. Chem Immunol Allergy. 2014;99:170–179. | |
Satchi-Fainaro R, Puder M, Davies JW, et al. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat Med. 2004;10(3):255–261. | |
Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2004;3(3):233–244. | |
Koh MY, Spivak-Kroizman T, Venturini S, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008;7(1):90–100. | |
Zhang H, Qian DZ, Tan YS, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105(50):19579–19586. | |
Rahtu-Korpela L, Karsikas S, Hörkkö S, et al. HIF prolyl 4-hydroxylase-2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes. Epub May 1,2014. | |
Ban HS, Uto Y, Nakamura H. Hypoxia-inducible factor inhibitors: a survey of recent patented compounds (2004–2010). Expert Opin Ther Pat. 2011;21(2):131–146. | |
Belozerov VE, Van Meir EG. Inhibitors of hypoxia-inducible factor-1 signaling. Curr Opin Investig Drugs. 2006;7(12):1067–1076. | |
Xia Y, Choi HK, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem. 2012;49:24–40. | |
Martens K, Bottelbergs A, Baes M. Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cremice: implications for metabolism research. FEBS Lett. 2010;584(5):1054–1058. | |
Urs S, Harrington A, Liaw L, Small D. Selective expression of an aP2/Fatty Acid Binding Protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res. 2006;15(5):647–653. | |
Zhang J, Wang Y, Gao Z, Yun Z, Ye J. Hypoxia-inducible factor 1 activation from adipose protein 2-cre mediated knockout of von Hippel-Lindau gene leads to embryonic lethality. Clin Exp Pharmacol Physiol. 2012;39(2):145–150. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.