Back to Journals » International Journal of Nanomedicine » Volume 15
Regulating Interactions Between Targeted Nanocarriers and Mononuclear Phagocyte System via an Esomeprazole-Based Preconditioning Strategy
Authors Belhadj Z ![]() , He B, Fu J
, He B, Fu J ![]() , Zhang H, Wang X, Dai W
, Zhang H, Wang X, Dai W ![]() , Zhang Q
, Zhang Q
Received 13 April 2020
Accepted for publication 5 August 2020
Published 25 August 2020 Volume 2020:15 Pages 6385—6399
DOI https://doi.org/10.2147/IJN.S258054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Zakia Belhadj,1 Bing He,1 Jijun Fu,2 Hua Zhang,1 Xueqing Wang,1 Wenbing Dai,1 Qiang Zhang1
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, People’s Republic of China; 2School of Pharmaceutical Sciences, Guangzhou Medical University, Guangzhou, 511436, People’s Republic of China
Correspondence: Qiang Zhang; Wenbing Dai Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, People’s Republic of China
Tel +86-10-82802791
; Tel +86-10-82802791
Email [email protected]; [email protected]
Purpose: The mononuclear phagocyte system (MPS) presents a formidable obstacle that hampers the delivery of various nanopreparations to tumors. Therefore, there is an urgent need to improve the off-MPS targeting ability of nanomedicines. In the present study, we present a novel preconditioning strategy to substantially increase the circulation times and tumor targeting of nanoparticles by regulating nanocarrier-MPS interactions.
Methods: In vitro, the effect of different vacuolar H+-ATPase inhibitors on macrophage uptake of targeted or nontargeted lipid vesicles was evaluated. Specifically, the clinically approved proton-pump inhibitor esomeprazole (ESO) was selected as a preconditioning agent. Then, we further investigated the blocking effect of ESO on the macrophage endocytosis of nanocarriers. In vivo, ESO was first intravenously administered into A549-tumor-bearing nude mice, and 24 h later, the c(RGDm7)-modified vesicles co-loaded with doxorubicin and gefitinib were intravenously injected.
Results: In vitro, ESO was found to reduce the interactions between macrophages and c(RGDm7)-modified vesicles by interfering with the latter’s lysosomal trafficking. Studies conducted in vivo confirmed that ESO pretreatment greatly decreased the liver and spleen distribution of the targeted vesicles, enhanced their tumor accumulation, and improved the therapeutic outcome of the drug-loaded nanomedicines.
Conclusion: Our findings indicate that ESO can regulate the nanoparticle-MPS interaction, which provides a feasible option for enhancing the off-MPS targeting of nanomedicines.
Keywords: mononuclear phagocyte system, preconditioning strategy, esomeprazole, V-ATPase inhibitors, targeted lipid vesicles
Introduction
A major challenge in the field of nanotechnology is the inability to deliver therapeutic moieties to target sites because of the tendency of phagocytic cells of the mononuclear phagocyte system (MPS) to rapidly sequester and clear nanoparticles (NPs) from the blood. Understanding MPS-NP interactions is crucial to developing effective nanotechnologies for therapeutic and in vivo imaging applications. Studies and meta-analyses have shown that more than 99% of injected anticancer agents are cleared by the organs of the MPS, primarily by Kupffer cells, leaving only a median of 0.7% (or 0.76%) to be delivered to solid tumors.1–4 An insufficient understanding of the interactions between NPs and biological systems, such as the formation of protein corona, NP-MPS interactions, and tumor uptake, explains the failure to produce NPs that can overcome these biological barriers.5–8
Various strategies to nanoparticle design have been developed to prolong circulation and enhance drug delivery to diseased tissues, several of which involve optimizing particles’ physicochemical characteristics, including size and architecture,9,10 and surface coating with hydrophilic polymer materials, such as polyethylene glycol (PEG).11,12 Another approach employs a reticuloendothelial system (RES, an older term for MPS) blockade strategy with blank conventional liposomes or D-self-peptide-labeled liposomes, or by depletion of MPS macrophages with an array of inorganic and organic materials.13–16 In addition, bioinspired strategies using nanocarriers from nature have been used to reduce nanoparticle uptake by the MPS.17–19 Although strategies based on unapproved drugs or complicated materials have made significant progress toward overcoming these biological barriers, translating that progress into clinical applications has proven difficult. As a result, there is growing interest in repositioning FDA-approved drugs that can minimize non-specific uptake of nanoparticles by MPS.
Proton-pump inhibitors (PPIs), the most widely administered medication worldwide for the reduction of stomach acid secretion, have proven to be safe and well-tolerated. Several reports have shown that PPIs inhibit H+/K+-ATPase in gastric parietal cells and vacuolar H+-ATPase (V-ATPase) overexpressed in tumor cells and liver macrophages (Kupffer cells).20,21 V-ATPases are localized in the membranes of acidic organelles, such as lysosomes, endosomes, secretory vesicles, and plasma membranes, and are important regulators of the transmembrane pH gradient.22–24 Because the macrophage endolysosomal system is active and exhibits high lysosomal enzyme expression levels, particularly V-ATPase,25 (the primary membrane channel involved in the drug sequestration), it is believed that inhibiting V-ATPase in the endolysosomal system prior to the injection of vesicles may decrease their MPS clearance and lead to their accumulation in tumors. V-ATPase inhibitors are weak bases that tend to be trapped in the acidic lysosomal lumen through protonation.26,27 Various approaches have been used to abolish lysosomal drug sequestration in hopes of sensitizing multidrug-resistant cells to chemotherapy drugs.28 PPIs pretreatment reportedly induces an increase in both extracellular and lysosomal pH via the inhibition of V-ATPase activity, which may sensitize cancer cells that are resistant to chemotherapy drugs.29 Bafilomycin A1, a V-ATPase inhibitor, has been reported to restore the sensitivity of weak chemotherapeutic bases to drug-resistant proximal tubule cells, but bafilomycin A1 has been shown to be toxic in in vivo studies.28 The efficacy of PPIs in combination with chemotherapeutic agents for cancer treatment has been evaluated in clinical trials. One Phase III clinical trial demonstrated that PPIs improved the antitumor effect of docetaxel in combination with cisplatin in metastatic breast cancer patients.30
In the current study, we sought to use clinically approved V-ATPase inhibitors as endocytosis inhibitors to minimize monocyte–macrophage uptake of nanoparticles and investigate the possibility of decreasing the MPS clearance and increasing the circulation time of targeted lipid vesicles. First, a cyclic homing peptide “c(RGDm7)” was modified on the phospholipid vesicles loaded with both a cytotoxic agent (DOX) and epidermal growth factor receptor (EGFR) inhibitor (GE) to achieve sequential release of these anticancer drugs (Figure 1). Then, we examined the effect of different V-ATPase inhibitors on macrophage uptake of targeted and plain vesicles to select the appropriate preconditioning agent. Accordingly, esomeprazole (ESO) was selected for further studies. The in vivo biodistribution and antitumor efficacy of targeted drug delivery systems after ESO pretreatment were then investigated.
 |
Figure 1 Schematic representation of ESO-based preconditioning strategy for reduction of nanoparticle-MPS interaction. |
Materials and Methods
Integrin αvβ3 Expression in A549 and MCF-7 Cells
The human lung carcinoma cells (A549) and human breast cancer cell line (MCF-7) were obtained from the Institute of Basic Medical Science, Chinese Academy of Medical Sciences (Beijing, China). The cells were seeded on glass bottom dishes for 24 h. The cells were then rinsed three times with PBS, and fixed with 4% paraformaldehyde. After being blocked with 5% BSA at 37 °C for 1 h, the cells were incubated with integrin αvβ3 rabbit monoclonal antibodies (ab75872, Abcam, UK), followed by incubation with secondary antibody Texas Red-conjugated Affinipure Goat Anti-rabbit IgG at 37 °C for 1 h. Cell nuclei were dyed with Hoechst 33258, and the cells were visualized using a confocal laser scanning microscope (CLSM, Heidelberg, Germany).
Synthesis and Characterization of c(RGDm7)-PEG-DSPE
Nucleophilic substitution reaction was used to conjugate c(RGDm7) with NHS-PEG2000-DSPE.31 c(RGDm7) was dissolved in anhydrous DMF and mixed with NHS-PEG2000-DSPE at a molar ratio of 1:2. The pH was titrated to 8.0 with triethylamine, and the reaction was maintained for 120 h at ambient temperature. After further purification using dialysis (MWCO 3500 Da) and lyophilization, c(RGDm7)-PEG-DSPE was collected and analyzed by MALDI-TOF-MS.
Preparation and Characterization of Lipid Vesicles Modified with c(RGDm7)-PEG-DSPE
Lipid vesicles were prepared according to a previously reported method.32 Briefly, the lipid composition of ligand-modified lipid vesicles (c(RGDm7)-LS) was EPC, cholesterol, mPEG-DSPE, and c(RGDm7)-PEG-DSPE (55:40:3:2 molar ratio), and EPC, cholesterol, mPEG-DSPE (55:40:5 molar ratio) for PEGylated vesicles known as liposomes (LS). For DiO-, and DiR-labeled vesicles, 1 mol % DiO or DiR was added to the lipid mixture. The solvent was rotary evaporated at 40 °C, and the lipid film dried overnight at room temperature. The resulting film was hydrated with normal saline solution at 60 °C for 30 min. After hydration, the mixture was serially extruded with polycarbonate membranes of 200, 100, and 50 nm on Avanti Mini-Extruder (Avanti Polar Lipids). For the preparation of dual drug-loaded vesicles, lipids and GE at a weight ratio of 20:1 were evaporated at 40 °C and dried under vacuum overnight at room temperature to form a thin film. This lipid film was hydrated with 0.32 M (NH4)2SO4 at 60 °C for 30 min, and extruded across a series of polycarbonate membranes (from 200 to 50 nm) using an Avanti Mini-Extruder (Avanti Polar Lipids). Gel filtration over a Sephadex G50 column was performed to form unilamellar vesicles. DOX was then actively remote-loaded into lipid vesicles using an ammonium sulfate gradient procedure as reported previously.33 The final concentrations of GE, DOX and phospholipid used in the formulation were approximately 0.6 mg/mL, 1 mg/mL, and 15 mg/mL, respectively. The particle size distribution and morphology of different vesicles were determined using a Malvern Zetasizer (Malvern Instruments, Malvern, UK) and transmission electron microscope (TEM) (H-7000, Hitachi, Japan), respectively.
In vitro Cell Viability
The murine macrophage RAW264.7 cell line was purchased from the Institute of Basic Medical Science, Chinese Academy of Medical Sciences (Beijing, China). The cell viability of different drugs (omeprazole, dexlansoprazole, ESO, enoxacin, and chloroquine) on RAW 264.7 cells was estimated by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay using the untreated group as a control. RAW 264.7 cells (3000 cells per well) were cultured in 96-well plates for 24 h. Serial concentrations of drugs were added to cells for 24, and 48 h incubation. Afterward, 10 μL of MTT solution (5 mg/mL) was added into each well, followed by incubation for another 4 h at 37 °C. The absorbance at 490 nm representing cell viability was determined using a microplate reader.
Cellular Uptake Study in vitro
Murine macrophage cells (RAW 264.7) were used to investigate the effect of V-ATPase inhibitors on macrophage uptake of a subsequent dose of vesicles. Briefly, cells grown overnight in 12-well plates were pretreated with various concentrations of omeprazole, dexlansoprazole, ESO, enoxacin, or chloroquine at 37 °C for 24 h. Lipid vesicles (LS-DiO, c(RGDm7)-LS-DiO) at a final concentration of 5 μM were then added into the wells and further incubated at 37 °C for 4 h. Cellular uptake was quantitatively analyzed using a FACSAria II flow cytometer (BD Biosciences).
Similarly, the cellular internalization of modified and unmodified vesicles by A549 cells was evaluated qualitatively by confocal fluorescence microscopy and quantitatively by flow cytometry. Briefly, cells were cultured in 12-well plates in DMEM at 37 °C for 24 h. LS-DiO or c(RGDm7)-LS-DiO (5 μM) were incubated with cells for 4 h. For qualitative cellular uptake measurements, cells were rinsed thrice with PBS and fixed with paraformaldehyde. Cells were then dyed with Hoechst 33258 for imaging under a CLSM (TCS SP5, Leica, Germany). Cells were also analyzed using a FACSAria II flow cytometer (BD Biosciences).
Mechanism of Blocking Nanoparticle Uptake
Lysosomal co-localization was performed to assess the blocking effect of ESO on macrophage uptake of nanoparticles. Briefly, RAW 264.7 cells were pretreated with ESO at a concentration of 200 μM for 24 h, then incubated with 5 μM LS-DiO or c(RGDm7)-LS-DiO for 4 h, followed by incubation with 50 nM LysoTracker for 60 min and 10 mM Hoechst 33258 for 10 min to observe lysosomes and nuclei, respectively. Cells were then gently rinsed thrice with ice-cold PBS for visualization under CLSM (TCS SP5, Leica, Germany).
In vitro Cytotoxicity
In vitro cytotoxicity of nanoformulations was evaluated using MTT assay. After 24 h of culture, A549 cells (3× 103 cells per well) in 96-well culture plates were incubated with various formulations at a series of DOX concentrations. After 72 h, the absorbance was measured at 490 nm using a plate reader.
Biodistribution of Nanocarriers Pretreated with ESO
Peking University Health Science Center approved the animal experiments and were performed in compliance with guidelines evaluated by the Ethics Committee of Peking University. A549 cells (5 × 106, 100 μL) were injected subcutaneously into the right armpit of each nude mouse to establish an animal lung tumor model. To evaluate the effect of ESO on vesicle biodistribution, nude mice were pre-injected with ESO at a dose of 3 mg/kg. After 24 h, 100 μL DiR-labeled vesicles were intravenously injected to animals. Four hours later, the major organs and tumor tissues were carefully collected, rinsed with cold PBS for ex vivo imaging.
The immunofluorescence study was further performed by intravenously injecting various DiO-loaded vesicles pretreated with or without ESO into A549 tumor-bearing nude mice 14 days post-implantation. Four hours after injection, mice were sacrificed, and tumor tissues were collected and frozen in Tissue-Tek® O.C.T. compound and fixed in paraformaldehyde. Samples were then cut into 3 μm slices, followed by incubation with anti-CD31 antibody overnight to visualize tumor angiogenesis. Slides were stained with secondary antibody and Hoechst, and then observed under CLSM.
Pharmacokinetics Study
SD rats were used to investigate the efficiency of ESO pretreatment on pharmacokinetic properties of lipid vesicles. Briefly, rats were stochastically divided into four groups (6 rats per group) and administrated LS-GE/DOX, c(RGDm7)-LS-GE/DOX, ESO + LS-GE/DOX and ESO + c(RGDm7)-LS-GE/DOX at a DOX dose of 6 mg/kg and a GE dose of 2.4 mg/kg. At predetermined time points (0.083, 0.167, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h post-i.v. injection), blood samples were collected, centrifuged, and the separated plasma was analyzed by HPLC. Pharmacokinetics parameters (area-under curve (AUC(0−t)), mean residence time (MRT(0−t)), and plasma half-life (t1/2) were determined using a non-compartmental model by Kinetica 4.4 (Thermo, USA).
In vivo Antitumor Efficacy Studies
Xenograft models for lung cancer were established by subcutaneously injecting 5 × 106 A549 cells into the right flank of BALB/c nude mice. Five groups of 8 mice each were intravenously administrated with 100 μL of saline, LS-GE/DOX, c(RGDm7)-LS-GE/DOX, LS-GE/DOX (pretreated with 3 mg/kg of ESO for 24 h), or c(RGDm7)-LS-GE/DOX (pretreated with 3 mg/kg of ESO for 24 h) at a DOX dose of 2 mg/kg and a GE dose of 0.8 mg/kg for 10 days. Tumor sizes were measured with a digital caliper every two days, and tumor volumes were calculated using the formula (L ×W2)/2 where L is the largest and W is the smallest diameter, respectively. Relative body weight change was monitored throughout the experiment. At day 12 after treatment, some mice were sacrificed by cervical dislocation, the tumors and main organs (heart, liver, spleen, lung, and kidney) were harvested. The fixed tumors sections were subjected to TUNEL apoptosis and CD31 staining. The other organs were sectioned and stained with hematoxylin and eosin (H&E). Animals were euthanized when tumor size was >2000 mm3, which was considered as the endpoint of survival data.
Biological Safety Evaluation
In vivo toxicity of nanocarriers was examined on healthy normal mice randomized into five groups (n = 3) and treated as previously described. At the 12th day after treatment, blood samples were obtained from all mice for the measurement of serum enzyme markers of heart (CK: creatinine kinase, LDH: lactate dehydrogenase), liver (ALT: alanine aminotransferase, AST: aspartate aminotransferase), and renal function tests (CREA: creatinine and BUN: blood urea nitrogen).
Statistical Analysis
Quantitative data are reported as mean ± SD. Student’s t-test was used to compare two groups, and one-way ANOVA was used to compare multiple groups. Data were analyzed using GraphPad Prism 6.0 software. The level of significance was set at p < 0.05.
Results and Discussion
Receptor Expression and Characterization of c(RGDm7)-PEG-DSPE
A receptor expression examination showed that A549 cells were αvβ3 integrin-positive, while MCF-7 cells expressed lower integrin levels (Figure S1). Then the ligand c(RGDm7) was attached to the distal end of NHS-PEG-DSPE through the reaction of amino groups with active groups of NHS. MALDI-TOF-MS indicated that the NHS-PEG-DSPE peak (with an average molecular weight of 2800 Da) was right-shifted after c(RGDm7) attachment. The analyzed molecular weight of the c(RGDm7)-PEG-DSPE was approximately 3400 Da, and the difference in mass was consistent with the theoretical molecular weight of the c(RGDm7) (Figure S2). This analysis proved that c(RGDm7)-PEG-DSPE was successfully synthesized.
Characterization of Lipid Vesicles
Dynamic light scattering was used to determine the mean particle size of different lipid vesicles. Both plain and modified vesicles displayed similar particle size distributions, indicating that the presence of a ligand did not affect their physical characteristics. More than 90% of DOX and 50% of GE were encapsulated in lipid vesicles. The precipitation of GE due to lower-energy macroscopic crystals in water resulted in relatively lower encapsulation efficiency compared to that of DOX.34 Additionally, TEM revealed that the particles were spherical with nanometer-sizes (Figure 2).
 |
Figure 2 Characterization of different nanoformulations. Size distribution (A) and TEM (B) of c(RGDm7)-LS-GE/DOX, c(RGDm7)-LS-DOX, c(RGDm7)-LS-GE, and LS-GE/DOX. |
In vitro Cell Viability
The cell viability of RAW 264.7 cells after treatment with V-ATPase inhibitors (omeprazole, dexlansoprazole, ESO, enoxacin) and antimalarial drug (chloroquine) was assessed to confirm their biocompatibility and toxic dose ranges. Following 24 h of incubation, a slight decrease was observed in the viability of RAW 264.7 cells treated with various V-ATPase inhibitors, while a significant toxicity was observed with even 100 μM chloroquine when compared with the control at all drug concentrations. Following 48 h of treatment, a decrease in cell viability was detected when the concentration of V-ATPase inhibitor was 150 μM or higher (Figure 3). These results indicated the biosafety of V-ATPase inhibitors which did not damage macrophage cells even at higher concentrations. Based on the cell viability analysis, a concentration ranged from 5 to 200 μM with an incubation time of 24 h was selected for further study.
 |
Figure 3 Effect of different drugs on the cell viability of RAW 264.7 cells. MTT assay was used to assess the viability of RAW 264.7 cells after 24 h (A), and 48 h (B) of incubation; data are shown as mean ± SD (n = 3). |
Cellular Uptake and Lysosomal Localization
The effect of V-ATPase inhibitors on vesicle uptake in RAW 264.7 cells was assessed. Flow cytometry analysis showed that drug concentrations of 25 μM and 50 μM had a negligible influence on the cellular uptake of vesicles (Figure 4). It was found that ESO was more effective than other V-ATPase inhibitors, and its effect on macrophage uptake of targeted and nontargeted vesicles was concentration-dependent. As the concentration increased to 200 μM, ESO pretreatment effectively reduced macrophage endocytosis of both c(RGDm7)-LS and unmodified vesicles.
 |
Figure 4 Effect of different V-ATPase inhibitors on macrophage uptake of c(RGDm7)-LS (A), and LS (B). RAW 264.7 cells were pretreated with different concentrations of V-ATPase inhibitors at 37 °C for 24 h. Nanocarriers (LS-DiO, c(RGDm7)-LS-DiO) at a final concentration of 5 μM were incubated with cells at 37 °C for 4 h. Data were mean ± SD (n = 3, **p < 0.01, ***p < 0.001). |
Fluorescence microscopy (Figure 5) revealed that the vesicles could co-localize with lysosomes in RAW 264.7 cells. However, pretreatment with ESO significantly decreased their co-localization with lysosomes. The main function of Kupffer cells is to sequester nanoparticles circulating from the bloodstream. Upon internalization by Kupffer cells, NPs are delivered to early endosomes, late endosomes and lysosomes, respectively, through an intracellular trafficking process, and finally degraded in lysosomes.35–38 As previously reported, an alkaline lysosome environment induced by PPIs pretreatment can impair lysosomal enzymes which are active at a lower pH optimum of approximately 4.80.39,40 Previous study has clarified that PPIs are enriched in acidic organelles, where they are activated and affect V-ATPases.21 In the present study, ESO could alter the phagocytic ability of macrophages by inhibiting lysosomal enzymes which could in turn decrease the macrophage uptake of the subsequently injected NPs. Therefore, macrophage sequestration of NPs will be impaired by ESO pretreatment. Moreover, PPIs pretreatment has been shown to elevate the sensitivity of various cell lines to chemotherapeutic drugs.41–43 On the one hand, ESO can decrease macrophage uptake of NPs; on the other, it can enhance uptake in tumor cells, making it an attractive element in combination antitumor drug therapy.
 |
Figure 5 Influence of ESO on lysosomal co-localization of DiO-loaded LS in RAW 264.7 cells. Scale bar =10 μm. |
The cellular uptake of c(RGDm7)-LS on human non-small cell lung cancer (NSCLC) A549 cells was also investigated. The c(RGDm7)-LS could be preferentially taken up by A549 cells via interaction between RGD and integrin αvβ3 overexpressed on tumor cells. This interaction increased the endocytosis of targeted vesicles by 4.65-fold compared with nontargeted vesicles. The findings by confocal microscopy images were consistent with that by flow cytometry analysis (Figure 6).
 |
Figure 6 Cellular uptake of DiO-loaded LS in A549 cells. (A) CLSM images of A549 cells incubated with different formulations at 37 °C for 4 h. (B) Flow cytometry analysis of cell uptake. ***p < 0.001 (Scale bar =10 μm). |
Cytotoxicity of c(RGDm7)-LS-GE/DOX
The cell viabilities of free DOX and different vesicular formulations on A549 cells were quantified by MTT assay (Figure 7). The anti-proliferation effect of different treatments exhibited a concentration-dependent pattern. Free DOX showed the best antiproliferative effect in A549 cells due to quick internalization by the cells.44 The c(RGDm7)-LS-GE/DOX yielded the lowest IC50 value among all the vesicular formulations, which could be attributed to the markedly enhanced cellular internalization by the c(RGDm7) modification.
 |
Figure 7 Cytotoxic effect of various nanoformulations on A549 cells. Cell viability was investigated using MTT assay after 72 h of incubation (mean ± SD, n = 3). |
Preconditioning Strategy in vivo
In cellular studies, ESO was the most effective preconditioning drug for blocking macrophages. To further investigate whether ESO pretreatment is the optimal preconditioning strategy in vivo, we performed the in vivo and ex vivo distribution of various DiR-labeled vesicles in nude mice bearing subcutaneous A549 tumor xenografts. It was proved that pretreatment with ESO reduced the accumulation of labeled vesicles in liver and spleen. Moreover, the tumoritropic delivery of c(RGDm7)-LS was enhanced following ESO pretreatment. As shown in Figure 8, the ESO + c(RGDm7)-LS group exhibited the highest fluorescence signal in the tumor region compared with the c(RGDm7)-LS group and unmodified vesicles with or without ESO pretreatment. The tumor accumulation was approximately 5.35-fold higher in the ESO + c(RGDm7)-LS group than that in the LS group, which might be due to the inhibiting of lysosomal trafficking by ESO, leading to decreased liver and spleen accumulation of c(RGDm7)-LS. Although nontargeted vesicles did not exhibit higher liver biodistribution, tumor accumulation was lower compared with targeted nanoparticles. This indicated that the preconditioning strategy and ligand modification together could decrease MPS uptake of c(RGDm7)-LS and enhance its tumor accumulation.
 |
Figure 8 Effect of ESO on the biodistribution of nanocarriers. (A) In vivo fluorescence images of A549 tumor-bearing nude mice 4 h post-injection of different formulations. (B) Quantitative analysis of the mean fluorescence intensity in the main organs 4 h after i.v. administration of nanocarriers. (C) Ex vivo images of harvested main organs and tumors after intravenous injection of different formulations. (D) Mean fluorescence intensity from DiR-labeled vesicles in tumors. Data are presented as mean ± SD (n=3, **p < 0.01, ***p < 0.001). |
Acidification of the different cellular compartments by V-ATPase is reportedly necessary for intracellular trafficking within the endolysosomal system.45–47 Because macrophages express proton-pump V-ATPases at high levels on their plasma membranes and specialized intracellular compartments, particularly in lysosomal acidification mechanisms,25,48 it is expected that ESO could reduce lysosomal trafficking and consequently decrease intracellular nanoparticle sequestration by inhibiting V-ATPase-dependent lysosome acidification. Integrin αvβ3 is also expressed on the tumor vasculature, which increases the tumor retention of nanocarriers.49,50 RGD peptides have been demonstrated to promote the transcytosis of nanocarriers into tumors.51,52 As mentioned above, integrin αvβ3 receptors were highly expressed on lung tumor cells, and the cellular uptake of targeted vesicles by A549 cells was evident. The attachment of (cRGD) peptides onto polymeric micelles with plasmid DNA has been previously shown to treat intractable pancreatic tumors through the ligand and integrin interaction on tumor vasculature.53
An immunofluorescence study was also carried out to assess the feasibility of the preconditioning strategy (Figure S3). The ESO + c(RGDm7)-LS group tended to accumulate more in tumor regions compared with the other three groups, which is consistent with in vivo biodistribution. Furthermore, ESO + c(RGDm7)-LS co-localized well with CD31-labeled blood vessels, demonstrating an ability to target tumor neovasculature.
In vivo Circulation Studies
A pharmacokinetics study demonstrated that ESO pretreatment could prolong the circulation time of c(RGDm7)-LS-GE/DOX (Figure 9), which is of considerable significance to drug delivery. A preconditioning strategy was observed to increase the area under the curve (AUC), blood-circulation half-life (t1/2), and mean residence time (MRT) compared with those of c(RGDm7)-LS-GE/DOX (Table S1-S2). The plasma concentration of DOX was maintained at high levels even at 72 h, but the level of GE could only be detected within 24 h. Compared with the c(RGDm7)-LS group, the ESO + c(RGDm7)-LS-GE/DOX group was 1.86/2.12-fold higher in AUC, 1.23/1.91-fold higher in t1/2 and 1.21/1.98-fold higher in MRT, based on the analysis of DOX and GE, respectively. This confirmed that a preconditioning strategy using ESO enabled effective blood persistence of DOX and GE.
 |
Figure 9 In vivo pharmacokinetics of nanocarriers with and without ESO pretreatment. Plasma DOX (A) and GE (B) concentration-time profiles of nanoformulations in rats (n = 6). |
In vivo Antitumor Efficacy
After demonstrating the efficiency of ESO pretreatment in enhancing tumor targeting of c(RGDm7)-LS, we then examined the antitumor efficacy in five groups of mice (n = 8 per group) bearing subcutaneous A549 tumors. Both tumor volume and body weights were recorded after the different treatments were initiated (Figure 10A and C). Remarkably, ESO + c(RGDm7)-LS-GE/DOX showed the most significant antitumor efficacy in reducing tumor growth in comparison with the other treatment groups. The therapeutic efficacy was also checked by monitoring survival time of mice (Figure 10B). ESO + c(RGDm7)-LS-GE/DOX significantly extended the overall survival of A549 tumor–bearing mice, with a median survival time of 35 days compared with that of the saline group (21 days, p < 0.001), LS-GE/DOX group (24 days, p < 0.001), c(RGDm7)-LS-GE/DOX group (28 days, p < 0.01), and ESO + LS-GE/DOX group (30 days, p < 0.005). These data confirmed the enhanced therapeutic efficiency of c(RGDm7)-LS-GE/DOX pretreated with ESO. Taken together, ESO could inhibit V-ATPase in lysosomes of Kupffer cells and then decrease the sequestration of NPs. Moreover, ESO might sensitize lung tumor cells to DOX and GE, leading to a stronger therapeutic effect of c(RGDm7)-LS-GE/DOX. More detailed information about tumors and corresponding immunohistochemical studies (TUNEL, CD31 staining) in different groups was conducted (Figure 10D and E), respectively. Collectively, consistent with previous data, ESO + c(RGDm7)-LS-GE/DOX induced the smallest tumor size, the greatest cell apoptosis, the best inhibitory effect on angiogenesis, and the longest survival time. All these indicated that the ligand modification and preconditioning strategy could significantly improve the therapy outcome of tumor.
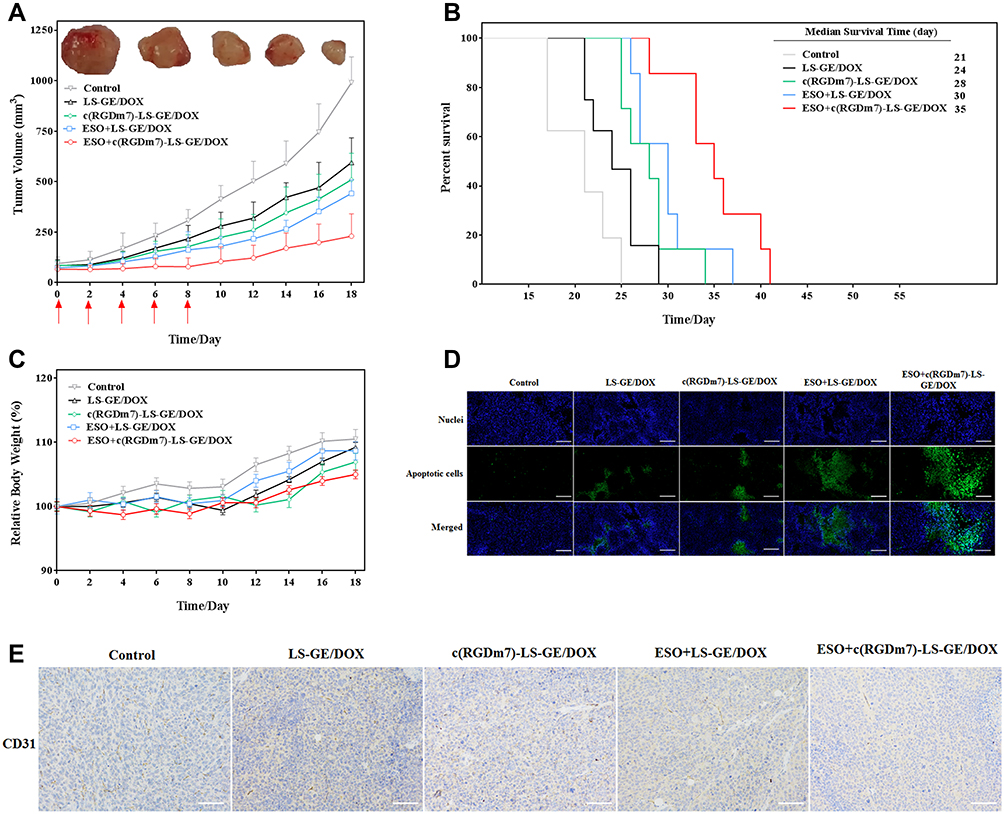 |
Figure 10 In vivo antitumor efficacy of ESO + c(RGDm7)-LS-GE/DOX. (A) Tumor growth curves and images of excised A549 tumors (n = 8). Red arrows indicate treatment. (B) Survival of the mice over time (n = 7). (C) Relative body weight of treated tumor-bearing mice over time (n = 8; mean ± SD). (D) TUNEL apoptosis and (E) CD31 staining performed on A549 tumor sections. Scale bar = 200 μm. |
Biosafety Evaluation
The toxicity of nanocarriers was investigated by H&E stained histology analysis, and blood biochemistry. Histological examinations showed no obvious signs of damage on the main organs in the treated groups (Figure S4A). In addition, no noticeable heart, hepatic, nor renal toxicity was observed in all treated groups, as indicated by the corresponding values of function markers for heart (CK, LDH), liver (ALT, AST), and kidney (CREA, BUN) (Figure S4B). We found that ESO-based preconditioning strategy in combination with a lipid vesicular drug delivery system produced a safe and biocompatible treatment without significant adverse effects.
Conclusion
In conclusion, we demonstrated that an ESO-based preconditioning strategy is a promising approach to reducing MPS uptake and improving the targeting efficiency of nanocarriers. In vitro experiments indicated that ESO pretreatment could significantly decrease the uptake of targeted vesicles by macrophage cells compared with those without pretreatment. In vivo data showed that a preconditioning strategy with c(RGDm7) modification resulted in lower MPS distribution and higher tumor accumulation of the ESO + c(RGDm7)-LS group. Moreover, ESO + c(RGDm7)-LS-GE/DOX achieved outstanding antitumor activity. This preconditioning strategy based on an FDA-approved drug may expand the range of applications of various nanoparticles, and open the door to the discovery of other types of preconditioning agents. This study may lead to a viable strategy to improve the efficiency of targeted drug delivery systems.
Abbreviations
MPS, mononuclear phagocyte system; ESO, esomeprazole; PPI, proton-pump inhibitor; V-ATPase, vacuolar H+-ATPase; DOX, doxorubicin; GE, gefitinib; CLSM, confocal laser microscope; TEM, transmission electron microscope.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. doi:10.1016/j.jconrel.2016.01.020
2. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014. doi:10.1038/natrevmats.2016.14
3. MacParland SA, Tsoi KM, Ouyang B, et al. Phenotype determines nanoparticle uptake by human macrophages from liver and blood. ACS Nano. 2017;11(3):2428–2443. doi:10.1021/acsnano.6b06245
4. Cheng YH, He C, Riviere JE, Monteiro-Riviere NA, Lin Z. Meta-analysis of nanoparticle delivery to tumors using a physiologically based pharmacokinetic modeling and simulation approach. ACS Nano. 2020;14(3):3075–3095. doi:10.1021/acsnano.9b08142
5. Tsoi KM, MacParland SA, Ma X-Z, et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15(11):1212. doi:10.1038/nmat4718
6. Pelaz B, Del Pino P, Maffre P, et al. Surface functionalization of nanoparticles with polyethylene glycol: effects on protein adsorption and cellular uptake. ACS Nano. 2015;9(7):6996–7008. doi:610.1021/acsnano.5b01326
7. Treuel L, Brandholt S, Maffre P, Wiegele S, Shang L, Nienhaus GU. Impact of protein modification on the protein corona on nanoparticles and nanoparticle-cell interactions. ACS Nano. 2014;8(1):503–513. doi:10.1021/nn405019v
8. Nel AE, Madler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat2442
9. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
10. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532. doi:10.1002/smll.201201390
11. Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–2266. doi:10.1016/j.bbamem.2009.06.022
12. Kong L, Campbell F, Kros A. DePEGylation strategies to increase cancer nanomedicine efficacy. Nanoscale Horiz. 2019;4:378. doi:10.1039/C8NH00417J
13. Liu T, Choi H, Zhou R, Chen IW. RES blockade: a strategy for boosting efficiency of nanoparticle drug. Nano Today. 2015;10(1):11–21. doi:10.1016/j.nantod.2014.12.003
14. Sun X, Yan X, Jacobson O, et al. Improved tumor uptake by optimizing liposome based RES blockade strategy. Theranostics. 2017;7(2):319–328. doi:10.7150/thno.18078
15. Tavares AJ, Poon W, Zhang Y-N, et al. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc Natl Acad Sci U S A. 2017;114(51):10871–E10880. doi:10.1073/pnas.1713390114
16. Tang Y, Wang X, Li J, et al. Overcoming the reticuloendothelial system barrier to drug delivery with a “don’t-eat-us” strategy. ACS Nano. 2019;13:13015–13026. doi:10.1021/acsnano.9b05679
17. Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10(7):521–535. doi:10.1038/nrd3499
18. Anselmo AC, Gupta V, Zern BJ, et al. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano. 2013;7(12):11129–11137. doi:10.1021/nn404853z
19. Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi:10.1038/nnano.2012.1212
20. Kuchuk O, Tuccitto A, Citterio D, et al. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology. 2018;7(7):e1445452. doi:10.1080/2162402x.2018.1445452
21. Wu D, Qiu T, Zhang Q, et al. Systematic toxicity mechanism analysis of proton pump inhibitors: an in silico study. Chem Res Toxicol. 2015;28(3):419–430. doi:10.1021/tx5003782
22. Tarrado-Castellarnau M, de Atauri P, Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget. 2016;7(38):62726–62753. doi:10.18632/oncotarget.10911
23. Koltai T. Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther. 2016;9:6343–6360. doi:10.2147/ott.s115438
24. Nishi T, Forgac M. The vacuolar (H+)-ATPases-nature’s most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3(2):94–103. doi:10.1038/nrm729
25. Wang SP, Krits I, Bai S, Lee BS. Regulation of enhanced vacuolar H+-ATPase expression in macrophages. J Biol Chem. 2002;277(11):8827–8834. doi:10.1074/jbc.M111959200
26. Logan R, Kong AC, Axcell E, Krise JP. Amine-containing molecules and the induction of an expanded lysosomal volume phenotype: a structure-activity relationship study. J Pharm Sci. 2014;103(5):1572–1580. doi:10.1002/jps.23949
27. Funk RS, Krise JP. Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Mol Pharmaceutics. 2012;9(5):1384–1395. doi:10.1021/mp200641e
28. Ouar Z, Bens M, Vignes C, et al. Inhibitors of vacuolar H+-ATPase impair the preferential accumulation of daunomycin in lysosomes and reverse the resistance to anthracyclines in drug-resistant renal epithelial cells. Biochem J. 2003;370(Pt 1):185–193. doi:10.1042/bj20021411
29. Luciani F, Spada M, De Milito A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96(22):1702–1713. doi:10.1093/jnci/djh305
30. Wang BY, Zhang J, Wang JL, et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J Exp Clin Cancer Res. 2015;34:85. doi:10.1186/s13046-015-0194-x
31. Du W, Fan Y, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials. 2013;34(3):794–806. doi:10.1016/j.biomaterials.2012.10.003
32. Yan Z, Wang F, Wen Z, et al. LyP-1-conjugated PEGylated liposomes: a carrier system for targeted therapy of lymphatic metastatic tumor. J Control Release. 2012;157(1):118–125. doi:10.1016/j.jconrel.2011.07.034
33. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta Biochim Biophys Acta. 1993;1151(2):201–215. doi:10.1016/0005-2736(93)90105-9
34. Pinkerton NM, Grandeury A, Fisch A, Brozio J, Riebesehl BU, Prud’homme RK. Formation of stable nanocarriers by in situ ion pairing during block-copolymer-directed rapid precipitation. Mol Pharmaceutics. 2013;10(1):319–328. doi:10.1021/mp300452g
35. Chou LY, Ming K, Chan WC. Strategies for the intracellular delivery of nanoparticles. Chem Soc Rev. 2011;40(1):233–245. doi:10.1039/c0cs00003e
36. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nature Rev Mol Cell Biol. 2009;10(9):597–608. doi:10.1038/nrm2755
37. Park M, Salgado JM, Ostroff L, et al. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52(5):817–830. doi:10.1016/j.neuron.2006.09.040
38. Venkatachalam K, Wong CO, Zhu MX. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium. 2015;58(1):48–56. doi:10.1016/j.ceca.2014.10.008
39. Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978;75(7):3327–3331. doi:10.1073/pnas.75.7.3327
40. Liu W, Baker SS, Trinidad J, et al. Inhibition of lysosomal enzyme activities by proton pump inhibitors. J Gastroenterol. 2013;48(12):1343–1352. doi:10.1007/s00535-013-0774-5
41. Chen M, Huang SL, Zhang XQ, et al. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1alpha/P-gp and MRP1 signaling pathway in vitro and in vivo. J Cell Biochem. 2012;113(7):2474–2487. doi:10.1002/jcb.24122
42. Lindner K, Borchardt C, Schopp M, et al. Proton pump inhibitors (PPIs) impact on tumour cell survival, metastatic potential and chemotherapy resistance, and affect expression of resistance-relevant miRNAs in esophageal cancer. J Exp Clin Cancer Res. 2014;33:73. doi:10.1186/s13046-014-0073-x
43. Yu M, Lee C, Wang M, Tannock IF. Influence of the proton pump inhibitor lansoprazole on distribution and activity of doxorubicin in solid tumors. Cancer Sci. 2015;106(10):1438–1447. doi:10.1111/cas.12756
44. Wei X, Gao J, Zhan C, et al. Liposome-based glioma targeted drug delivery enabled by stable peptide ligands. J Control Release. 2015;218:13–21. doi:10.1016/j.jconrel.2015.09.059
45. Repnik U, Cesen MH, Turk B. The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol. 2013;5(1):a008755. doi:10.1101/cshperspect.a008755
46. Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25(1):99–112. doi:10.14670/HH-25.99
47. Piper RC, Luzio JP. Late endosomes: sorting and partitioning in multivesicular bodies. Traffic. 2001;2(9):612–621. doi:10.1034/j.1600-0854.2001.20904.x
48. Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi:10.1146/annurev-physiol-012110-142317
49. Ji T, Zhao Y, Ding Y, Nie G. Using functional nanomaterials to target and regulate the tumor microenvironment: diagnostic and therapeutic applications. Adv Mater. 2013;25(26):3508–3525. doi:10.1002/adma.201300299
50. Miura Y, Takenaka T, Toh K, et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood–brain tumor barrier. ACS Nano. 2013;7(10):8583–8592. doi:10.1021/nn402662d
51. Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188(6):759–768. doi:10.1083/jcb.200910104
52. Nel A, Ruoslahti E, Meng H. New insights into “permeability” as in the enhanced permeability and retention effect of cancer nanotherapeutics. ACS Nano. 2017;11(10):9567–9569. doi:10.1021/acsnano.7b07214
53. Chen Q, Osada K, Ge Z, et al. Polyplex micelle installing intracellular self-processing functionalities without free catiomers for safe and efficient systemic gene therapy through tumor vasculature targeting. Biomaterials. 2017;113:253–265. doi:10.1016/j.biomaterials.2016.10.042
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.